
MMP-3 Knockout Induces Global Transcriptional Changes and Reduces Cerebral Infarction in Both Male and Female Models of Ischemic Stroke


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Genetic Knockout of MMP-3 Reduces Infarct Volume in Stroke Mouse Brains
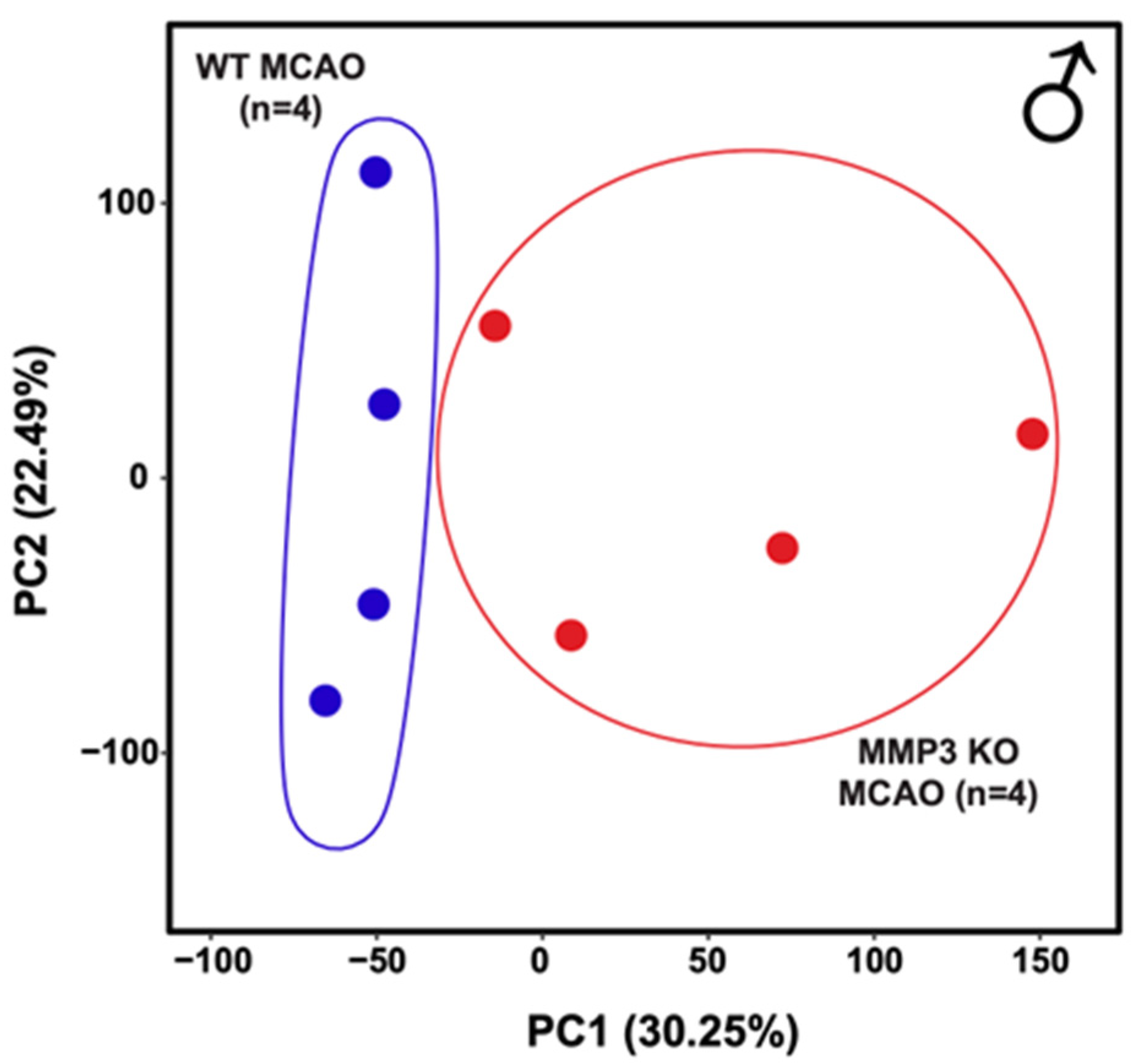
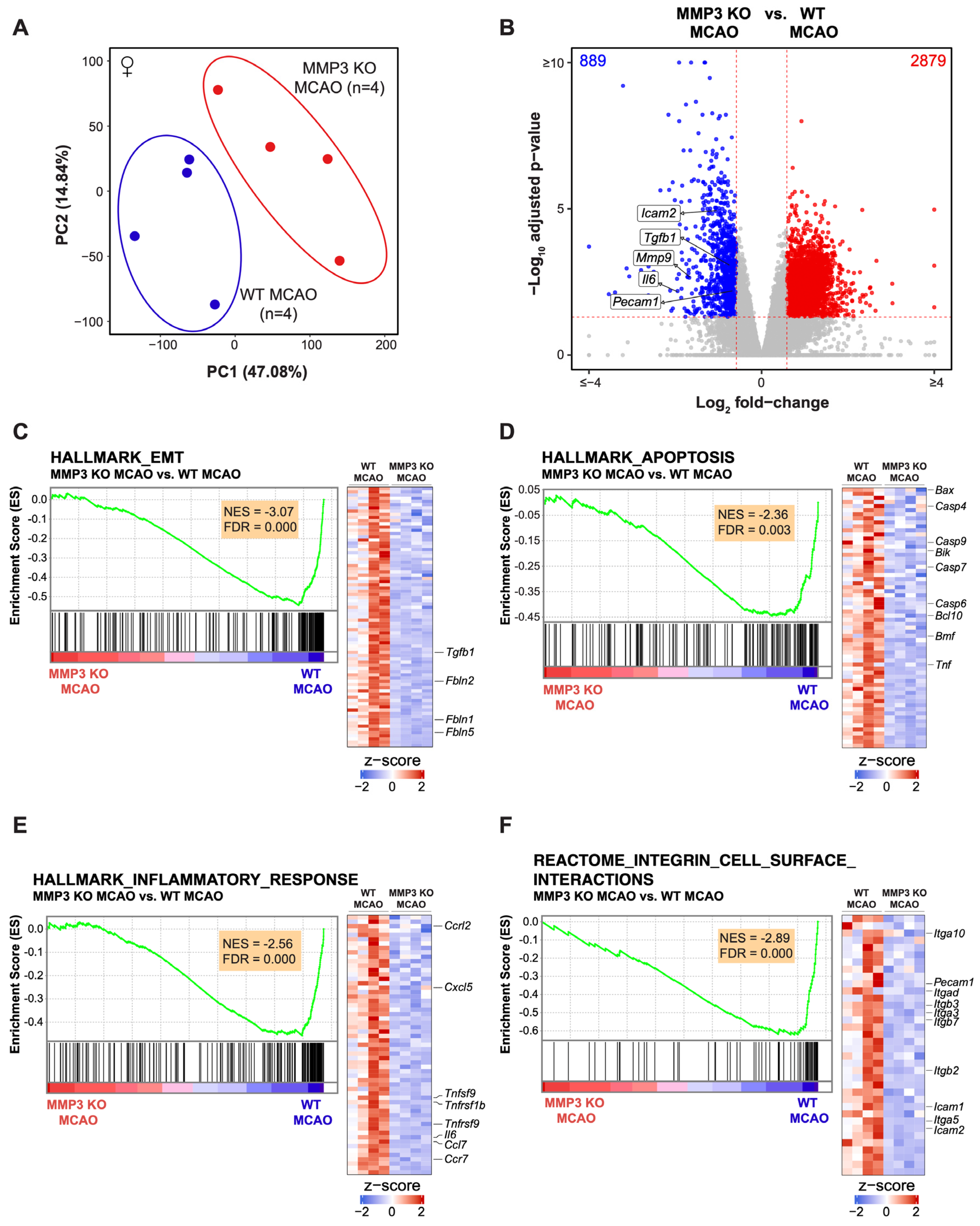
2.2. MMP-3 Deletion Induces Global Transcriptional Changes in the Brains of Male and Female Mice in the Subacute Stroke Phase
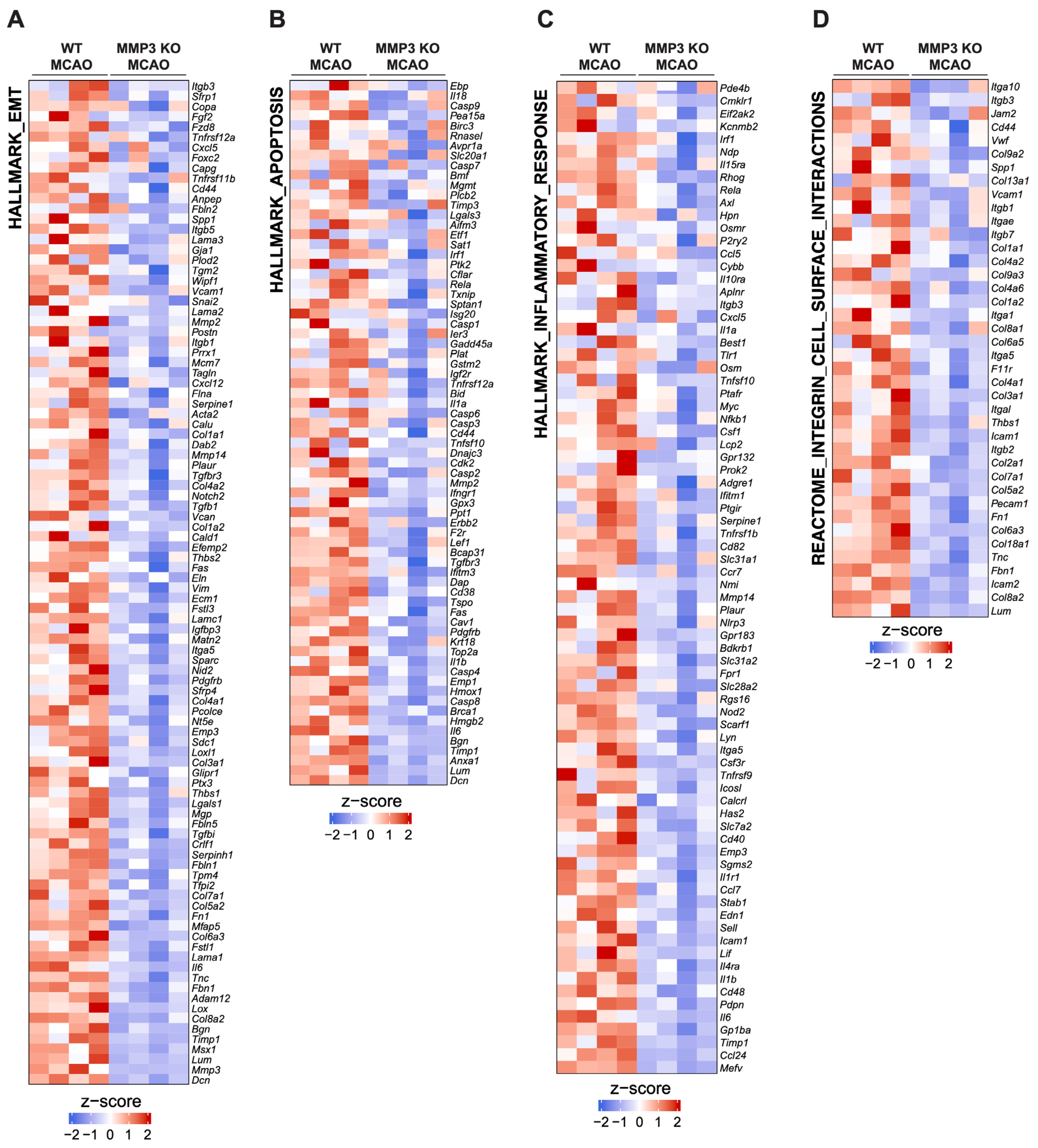
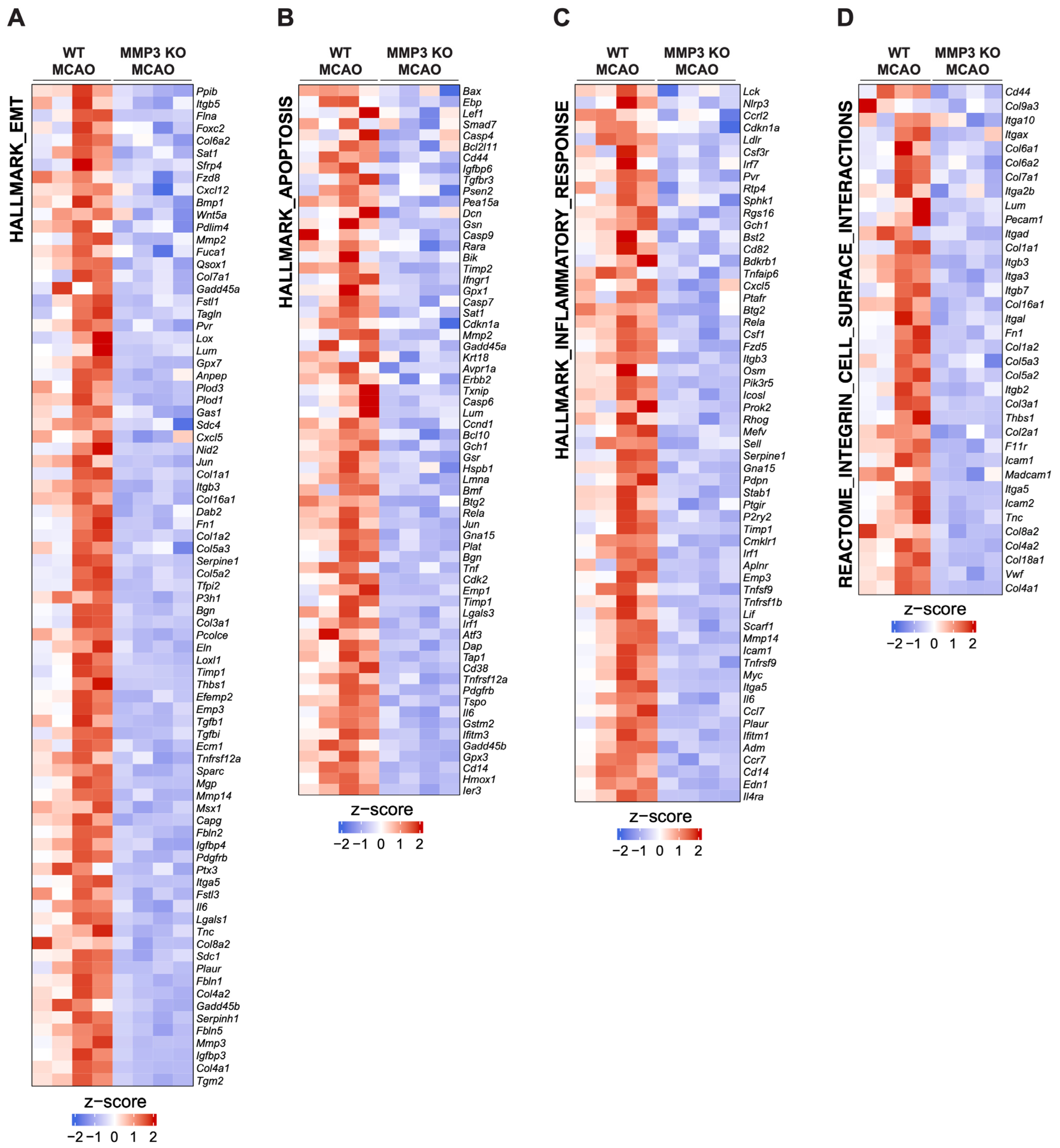
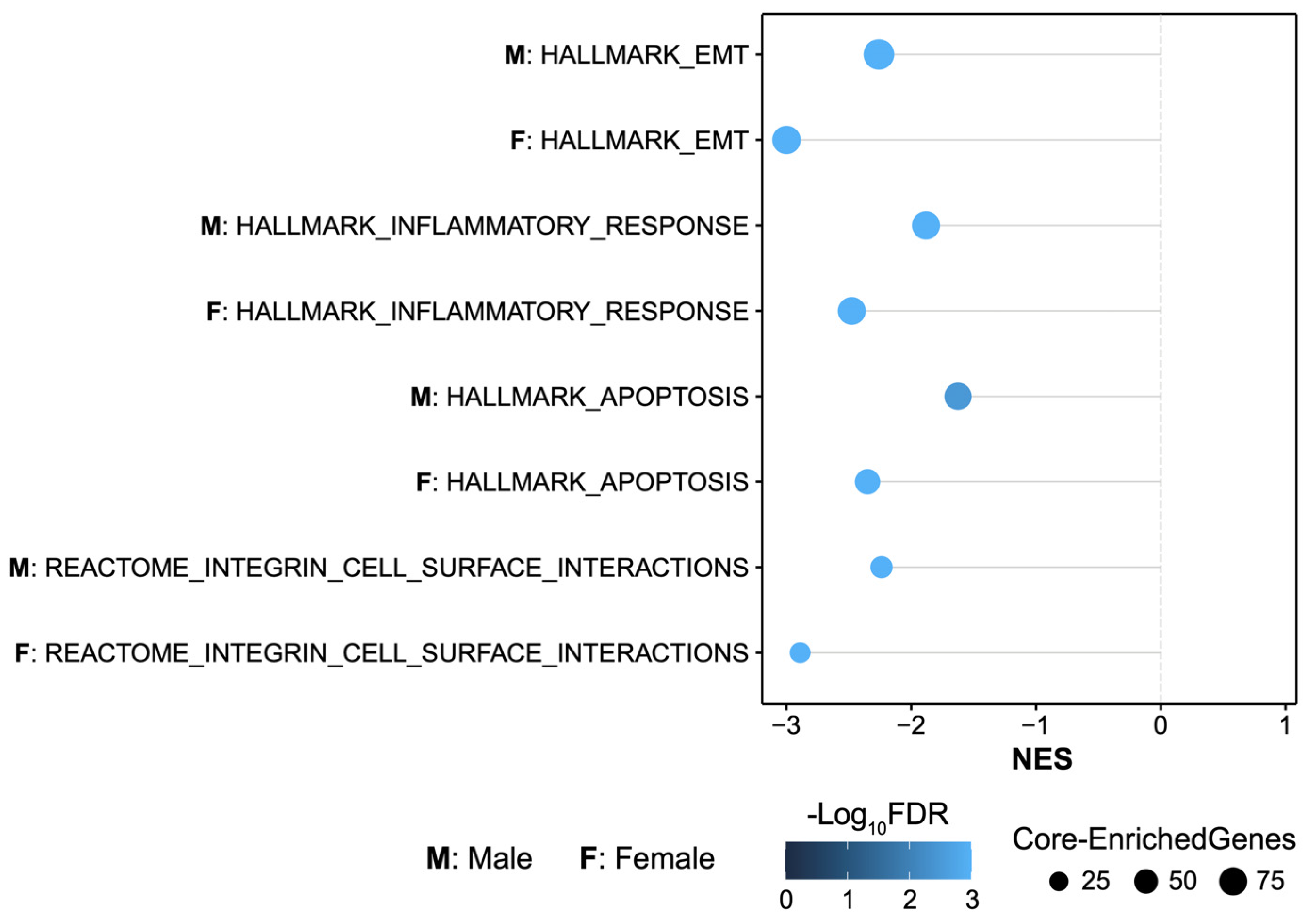
2.3. MMP-3 KO Decreases EMT Gene Expression in the Subacute Stroke Phase
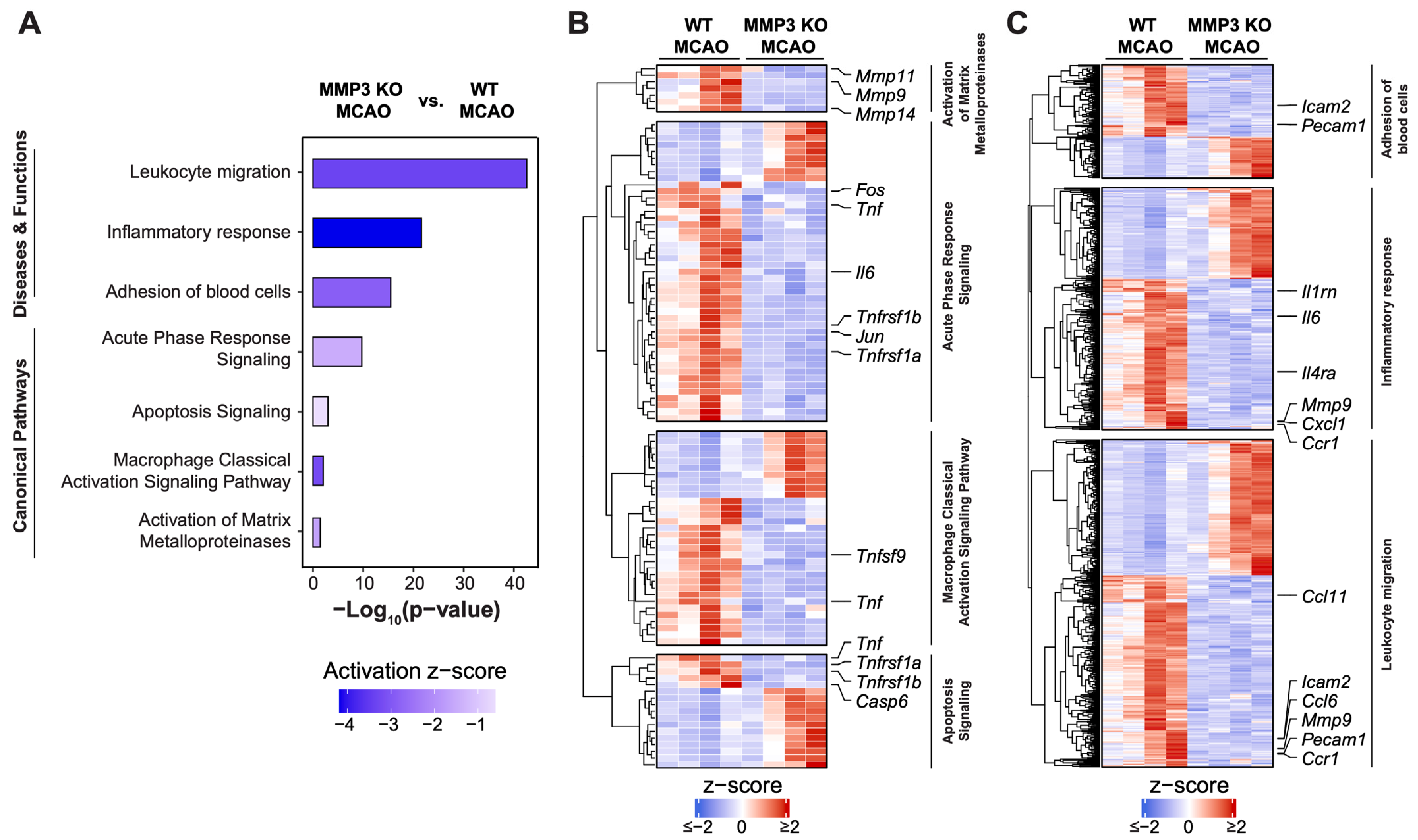
2.4. MMP-3 KO Attenuates Inflammatory Mediator Gene Expression in the Brain during the Subacute Stroke Phase
2.5. MMP-3 KO Reduces Apoptotic Gene Expression in the Brain during the Subacute Stroke Phase
2.6. MMP-3 KO Downregulates Expression of Genes Involved in Integrin Cell Surface Interactions
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Animal Model of Stroke
4.3. Quantification of Infarct Volume
4.4. RNA Sequencing (RNA-seq)
4.4.1. RNA-seq Data Processing
4.4.2. RNA-seq Quality Control and Quality Assurance (QC/QA)
4.4.3. Differential Gene Expression and Ingenuity Pathway Analysis (IPA)
4.4.4. Gene Set Enrichment Analysis (GSEA)
4.5. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
Appendix A

References
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef] [PubMed]
- Feigin, V.L.; Forouzanfar, M.H.; Krishnamurthi, R.; Mensah, G.A.; Connor, M.; Bennett, D.A.; Moran, A.E.; Sacco, R.L.; Anderson, L.; Truelsen, T.; et al. Global and regional burden of stroke during 1990–2010: Findings from the Global Burden of Disease Study 2010. Lancet 2014, 383, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Moskowitz, M.A.; Lo, E.H.; Iadecola, C. The Science of Stroke: Mechanisms in Search of Treatments. Neuron 2010, 67, 181–198. [Google Scholar] [CrossRef] [PubMed]
- Thom, T.; Haase, N.; Rosamond, W.; Howard, V.J.; Rumsfeld, J.; Manolio, T.; Zheng, Z.J.; Flegal, K.; O’Donnell, C.; Kittner, S.; et al. Heart disease and stroke statistics—2006 update: A report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2006, 113, e85–e151. [Google Scholar] [PubMed]
- Yamashita, T.; Deguchi, K.; Nagotani, S.; Abe, K. Vascular protection and restorative therapy in ischemic stroke. Cell Transplant. 2011, 20, 95–97. [Google Scholar] [CrossRef] [PubMed]
