
Catalytic Stability of S-1-(4-Hydroxyphenyl)-Ethanol Dehydrogenase from Aromatoleum aromaticum


Abstract
1. Introduction
2. Results and Discussion
2.1. Synthesis of Chiral Alcohols with Pure S-HPED
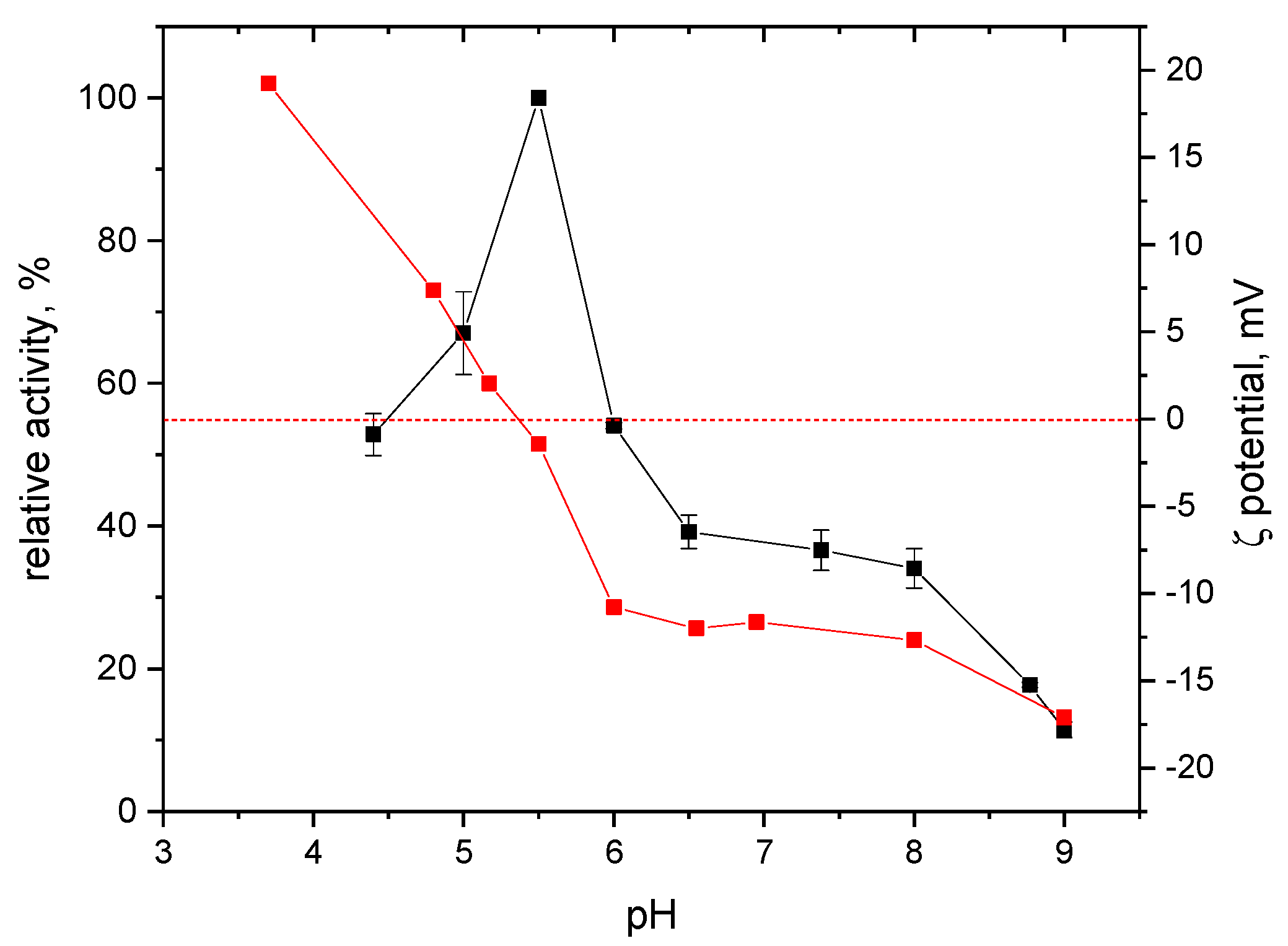
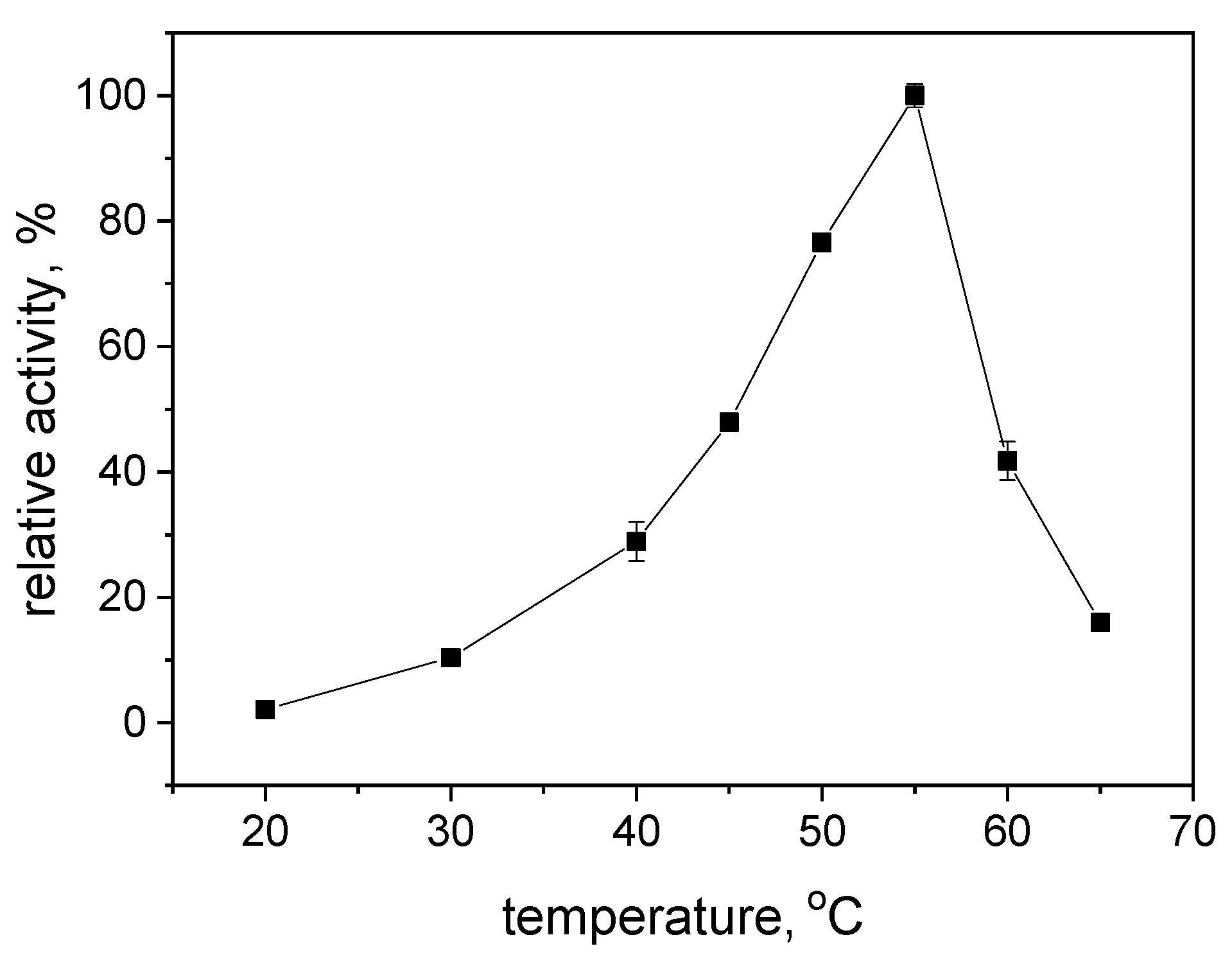
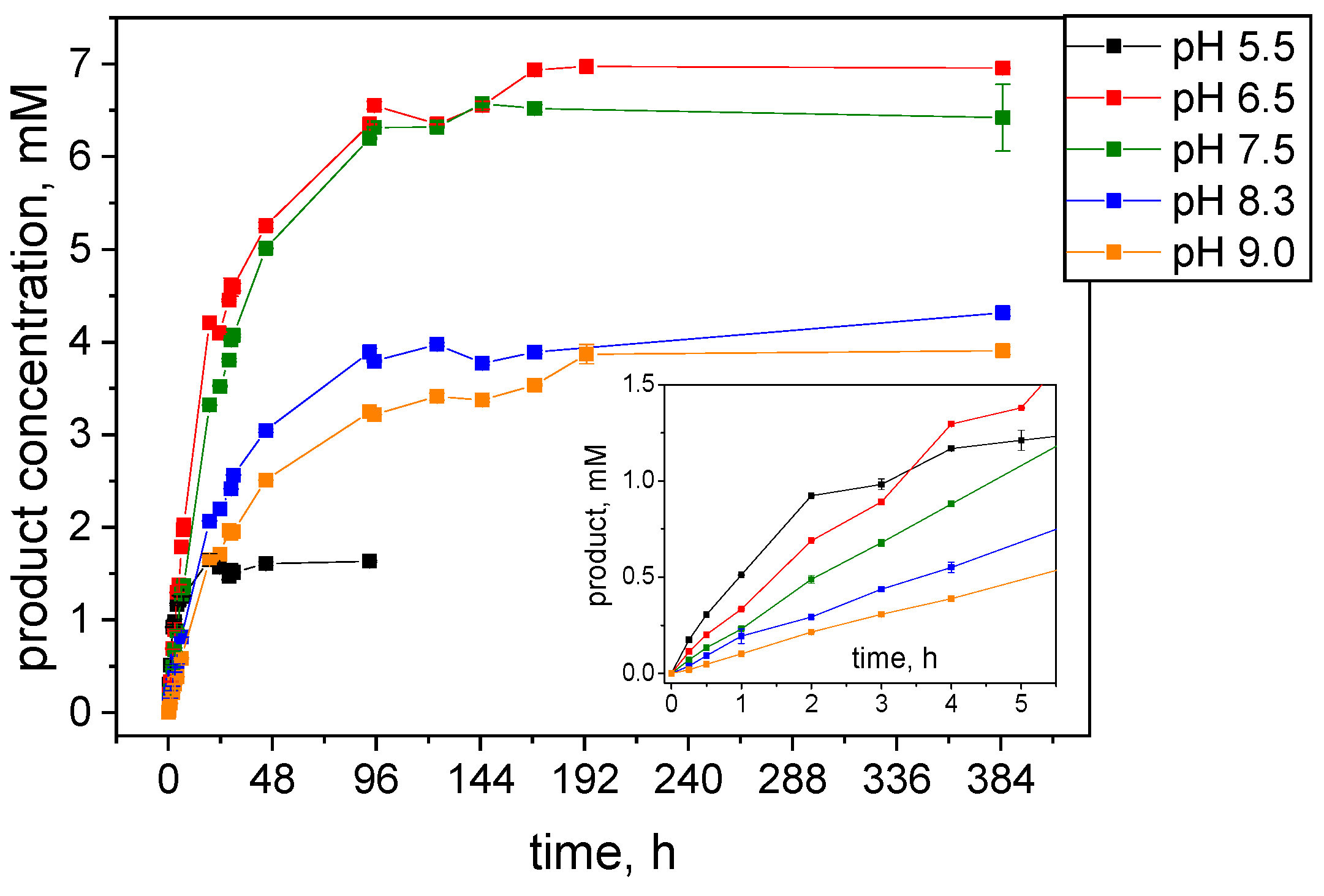
2.2. Effects of pH and Reaction Temperature on the Activity of S-HPED
2.2.1. Impact of Glucose
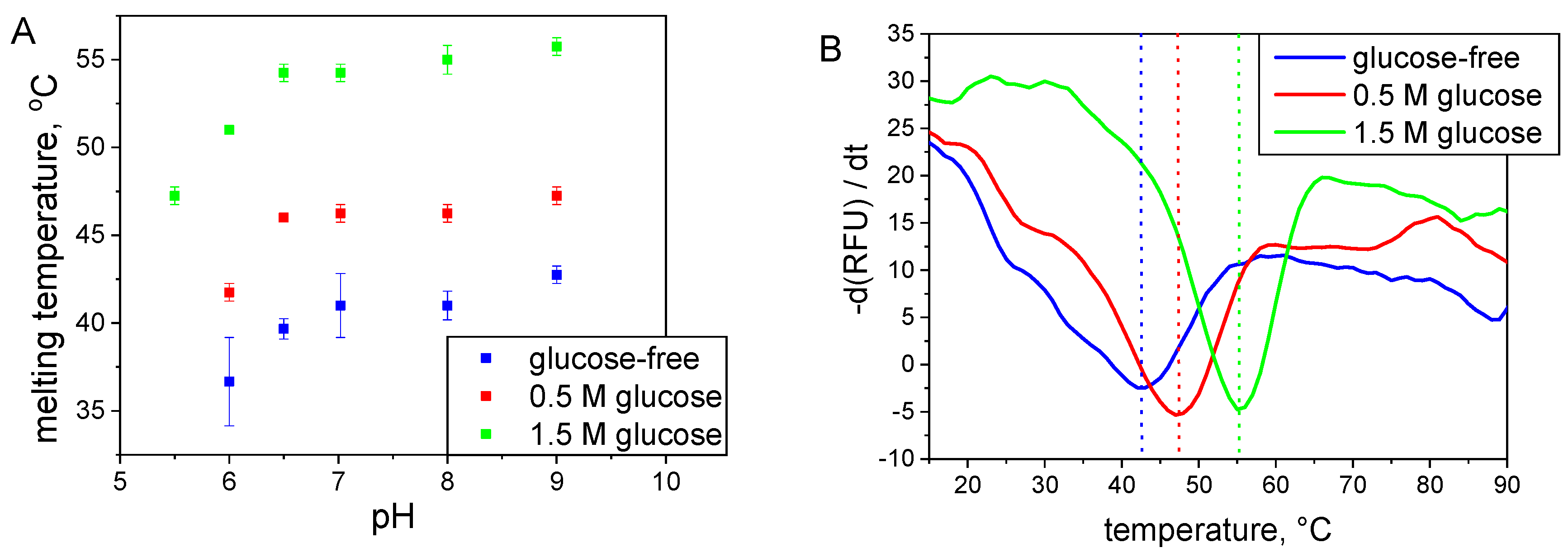
2.2.2. Thermal Shift Assay
2.3. Catalytic Stability and Aggregation of S-HPED at Elevated Temperatures
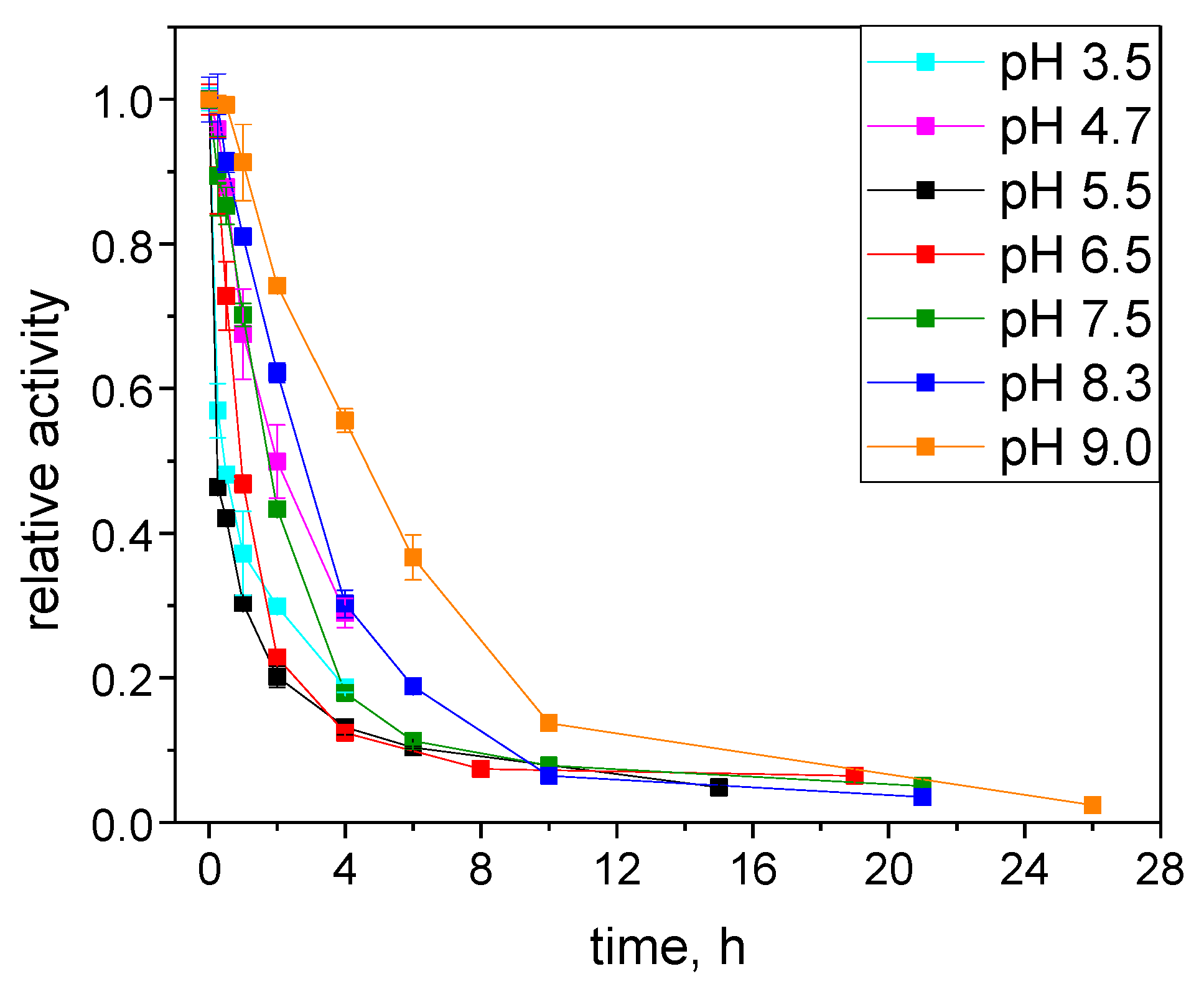
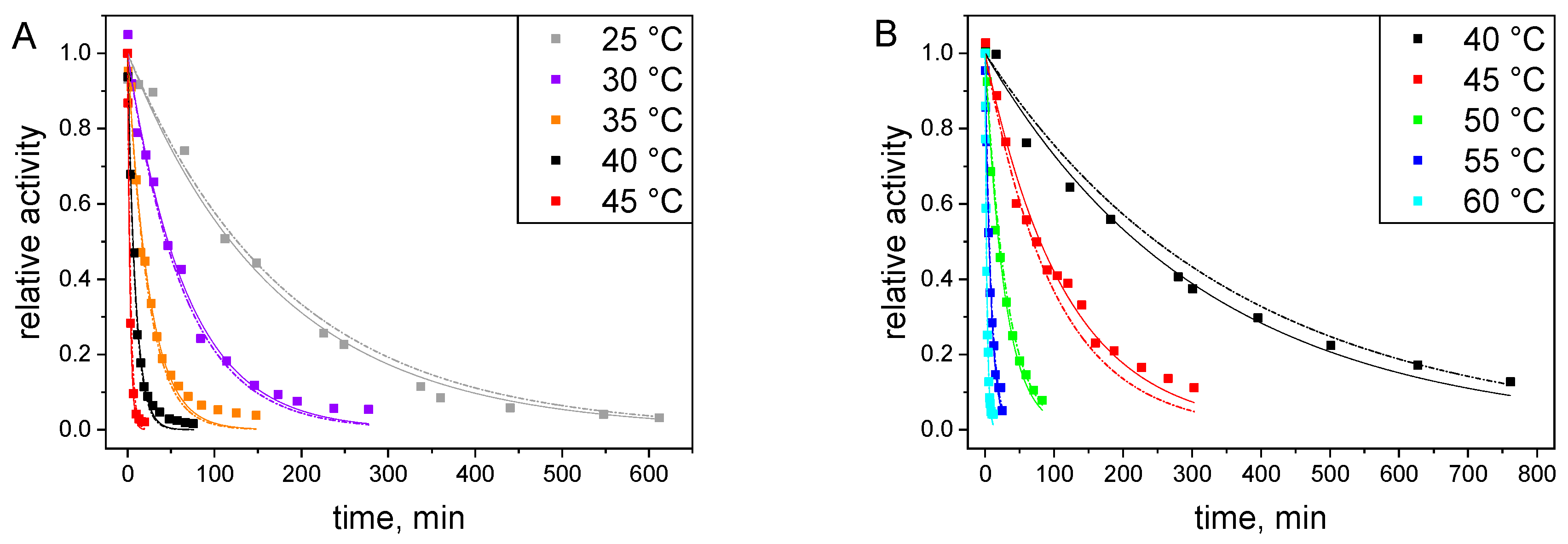
2.3.1. Catalytic Stability Study under Storage Conditions
2.3.2. Catalytic Stability Study under Reactor Conditions
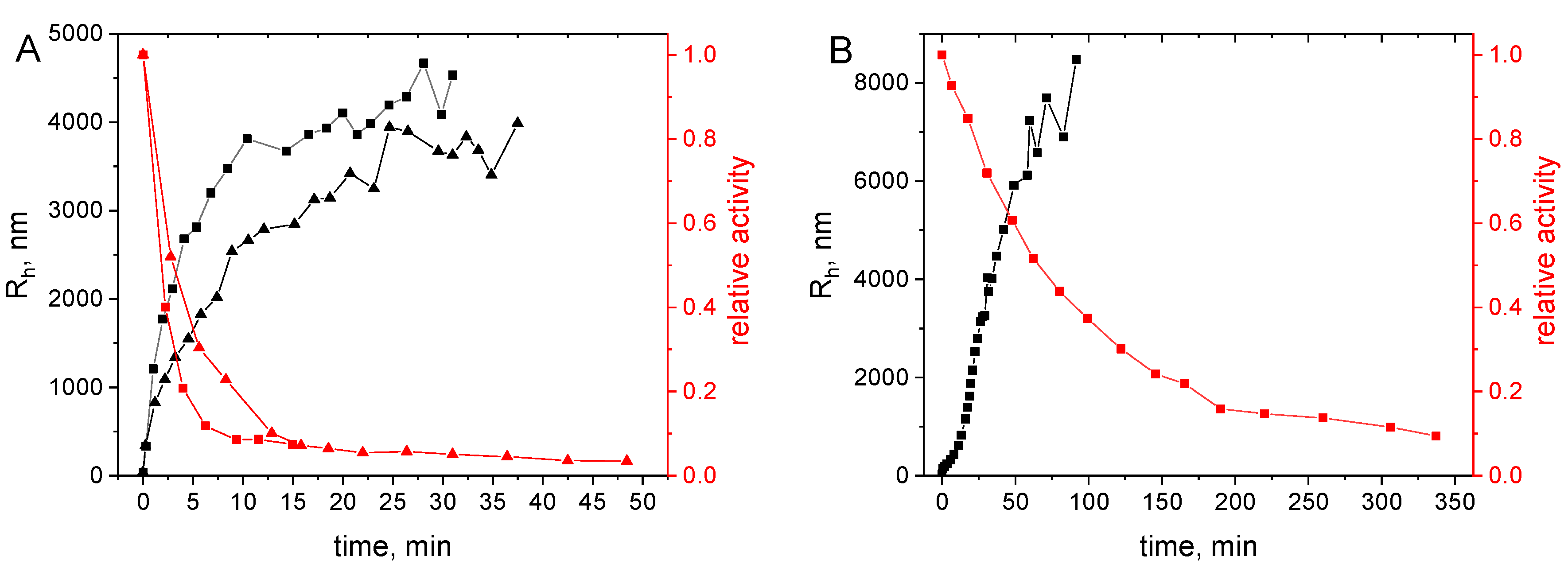
2.3.3. Aggregation under Storage Conditions
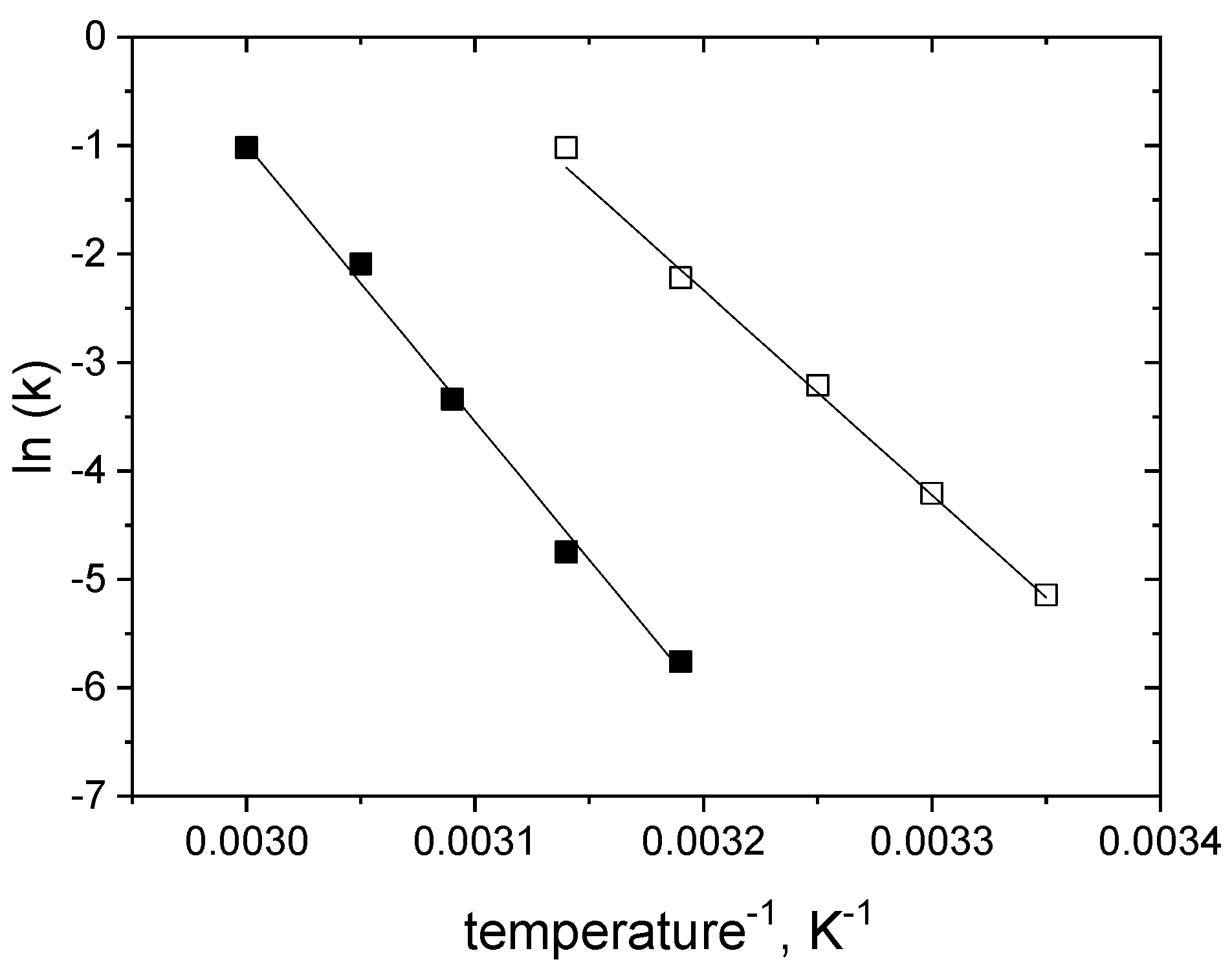
2.4. Modelling of Thermal Inactivation under Storage Conditions
3. Materials and Methods
3.1. Chemicals
3.2. Preparation of S-HPED
3.3. Synthesis of Chiral Alcohols with Pure S-HPED
3.4. Chiral Chromatographic Analysis
3.5. Effect of pH on the Initial Activity of S-HPED and Activity Decrease Rate
3.6. Effects of Temperature on the Activity of S-HPED
3.7. UV-Vis Activity Assay
3.8. S-HPED Stability Tests under Reaction Condition
3.9. Chromatographic Analyses—Reverse Phase (RP-HPLC)
3.10. Aggregation Measurements
3.11. Dynamic Light Scattering
3.12. Thermal Shift Assay
3.13. Thermal Inactivation Experiments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dudzik, A.; Snoch, W.; Borowiecki, P.; Opalinska-Piskorz, J.; Witko, M.; Heider, J.; Szaleniec, M. Asymmetric Reduction of Ketones and β-Keto Esters by (S)-1-Phenylethanol Dehydrogenase from Denitrifying Bacterium Aromatoleum aromaticum. Appl. Microbiol. Biotechnol. 2015, 99, 5055–5069. [Google Scholar] [CrossRef] [PubMed]
