
The Interaction of Vasopressin with Hormones of the Hypothalamo–Pituitary–Adrenal Axis: The Significance for Therapeutic Strategies in Cardiovascular and Metabolic Diseases

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Interactions of the Hypothalamo–Pituitary–Adrenal System with Vasopressin at the Cellular Level
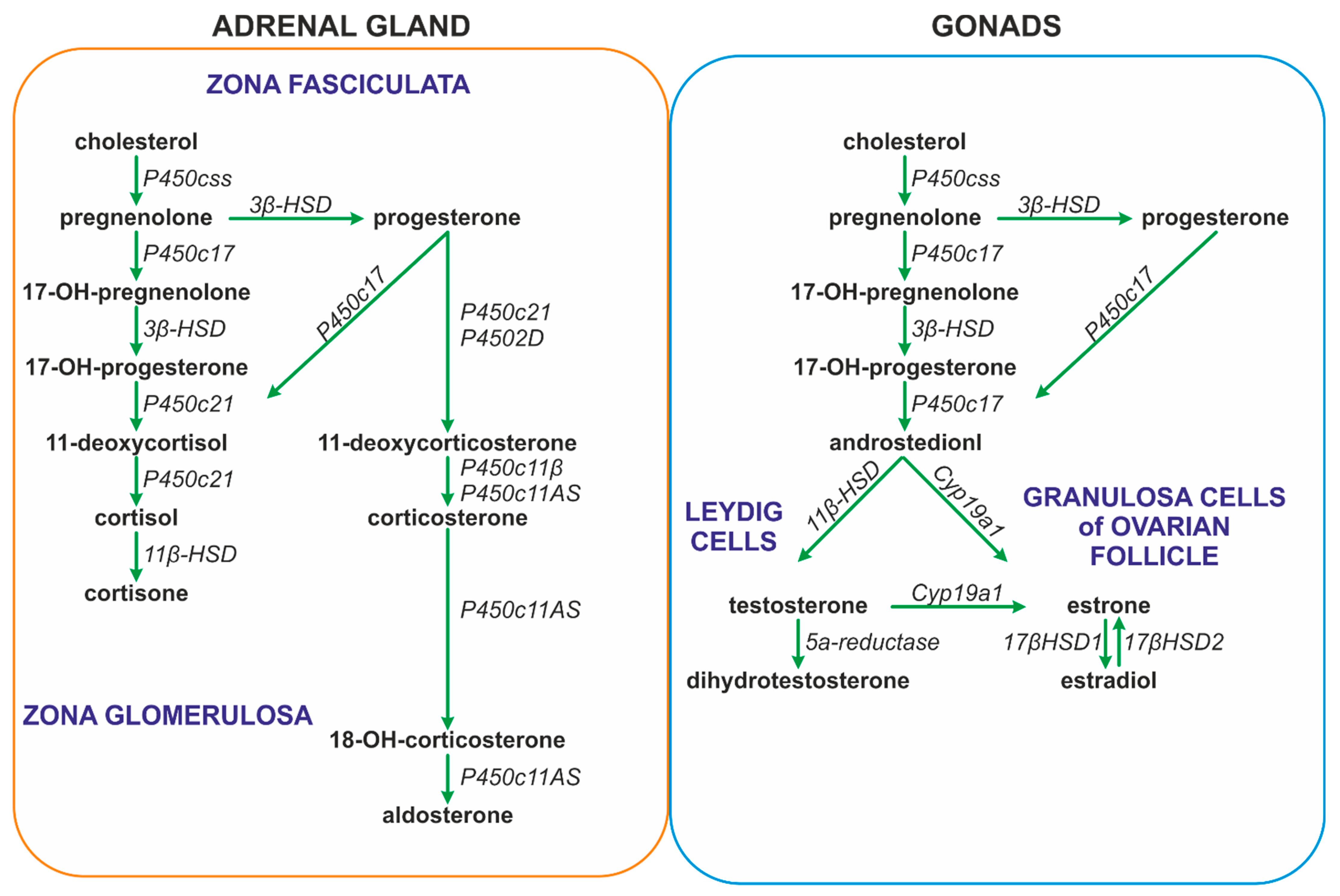
2.1. Genomic and Nongenomic Actions of Steroid Hormones
2.2. Genomic and Non-Genomic Effects of Vasopressin
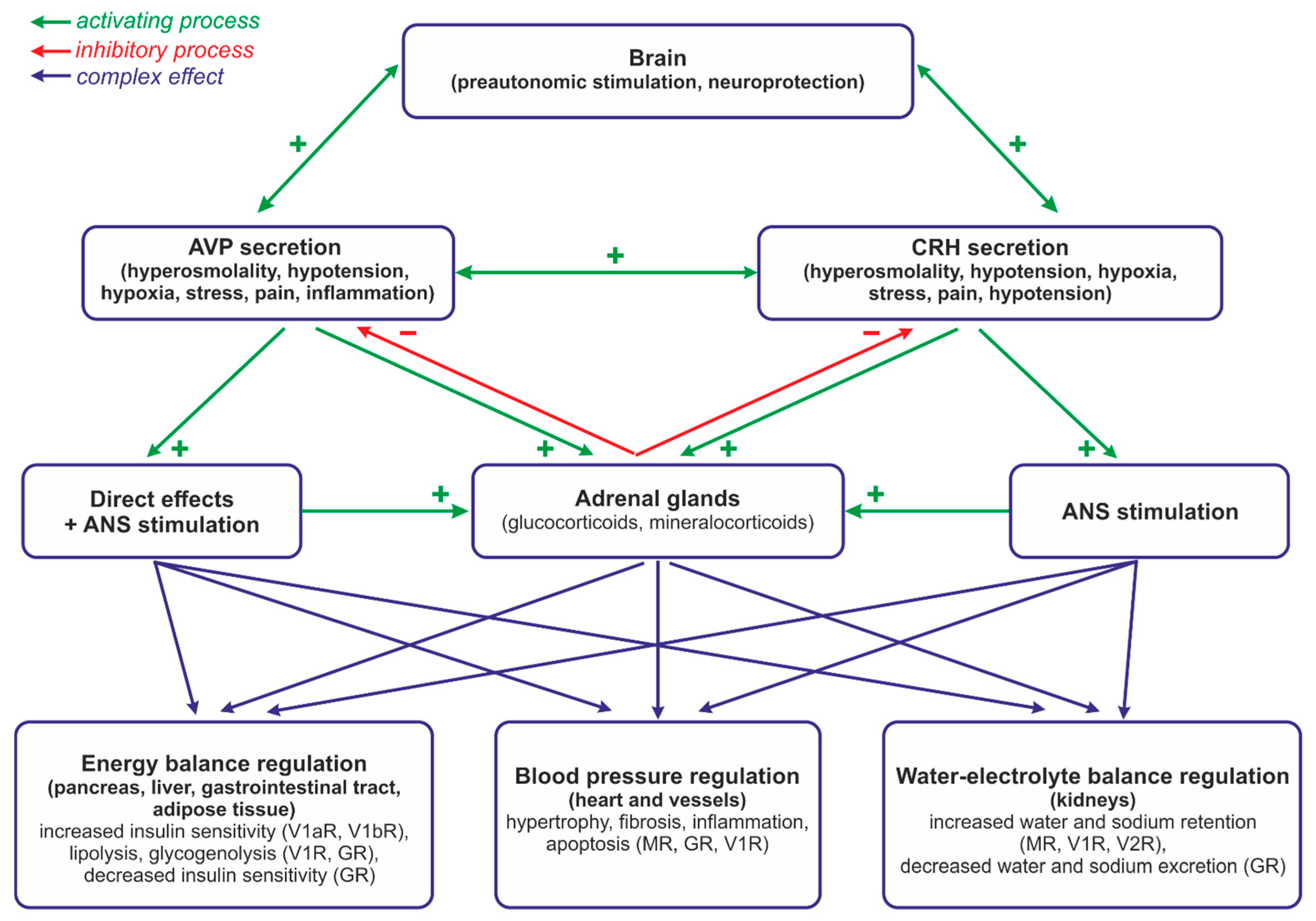
3. Role of the Hypothalamo–Pituitary–Adrenal System and Vasopressin in the Regulation of Energy Balance and Water–Electrolyte Balance at Rest and during Neurogenic Stress
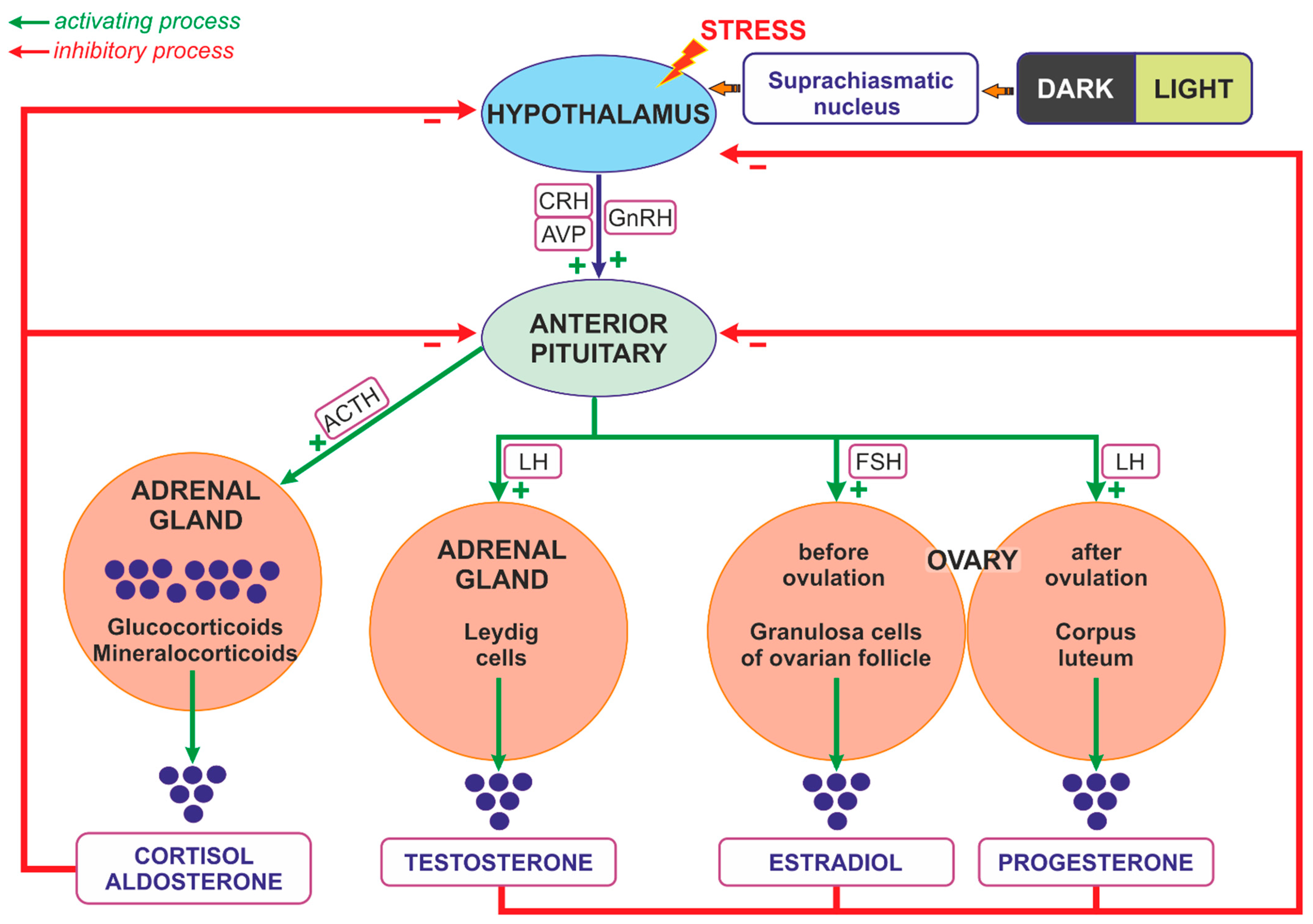
3.1. Regulation of Steroids Secretion
3.2. Interaction between Steroids and AVP in the Regulation of Energy Balance
3.2.1. Animal Studies
3.2.2. Human Studies
3.3. Interaction of AVP and Steroids in the Regulation of Water–Electrolyte Balance