- Sporns, P.B.; Minnerup, J.; Warneke, N.; Dziewas, R.; Hanning, U.; Berkemeyer, S.; Zoubi, T.; Heindel, W.; Schwindt, W.; Niederstadt, T. Impact of the Implementation of Thrombectomy with Stent Retrievers on the Frequency of Hemicraniectomy in Patients with Acute Ischemic Stroke. Clin. Neuroradiol. 2017, 27, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Wahlgren, N.; Moreira, T.; Michel, P.; Steiner, T.; Jansen, O.; Cognard, C.; Mattle, H.P.; van Zwam, W.; Holmin, S.; Tatlisumak, T.; et al. Mechanical thrombectomy in acute ischemic stroke: Consensus statement by ESO-Karolinska Stroke Update 2014/2015, supported by ESO, ESMINT, ESNR and EAN. Int. J. Stroke 2016, 11, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Roth, J.M. Recombinant tissue plasminogen activator for the treatment of acute ischemic stroke. Bayl. Univ. Med. Cent. Proc. 2011, 24, 257–259. [Google Scholar] [CrossRef]
- Kilic, E.; Bahr, M.; Hermann, D.M. Effects of recombinant tissue plasminogen activator after intraluminal thread occlusion in mice: Role of hemodynamic alterations. Stroke 2001, 32, 2641–2647. [Google Scholar] [CrossRef]
- Sumii, T.; Lo, E.H. Involvement of matrix metalloproteinase in thrombolysis-associated hemorrhagic transformation after embolic focal ischemia in rats. Stroke 2002, 33, 831–836. [Google Scholar] [CrossRef]
- Begley, D.J.; Brightman, M.W. Structural and functional aspects of the blood-brain barrier. Pept. Transp. Deliv. Into Cent. Nerv. Syst. 2003, 61, 39–78. [Google Scholar]
- Abbott, N.J. Astrocyte-endothelial interactions and blood-brain barrier permeability. J. Anat. 2002, 200, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Saunders, N.R.; Dreifuss, J.J.; Dziegielewska, K.M.; Johansson, P.A.; Habgood, M.D.; Mollgard, K.; Bauer, H.C. The rights and wrongs of blood-brain barrier permeability studies: A walk through 100 years of history. Front. Neurosci. 2014, 8, 404. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [PubMed]
- Lochhead, J.J.; McCaffrey, G.; Quigley, C.E.; Finch, J.; DeMarco, K.M.; Nametz, N.; Davis, T.P. Oxidative stress increases blood-brain barrier permeability and induces alterations in occludin during hypoxia-reoxygenation. J. Cereb. Blood Flow. Metab. 2010, 30, 1625–1636. [Google Scholar] [CrossRef] [PubMed]
- Yemisci, M.; Gursoy-Ozdemir, Y.; Vural, A.; Can, A.; Topalkara, K.; Dalkara, T. Pericyte contraction induced by oxidative-nitrative stress impairs capillary reflow despite successful opening of an occluded cerebral artery. Nat. Med. 2009, 15, 1031–1037. [Google Scholar] [CrossRef] [PubMed]
- Kuroiwa, T.; Ting, P.; Martinez, H.; Klatzo, I. The biphasic opening of the blood-brain barrier to proteins following temporary middle cerebral artery occlusion. Acta Neuropathol. 1985, 68, 122–129. [Google Scholar] [CrossRef]