- Borowiecki, P.; Telatycka, N.; Tataruch, M.; Żądło-Dobrowolska, A.; Reiter, T.; Schühle, K.; Heider, J.; Szaleniec, M.; Kroutil, W. Biocatalytic Asymmetric Reduction of γ-Keto Esters to Access Optically Active γ-Aryl-γ-Butyrolactones. Adv. Synth. Catal. 2020, 362, 2012–2029. [Google Scholar] [CrossRef]
- de Miranda, A.S.; Milagre, C.D.F.; Hollmann, F. Alcohol Dehydrogenases as Catalysts in Organic Synthesis. Front. Catal. 2022, 2, 900554. [Google Scholar] [CrossRef]
- Musa, M.M. Alcohol Dehydrogenases with Anti-Prelog Stereopreference in Synthesis of Enantiopure Alcohols. ChemistryOpen 2022, 11, e202100251. [Google Scholar] [CrossRef] [PubMed]
- Rudzka, A.; Zdun, B.; Antos, N.; Montero, L.M.; Reiter, T.; Kroutil, W.; Borowiecki, P. Biocatalytic Characterization of an Alcohol Dehydrogenase Variant Deduced from Lactobacillus Kefir in Asymmetric Hydrogen Transfer. Commun. Chem. 2023, 6, 217. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.G.; Yin, H.H.; Yu, D.F.; Chen, X.; Tang, X.L.; Zhang, X.J.; Xue, Y.P.; Wang, Y.J.; Liu, Z.Q. Recent Advances in Biotechnological Applications of Alcohol Dehydrogenases. Appl. Microbiol. Biotechnol. 2017, 101, 987–1001. [Google Scholar] [CrossRef]
- Chadha, A.; Padhi, S.K.; Stella, S.; Venkataraman, S.; Saravanan, T. Microbial Alcohol Dehydrogenases: Recent Developments and Applications in Asymmetric Synthesis. Org. Biomol. Chem. 2024, 22, 228–251. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Snajdrova, R.; Moore, J.C.; Baldenius, K.; Bornscheuer, U.T. Biocatalysis: Enzymatic Synthesis for Industrial Applications. Angew. Chemie Int. Ed. 2021, 60, 88–119. [Google Scholar] [CrossRef] [PubMed]
- Becker, P.; Wünsch, D.; Wöhlbrand, L.; Neumann-Schaal, M.; Schomburg, D.; Rabus, R. The Catabolic Network of Aromatoleum aromaticum EbN1T. Microb. Physiol. 2024, 34, 1–77. [Google Scholar] [CrossRef]
- Heider, J.; Szaleniec, M.; Sünwoldt, K.; Boll, M. Ethylbenzene Dehydrogenase and Related Molybdenum Enzymes Involved in Oxygen-Independent Alkyl Chain Hydroxylation. J. Mol. Microbiol. Biotechnol. 2016, 26, 45–62. [Google Scholar] [CrossRef]