Studies on Animals and Human Subjects
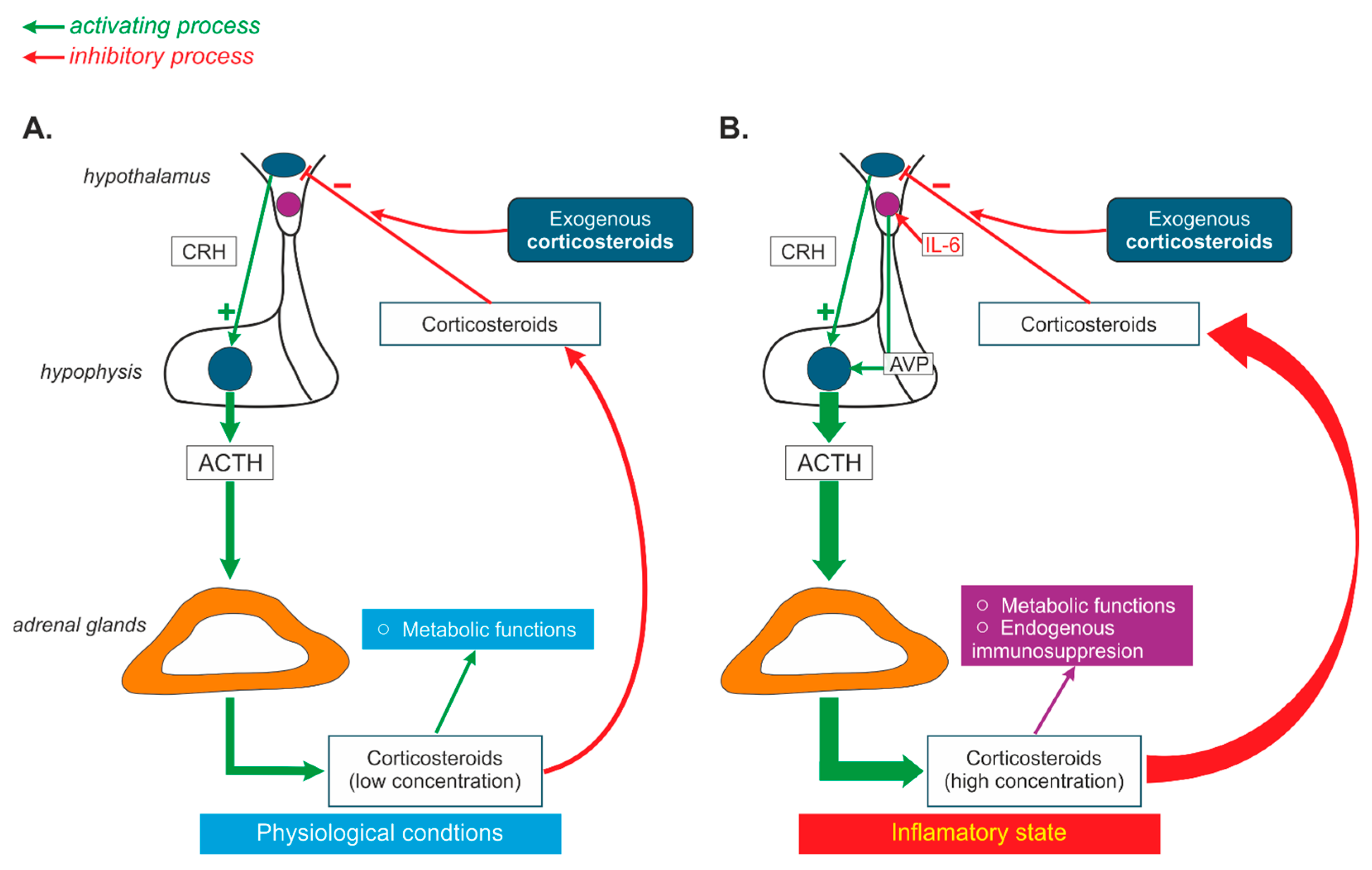
3.4. Interaction of AVP and Steroids in Neurogenic Stress
Sex Differences
4. Altered Interactions of Vasopressin with the Hypothalamo–Pituitary–Adrenal System in Cardiovascular and Metabolic Diseases
4.1. Cardiovascular Diseases
4.2. Metabolic Diseases
5. Impact of Therapeutic Interventions on Interactions of Vasopressin with the Hypothalamo–Pituitary–Adrenal System in Health and in Cardiovascular and Metabolic Diseases
5.1. Steroids and Vasopressin Treatments in Cardiovascular Diseases
5.2. Impact of Anti-Depressive and Neuroleptic Treatments on Vasopressin–HPA Interactions
6. Summary
7. Future Directions
8. Conclusions
- Vasopressin (AVP) and steroid hormones are frequently released together and closely cooperate in the regulation of blood pressure, metabolism, water–electrolyte balance, and behavior.
- Vasopressin interacts with specific components of the hypothalamo–pituitary–adrenal axis in the brain and in several peripheral organs and tissues, including the heart, vessels, kidneys, and adipose tissue.
- Appropriate interactions of AVP with the HPA are essential for the efficient regulation of water–electrolyte balance, blood pressure, and energy balance, and it is justified to consider vasopressin and the hypothalamo–pituitary axis as a highly coordinated, functional AVP-HPA system.
- Interactions between AVP and HPA are significantly altered in cardiovascular, respiratory, and metabolic diseases and during inflammation and neurogenic stress.
- Inappropriate interactions of AVP and steroids may initiate or intensify cardiovascular complications in metabolic diseases.
- The interplay of vasopressin and steroid hormones is not yet fully recognized and further studies are needed to determine the potentially beneficial or harmful consequences of interference with these factors in the treatment of specific pathological states.