- Shi, Y.; Zhang, L.; Pu, H.; Mao, L.; Hu, X.; Jiang, X.; Xu, N.; Stetler, R.A.; Zhang, F.; Liu, X.; et al. Rapid endothelial cytoskeletal reorganization enables early blood-brain barrier disruption and long-term ischaemic reperfusion brain injury. Nat. Commun. 2016, 7, 10523. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Rosenberg, G.A. Blood-brain barrier breakdown in acute and chronic cerebrovascular disease. Stroke 2011, 42, 3323–3328. [Google Scholar] [CrossRef]
- Defilippi, P.; Silengo, L.; Tarone, G. Alpha 6.beta 1 integrin (laminin receptor) is down-regulated by tumor necrosis factor alpha and interleukin-1 beta in human endothelial cells. J. Biol. Chem. 1992, 267, 18303–18307. [Google Scholar] [CrossRef]
- Gasche, Y.; Fujimura, M.; Morita-Fujimura, Y.; Copin, J.C.; Kawase, M.; Massengale, J.; Chan, P.H. Early appearance of activated matrix metalloproteinase-9 after focal cerebral ischemia in mice: A possible role in blood-brain barrier dysfunction. J. Cereb. Blood Flow. Metab. 1999, 19, 1020–1028. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Rutledge, B.J.; Gu, L.; Fiorillo, J.; Lukacs, N.W.; Kunkel, S.L.; North, R.; Gerard, C.; Rollins, B.J. Abnormalities in monocyte recruitment and cytokine expression in monocyte chemoattractant protein 1-deficient mice. J. Exp. Med. 1998, 187, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Pettigrew, L.C.; Kindy, M.S.; Scheff, S.; Springer, J.E.; Kryscio, R.J.; Li, Y.; Grass, D.S. Focal cerebral ischemia in the TNFalpha-transgenic rat. J. Neuroinflamm. 2008, 5, 47. [Google Scholar] [CrossRef] [PubMed]
- Rossi, B.; Angiari, S.; Zenaro, E.; Budui, S.L.; Constantin, G. Vascular inflammation in central nervous system diseases: Adhesion receptors controlling leukocyte-endothelial interactions. J. Leukoc. Biol. 2011, 89, 539–556. [Google Scholar] [CrossRef] [PubMed]
- Wolpe, S.D.; Davatelis, G.; Sherry, B.; Beutler, B.; Hesse, D.G.; Nguyen, H.T.; Moldawer, L.L.; Nathan, C.F.; Lowry, S.F.; Cerami, A. Macrophages secrete a novel heparin-binding protein with inflammatory and neutrophil chemokinetic properties. J. Exp. Med. 1988, 167, 570–581. [Google Scholar] [CrossRef]
- Chin, J.R.; Murphy, G.; Werb, Z. Stromelysin, a connective tissue-degrading metalloendopeptidase secreted by stimulated rabbit synovial fibroblasts in parallel with collagenase. Biosynthesis, isolation, characterization, and substrates. J. Biol. Chem. 1985, 260, 12367–12376. [Google Scholar] [CrossRef]
- Klein, T.; Bischoff, R. Physiology and pathophysiology of matrix metalloproteases. Amino Acids 2011, 41, 271–290. [Google Scholar] [CrossRef] [PubMed]
- Hahn-Dantona, E.; Ramos-DeSimone, N.; Sipley, J.; Nagase, H.; French, D.L.; Quigley, J.P. Activation of proMMP-9 by a plasmin/MMP-3 cascade in a tumor cell model. Regulation by tissue inhibitors of metalloproteinases. Ann. N. Y. Acad. Sci. 1999, 878, 372–387. [Google Scholar] [CrossRef]