- Dudzik, A.; Kozik, B.; Tataruch, M.; Wójcik, A.; Knack, D.; Borowski, T.; Heider, J.; Witko, M.; Szaleniec, M. The Reaction Mechanism of Chiral Hydroxylation of p -OH and p -NH 2 Substituted Compounds by Ethylbenzene Dehydrogenase. Can. J. Chem. 2013, 91, 775–786. [Google Scholar] [CrossRef]
- Tataruch, M.; Heider, J.; Bryjak, J.; Nowak, P.; Knack, D.; Czerniak, A.; Liesiene, J.; Szaleniec, M. Suitability of the Hydrocarbon-Hydroxylating Molybdenum-Enzyme Ethylbenzene Dehydrogenase for Industrial Chiral Alcohol Production. J. Biotechnol. 2014, 192, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Szaleniec, M.; Borowski, T.; Schühle, K.; Witko, M.; Heider, J. Ab Inito Modeling of Ethylbenzene Dehydrogenase Reaction Mechanism. J. Am. Chem. Soc. 2010, 132, 6014–6024. [Google Scholar] [CrossRef] [PubMed]
- Kniemeyer, O.; Heider, J. Ethylbenzene Dehydrogenase, a Novel Hydrocarbon-Oxidizing Molybdenum/Iron-Sulfur/Heme Enzyme. J. Biol. Chem. 2001, 276, 21381–21386. [Google Scholar] [CrossRef] [PubMed]
- Arndt, F.; Schmitt, G.; Winiarska, A.; Saft, M.; Seubert, A.; Kahnt, J.; Heider, J. Characterization of an Aldehyde Oxidoreductase from the Mesophilic Bacterium Aromatoleum aromaticum EbN1, a Member of a New Subfamily of Tungsten-Containing Enzymes. Front. Microbiol. 2019, 10, 71. [Google Scholar] [CrossRef]
- Winiarska, A.; Hege, D.; Gemmecker, Y.; Kryściak-Czerwenka, J.; Seubert, A.; Heider, J.; Szaleniec, M. Tungsten Enzyme Using Hydrogen as an Electron Donor to Reduce Carboxylic Acids and NAD+. ACS Catal. 2022, 12, 8707–8717. [Google Scholar] [CrossRef] [PubMed]
- Kalimuthu, P.; Hege, D.; Winiarska, A.; Gemmecker, Y.; Szaleniec, M.; Heider, J.; Bernhardt, P.V. Electrocatalytic Aldehyde Oxidation by a Tungsten Dependent Aldehyde Oxidoreductase from Aromatoleum aromaticum. Chem. A Eur. J. 2023, 29, e202203072. [Google Scholar] [CrossRef] [PubMed]
- Winiarska, A.; Ramírez-Amador, F.; Hege, D.; Gemmecker, Y.; Prinz, S.; Hochberg, G.; Heider, J.; Szaleniec, M.; Schuller, J.M. A Bacterial Tungsten-Containing Aldehyde Oxidoreductase Forms an Enzymatic Decorated Protein Nanowire. Sci. Adv. 2023, 9, eadg6689. [Google Scholar] [CrossRef]