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Hilton, J.G.; Scian, L.F.; Westermann, S.D.; Nakano, J.; Kruesi, O.R. Vasopressin stimulation of the isolated adrenal glands: Nature and mechanism of hydrocortisone secretion. Endocrinology 1960, 67, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Larsen, P.J.; Vrang, N.; Møller, M.; Jessop, D.S.; Lightman, S.L.; Chowdrey, H.S.; Mikkelsen, J.D. The diurnal expression of genes encoding vasopressin and vasoactive intestinal peptide within the rat suprachiasmatic nucleus is influenced by circulating glucocorticoids. Brain Res. Mol. Brain Res. 1994, 27, 342–346. [Google Scholar] [CrossRef] [PubMed]
- Mouri, T.; Itoi, K.; Takahashi, K.; Suda, T.; Murakami, O.; Yoshinaga, K.; Andoh, N.; Ohtani, H.; Masuda, T.; Sasano, N. Colocalization of corticotropin-releasing factor and vasopressin in the paraventricular nucleus of the human hypothalamus. Neuroendocrinology 1993, 57, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Otubo, A.; Kawakami, N.; Maejima, S.; Ueda, Y.; Morris, J.F.; Sakamoto, T.; Sakamoto, H. Vasopressin gene products are colocalised with corticotrophin-releasing factor within neurosecretory vesicles in the external zone of the median eminence of the Japanese macaque monkey (Macaca fuscata). J. Neuroendocr. 2020, 32, e12875. [Google Scholar] [CrossRef] [PubMed]
- Engler, D.; Pham, T.; Fullerton, M.J.; Ooi, G.; Funder, J.W.; Clarke, I.J. Studies of the secretion of corticotropin-releasing factor and arginine vasopressin into the hypophysial-portal circulation of the conscious sheep. I. Effect of an audiovisual stimulus and insulin-induced hypoglycemia. Neuroendocrinology 1989, 49, 367–381. [Google Scholar] [CrossRef] [PubMed]
- Familari, M.; Smith, A.I.; Smith, R.; Funder, J.W. Arginine vasopressin is a much more potent stimulus to ACTH release from ovine anterior pituitary cells than ovine corticotropin-releasing factor. 1. In vitro studies. Neuroendocrinology 1989, 50, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Labrie, F.; Giguere, V.; Proulx, L.; Lefevre, G. Interactions between CRF, epinephrine, vasopressin and glucocorticoids in the control of ACTH secretion. J. Steroid Biochem. 1984, 20, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Veldhuis, H.D.; de Kloet, E.R. Vasopressin-related peptides increase the hippocampal corticosterone receptor capacity of diabetes insipidus (Brattleboro) rat. Endocrinology 1982, 110, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Papanek, P.E.; Sladek, C.D.; Raff, H. Corticosterone inhibition of osmotically stimulated vasopressin from hypothalamic-neurohypophysial explants. Am. J. Physiol. 1997, 272 Pt 2, R158–R162. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, C.A.; Chen, Y.Z. Nongenomic effect of glucocorticoid on the release of arginine vasopressin from hypothalamic slices in rats. Neuroendocrinology 1995, 62, 628–633. [Google Scholar] [CrossRef] [PubMed]
- Calogero, A.E.; Liapi, C.; Chrousos, G.P. Hypothalamic and suprahypothalamic effects of prolonged treatment with dexamethasone in the rat. J. Endocrinol. Investig. 1991, 14, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Woodcock, E.A.; Mcleod, J.K.; Johnston, C.I. Vasopressin stimulates phosphatidylinositol turnover and aldosterone synthesis in rat adrenal glomerulosa cells: Comparison with angiotensin II. Endocrinology 1986, 118, 2432–2436. [Google Scholar] [CrossRef] [PubMed]
- Saito, R.; Ishiharada, N.; Ban, Y.; Honda, K.; Takano, Y.; Kamiya, H. Vasopressin V1 receptor in rat hippocampus is regulated by adrenocortical functions. Brain Res. 1994, 646, 170–174. [Google Scholar] [CrossRef] [PubMed]
- Watters, J.J.; Poulin, P.; Dorsa, D.M. Steroid hormone regulation of vasopressinergic neurotransmission in the central nervous system. Prog. Brain Res. 1998, 119, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Watters, J.J.; Wilkinson, C.W.; Dorsa, D.M. Glucocorticoid regulation of vasopressin V1a receptors in rat forebrain. Brain Res. Mol. Brain Res. 1996, 38, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Rabadan-Diehl, C.; Makara, G.; Kiss, A.; Lolait, S.; Zelena, D.; Ochedalski, T.; Aguilera, G. Regulation of pituitary V1b vasopressin receptor messenger ribonucleic acid by adrenalectomy and glucocorticoid administration. Endocrinology 1997, 138, 5189–5194. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.B.; Tannahill, L.A.; Biswas, S.; Lightman, S.L. Release of corticotrophin-releasing factor-41, arginine vasopressin and oxytocin from rat fetal hypothalamic cells in culture: Response to activation of intracellular second messengers and to corticosteroids. J. Endocrinol. 1992, 132, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Currie, I.S.; Gillies, G.; Brooks, A.N. Modulation of arginine vasopressin secretion from cultured ovine hypothalamic cells by glucocorticoids and opioid peptides. Neuroendocrinology 1994, 60, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Kokras, N.; Hodes, G.E.; Bangasser, D.A.; Dalla, C. Sex differences in the hypothalamic-pituitary-adrenal axis: An obstacle to antidepressant drug development? Br. J. Pharmacol. 2019, 176, 4090–4106. [Google Scholar] [CrossRef] [PubMed]