- Candelario-Jalil, E.; Yang, Y.; Rosenberg, G.A. Diverse roles of matrix metalloproteinases and tissue inhibitors of metalloproteinases in neuroinflammation and cerebral ischemia. Neuroscience 2009, 158, 983–994. [Google Scholar] [CrossRef]
- Ramos-DeSimone, N.; Hahn-Dantona, E.; Sipley, J.; Nagase, H.; French, D.L.; Quigley, J.P. Activation of matrix metalloproteinase-9 (MMP-9) via a converging plasmin/stromelysin-1 cascade enhances tumor cell invasion. J. Biol. Chem. 1999, 274, 13066–13076. [Google Scholar] [CrossRef]
- Suzuki, Y.; Nagai, N.; Umemura, K.; Collen, D.; Lijnen, H.R. Stromelysin-1 (MMP-3) is critical for intracranial bleeding after t-PA treatment of stroke in mice. J. Thromb. Haemost. 2007, 5, 1732–1739. [Google Scholar] [CrossRef] [PubMed]
- Hafez, S.; Abdelsaid, M.; El-Shafey, S.; Johnson, M.H.; Fagan, S.C.; Ergul, A. Matrix Metalloprotease 3 Exacerbates Hemorrhagic Transformation and Worsens Functional Outcomes in Hyperglycemic Stroke. Stroke 2016, 47, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Hamblin, M.H.; Murad, R.; Yin, J.; Vallim, G.; Lee, J.P. Modulation of gene expression on a transcriptome-wide level following human neural stem cell transplantation in aged mouse stroke brains. Exp. Neurol. 2022, 347, 113913. [Google Scholar] [CrossRef] [PubMed]
- Sternlicht, M.D.; Werb, Z. How matrix metalloproteinases regulate cell behavior. Annu. Rev. Cell Dev. Biol. 2001, 17, 463–516. [Google Scholar] [CrossRef] [PubMed]
- Sternlicht, M.D.; Lochter, A.; Sympson, C.J.; Huey, B.; Rougier, J.P.; Gray, J.W.; Pinkel, D.; Bissell, M.J.; Werb, Z. The stromal proteinase MMP3/stromelysin-1 promotes mammary carcinogenesis. Cell 1999, 98, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Lochter, A.; Srebrow, A.; Sympson, C.J.; Terracio, N.; Werb, Z.; Bissell, M.J. Misregulation of stromelysin-1 expression in mouse mammary tumor cells accompanies acquisition of stromelysin-1-dependent invasive properties. J. Biol. Chem. 1997, 272, 5007–5015. [Google Scholar] [CrossRef] [PubMed]
- Lochter, A.; Galosy, S.; Muschler, J.; Freedman, N.; Werb, Z.; Bissell, M.J. Matrix metalloproteinase stromelysin-1 triggers a cascade of molecular alterations that leads to stable epithelial-to-mesenchymal conversion and a premalignant phenotype in mammary epithelial cells. J. Cell Biol. 1997, 139, 1861–1872. [Google Scholar] [CrossRef] [PubMed]