- Höffken, H.W.; Duong, M.; Friedrich, T.; Breuer, M.; Hauer, B.; Reinhardt, R.; Rabus, R.; Heider, J. Crystal Structure and Enzyme Kinetics of the (S)-Specific 1-Phenylethanol Dehydrogenase of the Denitrifying Bacterium Strain EbN1. Biochemistry 2006, 45, 82–93. [Google Scholar] [CrossRef]
- Kniemeyer, O.; Heider, J. (S)-1-Phenylethanol Dehydrogenase of Azoarcus sp. Strain EbN1, an Enzyme of Anaerobic Ethylbenzene Catabolism. Arch. Microbiol. 2001, 176, 129–135. [Google Scholar] [CrossRef]
- Wu, W.; Yuan, X.; Gao, X.; Tan, C.; Li, S.; Xu, D. Production of ρ-Hydroxyacetophenone by Engineered Escherichia coli Heterologously Expressing 1-(4-Hydroxyphenyl)-Ethanol Dehydrogenase. J. Microbiol. Biotechnol. 2024, 34, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Tataruch, M.; Illeová, V.; Miłaczewska, A.; Borowski, T.; Mihal’, M.; Polakovič, M. Inactivation and Aggregation of R-Specific 1-(4-Hydroxyphenyl)-Ethanol Dehydrogenase from Aromatoleum aromaticum. Int. J. Biol. Macromol. 2023, 234, 123772. [Google Scholar] [CrossRef] [PubMed]
- Büsing, I.; Höffken, H.W.; Breuer, M.; Wöhlbrand, L.; Hauer, B.; Rabus, R. Molecular Genetic and Crystal Structural Analysis of 1-(4-Hydroxyphenyl)-Ethanol Dehydrogenase from “Aromatoleum aromaticum” EbN1. J. Mol. Microbiol. Biotechnol. 2015, 25, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Muhr, E.; Schühle, K.; Clermont, L.; Sünwoldt, K.; Kleinsorge, D.; Seyhan, D.; Kahnt, J.; Schall, I.; Cordero, P.R.; Schmitt, G.; et al. Enzymes of Anaerobic Ethylbenzene and P-Ethylphenol Catabolism in ‘Aromatoleum aromaticum’: Differentiation and Differential Induction. Arch. Microbiol. 2015, 197, 1051–1062. [Google Scholar] [CrossRef] [PubMed]
- Wöhlbrand, L.; Wilkes, H.; Halder, T.; Rabus, R. Anaerobic Degradation of P-Ethylphenol by “Aromatoleum aromaticum” Strain EbNl: Pathway, Regulation, and Involved Proteins. J. Bacteriol. 2008, 190, 5699–5709. [Google Scholar] [CrossRef] [PubMed]