- Orshal, J.M.; Khalil, R.A. Gender, sex hormones, and vascular tone. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 286, R233–R249. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.Q.; Kawashima, H.; Iwasaki, Y.; Uchida, K.; Sugimoto, K.; Itoi, K. Differential effects of forced swim-stress on the corticotropin-releasing hormone and vasopressin gene transcription in the parvocellular division of the paraventricular nucleus of rat hypothalamus. Neurosci. Lett. 2004, 358, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Hlavacova, N.; Bakos, J.; Jezova, D. Eplerenone, a selective mineralocorticoid receptor blocker, exerts anxiolytic effects accompanied by changes in stress hormone release. J. Psychopharmacol. 2010, 24, 779–786. [Google Scholar] [CrossRef] [PubMed]
- de Kloet, E.R.; Meijer, O.C.; de Nicola, A.F.; de Rijk, R.H.; Joëls, M. Importance of the brain corticosteroid receptor balance in metaplasticity, cognitive performance and neuro-inflammation. Front. Neuroendocr. 2018, 49, 124–145. [Google Scholar] [CrossRef] [PubMed]
- Karst, H.; Berger, S.; Turiault, M.; Tronche, F.; Schütz, G.; Joëls, M. Mineralocorticoid receptors are indispensable for nongenomic modulation of hippocampal glutamate transmission by corticosterone. Proc. Natl. Acad. Sci. USA 2005, 102, 19204–19207. [Google Scholar] [CrossRef] [PubMed]
- Karst, H.; den Boon, F.S.; Vervoort, N.; Adrian, M.; Kapitein, L.C.; Joëls, M. Non-genomic steroid signaling through the mineralocorticoid receptor: Involvement of a membrane-associated receptor? Mol. Cell. Endocrinol. 2022, 541, 111501. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, N.; Pfaff, D.W. Non-genomic actions of estrogens and their interaction with genomic actions in the brain. Front. Neuroendocr. 2008, 29, 238–257. [Google Scholar] [CrossRef] [PubMed]
- Lachize, S.; Apostolakis, E.M.; van der Laan, S.; Tijssen, A.M.; Xu, J.; de Kloet, E.R.; Meijer, O.C. Steroid receptor coactivator-1 is necessary for regulation of corticotropin-releasing hormone by chronic stress and glucocorticoids. Proc. Natl. Acad. Sci. USA 2009, 106, 8038–8042. [Google Scholar] [CrossRef] [PubMed]
- van Weert, L.T.C.M.; Buurstede, J.C.; Mahfouz, A.; Braakhuis, P.S.M.; Polman, J.A.E.; Sips, H.C.M.; Roozendaal, B.; Balog, J.; de Kloet, E.R.; Datson, N.A.; et al. NeuroD Factors Discriminate Mineralocorticoid From Glucocorticoid Receptor DNA Binding in the Male Rat Brain. Endocrinology 2017, 158, 1511–1522. [Google Scholar] [CrossRef] [PubMed]
- Skowron, K.J.; Booker, K.; Cheng, C.; Creed, S.; David, B.P.; Lazzara, P.R.; Lian, A.; Siddiqui, Z.; Speltz, T.E.; Moore, T.W. Steroid receptor/coactivator binding inhibitors: An update. Mol. Cell. Endocrinol. 2019, 493, 110471. [Google Scholar] [CrossRef] [PubMed]
- Stashi, E.; York, B.; O’Malley, B.W. Steroid receptor coactivators: Servants and masters for control of systems metabolism. Trends Endocrinol. Metab. 2014, 25, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, C.; Almeida-Prieto, B.; Nolze, A.; Alvarez de la Rosa, D. Structural and molecular determinants of mineralocorticoid receptor signalling. Br. J. Pharmacol. 2022, 179, 3103–3118. [Google Scholar] [CrossRef] [PubMed]
- Vanderhaeghen, T.; Beyaert, R.; Libert, C. Bidirectional Crosstalk Between Hypoxia Inducible Factors and Glucocorticoid Signalling in Health and Disease. Front. Immunol. 2021, 12, 684085. [Google Scholar] [CrossRef] [PubMed]
- Fadel, L.; Dacic, M.; Fonda, V.; Sokolsky, B.A.; Quagliarini, F.; Rogatsky, I.; Uhlenhaut, N.H. Modulating glucocorticoid receptor actions in physiology and pathology: Insights from coregulators. Pharmacol. Ther. 2023, 251, 108531. [Google Scholar] [CrossRef] [PubMed]
- Vandevyver, S.; Dejager, L.; Libert, C. Comprehensive overview of the structure and regulation of the glucocorticoid receptor. Endocr. Rev. 2014, 35, 671–693. [Google Scholar] [CrossRef] [PubMed]
- Clayton, S.A.; Jones, S.W.; Kurowska-Stolarska, M.; Clark, A.R. The role of microRNAs in glucocorticoid action. J. Biol. Chem. 2018, 293, 1865–1874. [Google Scholar] [CrossRef] [PubMed]
- Knutti, D.; Kaul, A.; Kralli, A. A tissue-specific coactivator of steroid receptors, identified in a functional genetic screen. Mol. Cell. Biol. 2000, 20, 2411–2422. [Google Scholar] [CrossRef] [PubMed]
- Meijer, O.C.; Buurstede, J.C.; Schaaf, M.J.M. Corticosteroid Receptors in the Brain: Transcriptional Mechanisms for Specificity and Context-Dependent Effects. Cell Mol. Neurobiol. 2019, 39, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Oakley, R.H.; Cidlowski, J.A. The biology of the glucocorticoid receptor: New signaling mechanisms in health and disease. J. Allergy Clin. Immunol. 2013, 132, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Oakley, R.H.; Cruz-Topete, D.; He, B.; Foley, J.F.; Myers, P.H.; Xu, X.; Gomez-Sanchez, C.E.; Chambon, P.; Willis, M.S.; Cidlowski, J.A. Cardiomyocyte glucocorticoid and mineralocorticoid receptors directly and antagonistically regulate heart disease in mice. Sci. Signal. 2019, 12, eaau9685. [Google Scholar] [CrossRef] [PubMed]
- Koning, A.C.A.M.; Buurstede, J.C.; van Weert, L.T.C.M.; Meijer, O.C. Glucocorticoid and Mineralocorticoid Receptors in the Brain: A Transcriptional Perspective. J. Endocr. Soc. 2019, 3, 1917–1930. [Google Scholar] [CrossRef] [PubMed]
- Sacta, M.A.; Chinenov, Y.; Rogatsky, I. Glucocorticoid Signaling: An Update from a Genomic Perspective. Annu. Rev. Physiol. 2016, 78, 155–180. [Google Scholar] [CrossRef] [PubMed]