- Potenta, S.; Zeisberg, E.; Kalluri, R. The role of endothelial-to-mesenchymal transition in cancer progression. Br. J. Cancer 2008, 99, 1375–1379. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Yang, H.; Wan, Y.; Yang, S.; Wu, J.; Chen, S.; Li, Y.; Jin, H.; He, Q.; Zhu, D.Y.; et al. Endothelial ETS1 inhibition exacerbate blood-brain barrier dysfunction in multiple sclerosis through inducing endothelial-to-mesenchymal transition. Cell Death Dis. 2022, 13, 462. [Google Scholar] [CrossRef]
- Derada Troletti, C.; Fontijn, R.D.; Gowing, E.; Charabati, M.; van Het Hof, B.; Didouh, I.; van der Pol, S.M.A.; Geerts, D.; Prat, A.; van Horssen, J.; et al. Inflammation-induced endothelial to mesenchymal transition promotes brain endothelial cell dysfunction and occurs during multiple sclerosis pathophysiology. Cell Death Dis. 2019, 10, 45. [Google Scholar] [CrossRef]
- Chen, D.; Li, L.; Wang, Y.; Xu, R.; Peng, S.; Zhou, L.; Deng, Z. Ischemia-reperfusion injury of brain induces endothelial-mesenchymal transition and vascular fibrosis via activating let-7i/TGF-βR1 double-negative feedback loop. Faseb J 2020, 34, 7178–7191. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Yang, G.; Li, G. Inflammatory mechanisms in ischemic stroke: Role of inflammatory cells. J. Leukoc. Biol. 2010, 87, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Radak, D.; Katsiki, N.; Resanovic, I.; Jovanovic, A.; Sudar-Milovanovic, E.; Zafirovic, S.; Mousad, S.A.; Isenovic, E.R. Apoptosis and Acute Brain Ischemia in Ischemic Stroke. Curr. Vasc. Pharmacol. 2017, 15, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Edwards, D.N.; Bix, G.J. Roles of blood-brain barrier integrins and extracellular matrix in stroke. Am. J. Physiol. Cell Physiol. 2019, 316, C252–C263. [Google Scholar] [CrossRef] [PubMed]
- Takada, Y.; Ye, X.; Simon, S. The integrins. Genome Biol. 2007, 8, 215. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.J.; Wu, X.; Nurkiewicz, T.R.; Kawasaki, J.; Davis, G.E.; Hill, M.A.; Meininger, G.A. Integrins and mechanotransduction of the vascular myogenic response. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H1427-33. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Butler, J.P.; Ingber, D.E. Mechanotransduction across the cell surface and through the cytoskeleton. Science 1993, 260, 1124–1127. [Google Scholar] [CrossRef] [PubMed]
- Obermeier, B.; Daneman, R.; Ransohoff, R.M. Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 2013, 19, 1584–1596. [Google Scholar] [CrossRef] [PubMed]
- Rochfort, K.D.; Cummins, P.M. The blood-brain barrier endothelium: A target for pro-inflammatory cytokines. Biochem. Soc. Trans. 2015, 43, 702–706. [Google Scholar] [CrossRef]
- Pan, W.; Stone, K.P.; Hsuchou, H.; Manda, V.K.; Zhang, Y.; Kastin, A.J. Cytokine signaling modulates blood-brain barrier function. Curr. Pharm. Des. 2011, 17, 3729–3740. [Google Scholar] [CrossRef]