- Rabus, R.; Trautwein, K.; Wöhlbrand, L. Towards Habitat-Oriented Systems Biology of “Aromatoleum aromaticum” EbN1: Chemical Sensing, Catabolic Network Modulation and Growth Control in Anaerobic Aromatic Compound Degradation. Appl. Microbiol. Biotechnol. 2014, 98, 3371–3388. [Google Scholar] [CrossRef] [PubMed]
- Vosmanská, M.; Sýkora, D.; Fähnrich, J.; Kovářová, M.; Volka, K. Extraction of P-Hydroxyacetophenone and Catechin from Norway Spruce Needles. Comparison of Different Extraction Solvents. Anal. Bioanal. Chem. 2005, 382, 1135–1140. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Wang, X.; Zhou, L.; Liu, Q.; Zhang, G.; Xue, B.; Hu, C.; Shen, X.; Sun, X.; Yan, Y.; et al. Establishing Biosynthetic Pathway for the Production of P-Hydroxyacetophenone and Its Glucoside in Escherichia coli. Metab. Eng. 2023, 76, 110–119. [Google Scholar] [CrossRef]
- Chouke, P.B.; Chaudhary, R.G.; Ingle, V.N. Solvent free synthesis of p-hydroxyacetophenone in a situ using eco-friendly catalyst in Fries rearrangement. J. Chem. Pharm. Res. 2015, 7, 727–731. [Google Scholar]
- Aalbers, F.S.; Fürst, M.J.L.J.; Rovida, S.; Trajkovic, M.; Rubén Gómez Castellanos, J.; Bartsch, S.; Vogel, A.; Mattevi, A.; Fraaije, M.W. Approaching Boiling Point Stability of an Alcohol Dehydrogenase through Computationally-Guided Enzyme Engineering. Elife 2020, 9, e54639. [Google Scholar] [CrossRef]
- Zhu, L.; Song, Y.; Chang, C.; Ma, H.; Yang, L.; Deng, Z.; Deng, W.; Qu, X. Engineering Leifsonia Alcohol Dehydrogenase for Thermostability and Catalytic Efficiency by Enhancing Subunit Interactions. ChemBioChem 2021, 22, 3178–3183. [Google Scholar] [CrossRef] [PubMed]
- Armani, D.; Piccolo, O.; Petri, A. Immobilization of Alcohol Dehydrogenases on Silica-Based Supports and Their Application in Enantioselective Ketone Reductions. Catalysts 2024, 14, 148. [Google Scholar] [CrossRef]
- Vasić, K.; Knez, Ž.; Kumar, S.; Pandey, J.K.; Leitgeb, M. Epoxy Functionalized Carboxymethyl Dextran Magnetic Nanoparticles for Immobilization of Alcohol Dehydrogenase. Acta Chim. Slov. 2020, 67, 1172–1179. [Google Scholar] [CrossRef]