- Faulkner, J.L.; Belin de Chantemèle, E.J. Mineralocorticoid Receptor and Endothelial Dysfunction in Hypertension. Curr. Hypertens. Rep. 2019, 21, 78. [Google Scholar] [CrossRef] [PubMed]
- Kokkinopoulou, I.; Moutsatsou, P. Mitochondrial Glucocorticoid Receptors and Their Actions. Int. J. Mol. Sci. 2021, 22, 6054. [Google Scholar] [CrossRef] [PubMed]
- Viho, E.M.G.; Buurstede, J.C.; Mahfouz, A.; Koorneef, L.L.; van Weert, L.T.C.M.; Houtman, R.; Hunt, H.J.; Kroon, J.; Meijer, O.C. Corticosteroid Action in the Brain: The Potential of Selective Receptor Modulation. Neuroendocrinology 2019, 109, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Winnay, J.N.; Xu, J.; O’Malley, B.W.; Hammer, G.D. Steroid receptor coactivator-1-deficient mice exhibit altered hypothalamic-pituitary-adrenal axis function. Endocrinology 2006, 147, 1322–1332. [Google Scholar] [CrossRef] [PubMed]
- Yi, P.; Yu, X.; Wang, Z.; O’Malley, B.W. Steroid receptor-coregulator transcriptional complexes: New insights from CryoEM. Essays Biochem. 2021, 65, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Marchi, D.; van Eeden, F.J.M. Homeostatic Regulation of Glucocorticoid Receptor Activity by Hypoxia-Inducible Factor 1: From Physiology to Clinic. Cells 2021, 10, 3441. [Google Scholar] [CrossRef] [PubMed]
- Syed, A.P.; Greulich, F.; Ansari, S.A.; Uhlenhaut, N.H. Anti-inflammatory glucocorticoid action: Genomic insights and emerging concepts. Curr. Opin. Pharmacol. 2020, 53, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Kodama, T.; Shimizu, N.; Yoshikawa, N.; Makino, Y.; Ouchida, R.; Okamoto, K.; Hisada, T.; Nakamura, H.; Morimoto, C.; Tanaka, H. Role of the glucocorticoid receptor for regulation of hypoxia-dependent gene expression. J. Biol. Chem. 2003, 278, 33384–33391. [Google Scholar] [CrossRef] [PubMed]
- Callera, G.E.; Touyz, R.M.; Tostes, R.C.; Yogi, A.; He, Y.; Malkinson, S.; Schiffrin, E.L. Aldosterone activates vascular p38MAP kinase and NADPH oxidase via c-Src. Hypertension 2005, 45, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Zeyen, L.; Seternes, O.M.; Mikkola, I. Crosstalk between p38 MAPK and GR Signaling. Int. J. Mol. Sci. 2022, 23, 3322. [Google Scholar] [CrossRef] [PubMed]
- Quatrini, L.; Ugolini, S. New insights into the cell-and tissue-specificity of glucocorticoid actions. Cell. Mol. Immunol. 2021, 18, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Davel, A.P.; Anwar, I.J.; Jaffe, I.Z. The endothelial mineralocorticoid receptor: Mediator of the switch from vascular health to disease. Curr. Opin. Nephrol. Hypertens. 2017, 26, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Geerling, J.C.; Loewy, A.D. Aldosterone in the brain. Am. J. Physiol. Ren. Physiol. 2009, 297, F559–F576. [Google Scholar] [CrossRef] [PubMed]
- Haque, M.; Wilson, R.; Sharma, K.; Mills, N.J.; Teruyama, R. Localisation of 11β-Hydroxysteroid Dehydrogenase Type 2 in Mineralocorticoid Receptor Expressing Magnocellular Neurosecretory Neurones of the Rat Supraoptic and Paraventricular Nuclei. J. Neuroendocrinol. 2015, 27, 835–849. [Google Scholar] [CrossRef] [PubMed]
- Vassiliou, A.G.; Athanasiou, N.; Vassiliadi, D.A.; Jahaj, E.; Keskinidou, C.; Kotanidou, A.; Dimopoulou, I. Glucocorticoid and mineralocorticoid receptor expression in critical illness: A narrative review. World J. Crit. Care Med. 2021, 10, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.M.; Wertz, S.L.; Pollard, C.M.; Desimine, V.L.; Maning, J.; McCrink, K.A.; Lymperopoulos, A. Novel Insights into the Crosstalk between Mineralocorticoid Receptor and G Protein-Coupled Receptors in Heart Adverse Remodeling and Disease. Int. J. Mol. Sci. 2018, 19, 3764. [Google Scholar] [CrossRef] [PubMed]
- Perlstein, R.S.; Whitnall, M.H.; Abrams, J.S.; Mougey, E.H.; Neta, R. Synergistic roles of interleukin-6, interleukin-1, and tumor necrosis factor in the adrenocorticotropin response to bacterial lipopolysaccharide in vivo. Endocrinology 1993, 132, 946–952. [Google Scholar] [CrossRef] [PubMed]
- Terada, Y.; Ueda, S.; Hamada, K.; Shimamura, Y.; Ogata, K.; Inoue, K.; Taniguchi, Y.; Kagawa, T.; Horino, T.; Takao, T. Aldosterone stimulates nuclear factor-kappa B activity and transcription of intercellular adhesion molecule-1 and connective tissue growth factor in rat mesangial cells via serum- and glucocorticoid-inducible protein kinase-1. Clin. Exp. Nephrol. 2012, 16, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Maturana, A.; Lenglet, S.; Python, M.; Kuroda, S.; Rossier, M.F. Role of the T-type calcium channel CaV3.2 in the chronotropic action of corticosteroids in isolated rat ventricular myocytes. Endocrinology 2009, 150, 3726–3734. [Google Scholar] [CrossRef] [PubMed]
- Rossier, M.F. The Cardiac Mineralocorticoid Receptor (MR): A Therapeutic Target Against Ventricular Arrhythmias. Front. Endocrinol. 2021, 12, 694758. [Google Scholar] [CrossRef] [PubMed]
- Funder, J.W. Aldosterone and Mineralocorticoid Receptors-Physiology and Pathophysiology. Int. J. Mol. Sci. 2017, 18, 1032. [Google Scholar] [CrossRef] [PubMed]
- Igbekele, A.E.; Jia, G.; Hill, M.A.; Sowers, J.R.; Jia, G. Mineralocorticoid Receptor Activation in Vascular Insulin Resistance and Dysfunction. Int. J. Mol. Sci. 2022, 23, 8954. [Google Scholar] [CrossRef] [PubMed]