- Cuadrado, E.; Rosell, A.; Penalba, A.; Slevin, M.; Alvarez-Sabin, J.; Ortega-Aznar, A.; Montaner, J. Vascular MMP-9/TIMP-2 and neuronal MMP-10 up-regulation in human brain after stroke: A combined laser microdissection and protein array study. J. Proteome Res. 2009, 8, 3191–3197. [Google Scholar] [CrossRef] [PubMed]
- Abdul, Y.; Jamil, S.; Li, W.; Ergul, A. Cerebral microvascular matrix metalloproteinase-3 (MMP3) contributes to vascular injury after stroke in female diabetic rats. Neurochem. Int. 2023, 162, 105462. [Google Scholar] [CrossRef] [PubMed]
- Engel, O.; Kolodziej, S.; Dirnagl, U.; Prinz, V. Modeling Stroke in Mice—Middle Cerebral Artery Occlusion with the Filament Model. J. Vis. Exp. 2011, 2423. [Google Scholar]
- Wei, N.; Xiao, L.; Xue, R.; Zhang, D.; Zhou, J.; Ren, H.; Guo, S.; Xu, J. MicroRNA-9 Mediates the Cell Apoptosis by Targeting Bcl2l11 in Ischemic Stroke. Mol. Neurobiol. 2016, 53, 6809–6817. [Google Scholar] [CrossRef] [PubMed]
- Kono, H.; Rock, K.L. How dying cells alert the immune system to danger. Nat. Rev. Immunol. 2008, 8, 279–289. [Google Scholar] [CrossRef]
- Qiu, Y.-M.; Zhang, C.-L.; Chen, A.-Q.; Wang, H.-L.; Zhou, Y.-F.; Li, Y.-N.; Hu, B. Immune Cells in the BBB Disruption After Acute Ischemic Stroke: Targets for Immune Therapy? Front. Immunol. 2021, 12, 678744. [Google Scholar] [CrossRef] [PubMed]
- Gülke, E.; Gelderblom, M.; Magnus, T. Danger signals in stroke and their role on microglia activation after ischemia. Ther. Adv. Neurol. Disord. 2018, 11, 1756286418774254. [Google Scholar] [CrossRef] [PubMed]
- Murray, K.N.; Buggey, H.F.; Denes, A.; Allan, S.M. Systemic immune activation shapes stroke outcome. Mol. Cell Neurosci. 2013, 53, 14–25. [Google Scholar] [CrossRef]
- Shichita, T.; Ito, M.; Morita, R.; Komai, K.; Noguchi, Y.; Ooboshi, H.; Koshida, R.; Takahashi, S.; Kodama, T.; Yoshimura, A. MAFB prevents excess inflammation after ischemic stroke by accelerating clearance of damage signals through MSR1. Nat. Med. 2017, 23, 723–732. [Google Scholar] [CrossRef]
- Baumann, H.; Gauldie, J. The acute phase response. Immunol. Today 1994, 15, 74–80. [Google Scholar] [CrossRef]
- Dziedzic, T. Clinical significance of acute phase reaction in stroke patients. Front. Biosci. 2008, 13, 2922–2927. [Google Scholar] [CrossRef]
- Shaafi, S.; Sharifipour, E.; Rahmanifar, R.; Hejazi, S.; Andalib, S.; Nikanfar, M.; Baradarn, B.; Mehdizadeh, R. Interleukin-6, a reliable prognostic factor for ischemic stroke. Iran. J. Neurol. 2014, 13, 70–76. [Google Scholar] [PubMed]
- Tariq, M.B.; Lee, J.; McCullough, L.D. Sex differences in the inflammatory response to stroke. Semin. Immunopathol. 2023, 45, 295–313. [Google Scholar] [CrossRef]