- Shinde, P.; Musameh, M.; Gao, Y.; Robinson, A.J.; Kyratzis, I. (Louis) Immobilization and Stabilization of Alcohol Dehydrogenase on Polyvinyl Alcohol Fibre. Biotechnol. Rep. 2018, 19, e00260. [Google Scholar] [CrossRef]
- Holtmann, D.; Hollmann, F. Is Water the Best Solvent for Biocatalysis? Mol. Catal. 2022, 517, 112035. [Google Scholar] [CrossRef]
- Zohra, F.; Majdoub, I.; Lavandera, I.; Kreit, J. Stabilizing Effects of Deep Eutectic Solvents on Alcohol Dehydrogenase Mediated Systems. Int. J. Bioeng. Life Sci. 2016, 10, 825–830. [Google Scholar]
- Mensink, M.A.; Frijlink, H.W.; van der Voort Maarschalk, K.; Hinrichs, W.L.J. How Sugars Protect Proteins in the Solid State and during Drying (Review): Mechanisms of Stabilization in Relation to Stress Conditions. Eur. J. Pharm. Biopharm. 2017, 114, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Illeová, V.; Šefčík, J.; Polakovič, M. Thermal Inactivation of Jack Bean Urease. Int. J. Biol. Macromol. 2020, 151, 1084–1090. [Google Scholar] [CrossRef]
- Polakovič, M.; Vrábel, P. Analysis of the Mechanism and Kinetics of Thermal Inactivation of Enzymes: Critical Assessment of Isothermal Inactivation Experiments. Process Biochem. 1996, 31, 787–800. [Google Scholar] [CrossRef]
- Illeová, V.; Polakovič, M. Theoretical Study of Discrimination of Mechanisms of Thermal Inactivation of Urease by Monitoring Its Oligomeric Composition. Chem. Pap. 2004, 58, 64–69. [Google Scholar]
- Polakovič, M.; Bryjak, J. Modelling of the Kinetics of Thermal Inactivation of Glucoamylase from Aspergillus niger. J. Mol. Catal. B Enzym. 2002, 19–20, 443–450. [Google Scholar] [CrossRef]
- Grancic, P.; Illeova, V.; Polakovic, M.; Sefcik, J. Thermally Induced Inactivation and Aggregation of Urease: Experiments and Population Balance Modelling. Chem. Eng. Sci. 2012, 70, 14–21. [Google Scholar] [CrossRef]