- Vanderhaeghen, T.; Timmermans, S.; Watts, D.; Paakinaho, V.; Eggermont, M.; Vandewalle, J.; Wallaeys, C.; Van Wyngene, L.; Van Looveren, K.; Nuyttens, L.; et al. Reprogramming of glucocorticoid receptor function by hypoxia. EMBO Rep. 2022, 23, e53083. [Google Scholar] [CrossRef] [PubMed]
- McEown, K.; Treit, D. Mineralocorticoid receptors in the medial prefrontal cortex and hippocampus mediate rats’ unconditioned fear behaviour. Horm. Behav. 2011, 60, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Reul, J.M.; de Kloet, E.R. Two receptor systems for corticosterone in rat brain: Microdistribution and differential occupation. Endocrinology 1985, 117, 2505–2511. [Google Scholar] [CrossRef] [PubMed]
- Reul, J.M.; de Kloet, E.R.; van Sluijs, F.J.; Rijnberk, A.; Rothuizen, J. Binding characteristics of mineralocorticoid and glucocorticoid receptors in dog brain and pituitary. Endocrinology 1990, 127, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Ahmadpour, D.; Grange-Messent, V. Involvement of Testosterone Signaling in the Integrity of the Neurovascular Unit in the Male: Review of Evidence, Contradictions, and Hypothesis. Neuroendocrinology 2021, 111, 403–420. [Google Scholar] [CrossRef] [PubMed]
- Castelli, M.P.; Casti, A.; Casu, A.; Frau, R.; Bortolato, M.; Spiga, S.; Ennas, M.G. Regional distribution of 5α-reductase type 2 in the adult rat brain: An immunohistochemical analysis. Psychoneuroendocrinology 2013, 38, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Hosoya, T.; Onoe, K.; Takashima, T.; Tanaka, M.; Ishii, A.; Nakatomi, Y.; Tazawa, S.; Takahashi, K.; Doi, H.; et al. Association between aromatase in human brains and personality traits. Sci. Rep. 2018, 8, 16841. [Google Scholar] [CrossRef] [PubMed]
- Ghoumari, A.M.; Abi Ghanem, C.; Asbelaoui, N.; Schumacher, M.; Hussain, R. Roles of Progesterone, Testosterone and Their Nuclear Receptors in Central Nervous System Myelination and Remyelination. Int. J. Mol. Sci. 2020, 21, 3163. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.J.; Lee, T.Y.; Kim, N.S.; Kwon, J.S. The Role of Estrogen Receptors and Their Signaling across Psychiatric Disorders. Int. J. Mol. Sci. 2020, 22, 373. [Google Scholar] [CrossRef] [PubMed]
- Jiao, L.; Machuki, J.O.; Wu, Q.; Shi, M.; Fu, L.; Adekunle, A.O.; Tao, X.; Xu, C.; Hu, X.; Yin, Z.; et al. Estrogen and calcium handling proteins: New discoveries and mechanisms in cardiovascular diseases. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H820–H829. [Google Scholar] [CrossRef] [PubMed]
- Trenti, A.; Tedesco, S.; Boscaro, C.; Trevisi, L.; Bolego, C.; Cignarella, A. Estrogen, Angiogenesis, Immunity and Cell Metabolism: Solving the Puzzle. Int. J. Mol. Sci. 2018, 19, 859. [Google Scholar] [CrossRef] [PubMed]
- Coolen, R.L.; Cambier, J.C.; Spantidea, P.I.; van Asselt, E.; Blok, B.F.M. Androgen receptors in areas of the spinal cord and brainstem: A study in adult male cats. J. Anat. 2021, 239, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, S.R.; Kelleher, C.; Khalil, R.A. Gender-based research underscores sex differences in biological processes, clinical disorders and pharmacological interventions. Biochem. Pharmacol. 2023, 215, 115737. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, R.L.; Lumia, A.R.; McGinnis, M.Y. Androgen receptors, sex behavior, and aggression. Neuroendocrinology 2012, 96, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Watkins, S.C.; Walker, W.H. Testosterone activates mitogen-activated protein kinase via Src kinase and the epidermal growth factor receptor in sertoli cells. Endocrinology 2007, 148, 2066–2074. [Google Scholar] [CrossRef] [PubMed]
- Davey, R.A.; Grossmann, M. Androgen Receptor Structure, Function and Biology: From Bench to Bedside. Clin. Biochem. Rev. 2016, 37, 3–15. [Google Scholar] [PubMed]
- Thomas, P. Membrane Androgen Receptors Unrelated to Nuclear Steroid Receptors. Endocrinology 2019, 160, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, V.S.; Grossmann, M.; Zajac, J.D.; Davey, R.A. The role of the androgen receptor in the pathogenesis of obesity and its utility as a target for obesity treatments. Obes. Rev. 2022, 23, e13429. [Google Scholar] [CrossRef] [PubMed]
- Lucas-Herald, A.K.; Touyz, R.M. Androgens and Androgen Receptors as Determinants of Vascular Sex Differences Across the Lifespan. Can. J. Cardiol. 2022, 38, 1854–1864. [Google Scholar] [CrossRef] [PubMed]
- Arterburn, J.B.; Prossnitz, E.R. G Protein-Coupled Estrogen Receptor GPER: Molecular Pharmacology and Therapeutic Applications. Annu. Rev. Pharmacol. Toxicol. 2023, 63, 295–320. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, B.; Byemerwa, J.; Krebs, T.; Lim, F.; Chang, C.Y.; McDonnell, D.P. Estrogen Receptor Signaling in the Immune System. Endocr. Rev. 2023, 44, 117–141. [Google Scholar] [CrossRef] [PubMed]
- Gregorio, K.C.R.; Laurindo, C.P.; Machado, U.F. Estrogen and Glycemic Homeostasis: The Fundamental Role of Nuclear Estrogen Receptors ESR1/ESR2 in Glucose Transporter GLUT4 Regulation. Cells 2021, 10, 99. [Google Scholar] [CrossRef] [PubMed]
- Rzemieniec, J.; Castiglioni, L.; Gelosa, P.; Muluhie, M.; Mercuriali, B.; Sironi, L. Nuclear Receptors in Myocardial and Cerebral Ischemia-Mechanisms of Action and Therapeutic Strategies. Int. J. Mol. Sci. 2021, 22, 12326. [Google Scholar] [CrossRef] [PubMed]