- Aktas, O.; Ullrich, O.; Infante-Duarte, C.; Nitsch, R.; Zipp, F. Neuronal damage in brain inflammation. Arch. Neurol. 2007, 64, 185–189. [Google Scholar] [CrossRef]
- Rodrigues, S.F.; Granger, D.N. Blood cells and endothelial barrier function. Tissue Barriers 2015, 3, e978720. [Google Scholar] [CrossRef] [PubMed]
- Shulman, Z.; Shinder, V.; Klein, E.; Grabovsky, V.; Yeger, O.; Geron, E.; Montresor, A.; Bolomini-Vittori, M.; Feigelson, S.W.; Kirchhausen, T.; et al. Lymphocyte crawling and transendothelial migration require chemokine triggering of high-affinity LFA-1 integrin. Immunity 2009, 30, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Schenkel, A.R.; Chew, T.W.; Muller, W.A. Platelet endothelial cell adhesion molecule deficiency or blockade significantly reduces leukocyte emigration in a majority of mouse strains. J. Immunol. 2004, 173, 6403–6408. [Google Scholar] [CrossRef] [PubMed]
- Zera, K.A.; Buckwalter, M.S. The Local and Peripheral Immune Responses to Stroke: Implications for Therapeutic Development. Neurotherapeutics 2020, 17, 414–435. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.; de Hoog, L.; Bix, G.J. Mice deficient in endothelial α5 integrin are profoundly resistant to experimental ischemic stroke. J. Cereb. Blood Flow. Metab. 2017, 37, 85–96. [Google Scholar] [CrossRef]
- Okada, Y.; Copeland, B.R.; Hamann, G.F.; Koziol, J.A.; Cheresh, D.A.; del Zoppo, G.J. Integrin alphavbeta3 is expressed in selected microvessels after focal cerebral ischemia. Am. J. Pathol. 1996, 149, 37–44. [Google Scholar]
- Alghisi, G.C.; Ponsonnet, L.; Rüegg, C. The Integrin Antagonist Cilengitide Activates αVβ3, Disrupts VE-Cadherin Localization at Cell Junctions and Enhances Permeability in Endothelial Cells. PLoS ONE 2009, 4, e4449. [Google Scholar] [CrossRef] [PubMed]
- Bendeck, M.P.; Irvin, C.; Reidy, M.; Smith, L.; Mulholland, D.; Horton, M.; Giachelli, C.M. Smooth Muscle Cell Matrix Metalloproteinase Production Is Stimulated via αvβ3 Integrin. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1467–1472. [Google Scholar] [CrossRef] [PubMed]
- Kovacic, J.C.; Dimmeler, S.; Harvey, R.P.; Finkel, T.; Aikawa, E.; Krenning, G.; Baker, A.H. Endothelial to Mesenchymal Transition in Cardiovascular Disease: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 190–209. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Zhang, Y.; Han, B.; Yang, L.; Chen, X.; Huang, R.; Wu, F.; Chao, J.; Liu, P.; Hu, G.; et al. Circular RNA DLGAP4 Ameliorates Ischemic Stroke Outcomes by Targeting miR-143 to Regulate Endothelial-Mesenchymal Transition Associated with Blood–Brain Barrier Integrity. J. Neurosci. 2018, 38, 32–50. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, D.; Kancharla, S.; Kolli, P.; Sharma, A.K.; Singh, S.; Kumar, S.; Mohanty, A.K.; Jena, M.K. Role of Fibulins in Embryonic Stage Development and Their Involvement in Various Diseases. Biomolecules 2021, 11, 685. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, T.; Kubota, S.; Kawata, K.; Mukudai, Y.; Uehara, J.; Ohgawara, T.; Ibaragi, S.; Sasaki, A.; Kuboki, T.; Takigawa, M. Novel transcription-factor-like function of human matrix metalloproteinase 3 regulating the CTGF/CCN2 gene. Mol. Cell Biol. 2008, 28, 2391–2413. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Wong, S.; Snyder, E.Y.; Hamblin, M.H.; Lee, J.P. Human neural stem cells rapidly ameliorate symptomatic inflammation in early-stage ischemic-reperfusion cerebral injury. Stem Cell Res. Ther. 2014, 5, 129. [Google Scholar] [CrossRef] [PubMed]
- Eckert, A.; Huang, L.; Gonzalez, R.; Kim, H.S.; Hamblin, M.H.; Lee, J.P. Bystander Effect Fuels Human Induced Pluripotent Stem Cell-Derived Neural Stem Cells to Quickly Attenuate Early Stage Neurological Deficits After Stroke. Stem Cells Transl. Med. 2015, 4, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Boese, A.C.; Eckert, A.; Hamblin, M.H.; Lee, J.P. Human neural stem cells improve early stage stroke outcome in delayed tissue plasminogen activator-treated aged stroke brains. Exp. Neurol. 2020, 329, 113275. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Ewels, P.; Magnusson, M.; Lundin, S.; Kaller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamblin, M.H.; Boese, A.C.; Murad, R.; Lee, J.-P. MMP-3 Knockout Induces Global Transcriptional Changes and Reduces Cerebral Infarction in Both Male and Female Models of Ischemic Stroke. Int. J. Mol. Sci. 2024, 25, 7383. https://doi.org/10.3390/ijms25137383
Hamblin MH, Boese AC, Murad R, Lee J-P. MMP-3 Knockout Induces Global Transcriptional Changes and Reduces Cerebral Infarction in Both Male and Female Models of Ischemic Stroke. International Journal of Molecular Sciences. 2024; 25(13):7383. https://doi.org/10.3390/ijms25137383
Chicago/Turabian StyleHamblin, Milton H., Austin C. Boese, Rabi Murad, and Jean-Pyo Lee. 2024. "MMP-3 Knockout Induces Global Transcriptional Changes and Reduces Cerebral Infarction in Both Male and Female Models of Ischemic Stroke" International Journal of Molecular Sciences 25, no. 13: 7383. https://doi.org/10.3390/ijms25137383
APA StyleHamblin, M. H., Boese, A. C., Murad, R., & Lee, J.-P. (2024). MMP-3 Knockout Induces Global Transcriptional Changes and Reduces Cerebral Infarction in Both Male and Female Models of Ischemic Stroke. International Journal of Molecular Sciences, 25(13), 7383. https://doi.org/10.3390/ijms25137383