- Fazeli, A.; Haji-Abdolvahab, M.; Shojaosadati, S.A.; Schellekens, H.; Khalifeh, K.; Moosavi-Movahedi, A.A.; Fazeli, M.R. Effect of Arginine on Pre-Nucleus Stage of Interferon Beta-1b Aggregation. Ageing Int. 2014, 15, 1619–1629. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Substrate | Product | Product S (%) | Product R (%) | % ee | Relative Activity * (%) |
|---|---|---|---|---|---|---|
| 0 | acetophenone | 1-phenylethanol | 100 | 0 | 100 | 100 |
| 1 | 4′-chloroacetophenone | 1-(4-chlorophenyl)ethanol | 100 | 0 | 100 | 123 |
| 2 | 4′-metoxyacetophenone | 1-(4-methoxyphenyl)ethanol | 100 | 0 | 100 | 82 |
| 3 | 4′-fluoroacetophenone | 1-(4-fluorophenyl)ethanol | 100 | 0 | 100 | 108 |
| 4 | 4′-nitroacetophenone | 1-(4-nitrophenyl)ethanol | - | - | 100 | 201 |
| 5 | 3′-aminoacetophenone | 1-(3-aminophenyl)ethanol | 100 | 0 | 100 | 57 |
| 5 | 4′-aminoacetophenone | 1-(4-aminophenyl)ethanol | 96 | 4 | 92 | 7 |
| 7 | 4′-ethylacetophenone | 1-(4-ethylphenyl)ethanol | - | - | 100 | 70 |
| 8 | 4′-bromoacetophenone | 1-(4-bromophenyl)ethanol | 100 | 0 | 100 | 130 |
| 9 | 4-acetylbenzonitrile | 4-(1-hydroxyethyl)benzonitrile | - | - | 100 | 186 |
| 10 | 2,2-dichloroacetophenone | 2,2-dichloro-1-phenylethanol | - | - | 100 | 344 |
| 11 | 4′-acetylbiphenyl | 1-(biphenyl-4-yl)ethanol | - | - | 100 | 204 |
| 12 | 1-indanone | 1-indanol | - | - | - | 0 |
| 13 | 2,3-benzofuran | 2,3-dihydro-1-benzofuran-3-ol | - | - | - | 0 |
| 14 | 3-phenyl-1-indanone | 3-phenyl-1-indanol | - | - | - | 0 |
| 15 | 6-hydroxy-1-indanone | 2,3-dihydro-1H-indene-1,6-diol | - | - | - | 0 |
| Time | pH 5.5 | pH 6.5 | pH 7.5 | pH 8.3 | pH 9.0 | |
|---|---|---|---|---|---|---|
| without additives | 0 h | 100% | 100% | 100% | 100% | 100% |
| 0.5 h | 0.3% | 3.2% | 2.4% | 2.5% | 2% | |
| with 2 mM acetophenone | 0 h | 100% | 100% | 100% | 100% | 100% |
| 0.5 h | 1.4% | 2.6% | 2.4% | 2.1% | 1.2% | |
| with 1.5 M glucose | 0 h | 100% | 100% | 100% | 100% | 100% |
| 0.5 h | 9.3% | 62.5% | 38% | 48% | 42.3% | |
| 2 h | 3.5% | 10% | 8.7% | 9.8% | 15.6% | |
| 5 h | 2% | 5.4% | 3% | 2.1% | 9.3% |
| Temperature (°C) | Glucose-Free | in 1.5 M Glucose | ||||
|---|---|---|---|---|---|---|
| k (min−1) | sso | SD (%) | k (min−1) | sso | SD (%) | |
| 25 | 0.00583 ± 0.00022 | 0.01418 | 3.303 | --- | --- | --- |
| 30 | 0.01489 ± 0.00053 | 0.01373 | 3.132 | --- | --- | --- |
| 35 | 0.04029 ± 0.00144 | 0.01639 | 3.305 | --- | --- | --- |
| 40 | 0.10868 ± 0.00301 | 0.00563 | 2.082 | 0.00315 ± 0.00011 | 0.01119 | 3.19 |
| 45 | 0.36106 ± 0.01189 | 0.00155 | 1.488 | 0.00867 ± 0.00026 | 0.01694 | 3.36 |
| 50 | --- | --- | --- | 0.03558 ± 0.00134 | 0.01179 | 3.274 |
| 55 | --- | --- | --- | 0.12340 ± 0.00354 | 0.00661 | 2.451 |
| 60 | --- | --- | --- | 0.36083 ± 0.00673 | 0.00296 | 1.57 |
| Parameters/ Inactivationconditions | k0 (min−1) | Ea (kJ mol−1) | SD (%) | n |
|---|---|---|---|---|
| glucose-free | 0.04266 ± 0.00127 | 156.60 ± 4.46 | 3.27 | 65 |
| in 1.5 M glucose | 0.03447 ± 0.00127 | 211.55 ± 4.63 | 3.78 | 63 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tataruch, M.; Illeová, V.; Kluza, A.; Cabadaj, P.; Polakovič, M. Catalytic Stability of S-1-(4-Hydroxyphenyl)-Ethanol Dehydrogenase from Aromatoleum aromaticum. Int. J. Mol. Sci. 2024, 25, 7385. https://doi.org/10.3390/ijms25137385
Tataruch M, Illeová V, Kluza A, Cabadaj P, Polakovič M. Catalytic Stability of S-1-(4-Hydroxyphenyl)-Ethanol Dehydrogenase from Aromatoleum aromaticum. International Journal of Molecular Sciences. 2024; 25(13):7385. https://doi.org/10.3390/ijms25137385
Chicago/Turabian StyleTataruch, Mateusz, Viera Illeová, Anna Kluza, Patrik Cabadaj, and Milan Polakovič. 2024. "Catalytic Stability of S-1-(4-Hydroxyphenyl)-Ethanol Dehydrogenase from Aromatoleum aromaticum" International Journal of Molecular Sciences 25, no. 13: 7385. https://doi.org/10.3390/ijms25137385
APA StyleTataruch, M., Illeová, V., Kluza, A., Cabadaj, P., & Polakovič, M. (2024). Catalytic Stability of S-1-(4-Hydroxyphenyl)-Ethanol Dehydrogenase from Aromatoleum aromaticum. International Journal of Molecular Sciences, 25(13), 7385. https://doi.org/10.3390/ijms25137385