- Guajardo-Correa, E.; Silva-Agüero, J.F.; Calle, X.; Chiong, M.; Henríquez, M.; García-Rivas, G.; Latorre, M.; Parra, V. Estrogen signaling as a bridge between the nucleus and mitochondria in cardiovascular diseases. Front. Cell Dev. Biol. 2022, 10, 968373. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhao, Z.; Lin, M.; Groban, L. Activation of GPR30 inhibits cardiac fibroblast proliferation. Mol. Cell. Biochem. 2015, 405, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Klinge, C.M. Estrogenic control of mitochondrial function. Redox Biol. 2020, 31, 101435. [Google Scholar] [CrossRef] [PubMed]
- Kurmann, L.; Okoniewski, M.; Dubey, R.K. Estradiol Inhibits Human Brain Vascular Pericyte Migration Activity: A Functional and Transcriptomic Analysis. Cells 2021, 10, 2314. [Google Scholar] [CrossRef] [PubMed]
- Machuki, J.O.; Zhang, H.Y.; Harding, S.E.; Sun, H. Molecular pathways of oestrogen receptors and β-adrenergic receptors in cardiac cells: Recognition of their similarities, interactions and therapeutic value. Acta Physiol. 2018, 222, e12978. [Google Scholar] [CrossRef] [PubMed]
- da Silva, J.S.; Montagnoli, T.L.; Rocha, B.S.; Tacco, M.L.C.A.; Marinho, S.C.P.; Zapata-Sudo, G. Estrogen Receptors: Therapeutic Perspectives for the Treatment of Cardiac Dysfunction after Myocardial Infarction. Int. J. Mol. Sci. 2021, 22, 525. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, N.; Silveyra, P. Estrogen receptor signaling mechanisms. Adv. Protein Chem. Struct. Biol. 2019, 116, 135–170. [Google Scholar] [CrossRef] [PubMed]
- Tran, Q.K. Reciprocality Between Estrogen Biology and Calcium Signaling in the Cardiovascular System. Front. Endocrinol. 2020, 11, 568203. [Google Scholar] [CrossRef] [PubMed]
- Menazza, S.; Murphy, E. The Expanding Complexity of Estrogen Receptor Signaling in the Cardiovascular System. Circ. Res. 2016, 118, 994–1007. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, S.; Mäkelä, S.; Treuter, E.; Tujague, M.; Thomsen, J.; Andersson, G.; Enmark, E.; Pettersson, K.; Warner, M.; Gustafsson, J.A. Mechanisms of estrogen action. Physiol. Rev. 2001, 81, 1535–1565. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Aponte, A.M.; Menazza, S.; Gucek, M.; Steenbergen, C.; Murphy, E. Additive cardioprotection by pharmacological postconditioning with hydrogen sulfide and nitric oxide donors in mouse heart: S-sulfhydration vs. S-nitrosylation. Cardiovasc. Res. 2016, 110, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, M.A.; Budish, R.A.; Kashyap, S.; Lindsey, S.H. GPER-novel membrane oestrogen receptor. Clin. Sci. 2016, 130, 1005–1016. [Google Scholar] [CrossRef] [PubMed]
- Shughrue, P.J.; Lane, M.V.; Merchenthaler, I. Comparative distribution of estrogen receptor-alpha and -beta mRNA in the rat central nervous system. J. Comp. Neurol. 1997, 388, 507–525. [Google Scholar] [CrossRef] [PubMed]
- Prossnitz, E.R.; Barton, M. The G-protein-coupled estrogen receptor GPER in health and disease. Nat. Rev. Endocrinol. 2011, 7, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Brann, D.W.; Lu, Y.; Wang, J.; Zhang, Q.; Thakkar, R.; Sareddy, G.R.; Pratap, U.P.; Tekmal, R.R.; Vadlamudi, R.K. Brain-derived estrogen and neural function. Neurosci. Biobehav. Rev. 2022, 132, 793–817. [Google Scholar] [CrossRef] [PubMed]
- Aickareth, J.; Hawwar, M.; Sanchez, N.; Gnanasekaran, R.; Zhang, J. Membrane Progesterone Receptors (mPRs/PAQRs) Are Going beyond Its Initial Definitions. Membranes 2023, 13, 260. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.M.; Imami, N.; Johnson, M.R. Progesterone Modulation of Pregnancy-Related Immune Responses. Front. Immunol. 2018, 9, 1293. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Su, C.; Ng, S. Non-genomic mechanisms of progesterone action in the brain. Front. Neurosci. 2013, 7, 159. [Google Scholar] [CrossRef] [PubMed]
- Vítků, J.; Hampl, R. Steroid Conjugates and Their Physiological Role. Physiol. Res. 2023, 72, S317–S322. [Google Scholar] [CrossRef] [PubMed]
- Pinna, G. Allopregnanolone (1938–2019): A trajectory of 80 years of outstanding scientific achievements. Neurobiol. Stress 2020, 13, 100246. [Google Scholar] [CrossRef] [PubMed]
- Buijs, R.M.; Hermes, M.H.; Kalsbeek, A. The suprachiasmatic nucleus-paraventricular nucleus interactions: A bridge to the neuroendocrine and autonomic nervous system. Prog. Brain Res. 1998, 119, 365–382. [Google Scholar] [CrossRef] [PubMed]
- Szczepanska-Sadowska, E.; Czarzasta, K.; Cudnoch-Jedrzejewska, A. Dysregulation of the Renin-Angiotensin System and the Vasopressinergic System Interactions in Cardiovascular Disorders. Curr. Hypertens. Rep. 2018, 20, 19. [Google Scholar] [CrossRef] [PubMed]
- Szczepanska-Sadowska, E.; Zera, T.; Sosnowski, P.; Cudnoch-Jedrzejewska, A.; Puszko, A.; Misicka, A. Vasopressin and Related Peptides; Potential Value in Diagnosis, Prognosis and Treatment of Clinical Disorders. Curr. Drug Metab. 2017, 18, 306–345. [Google Scholar] [CrossRef] [PubMed]
- Amico, J.A.; Finn, F.M.; Haldar, J. Oxytocin and vasopressin are present in human and rat pancreas. Am. J. Med. Sci. 1988, 296, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Hupf, H.; Grimm, D.; Riegger, G.A.; Schunkert, H. Evidence for a vasopressin system in the rat heart. Circ. Res. 1999, 84, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Burbach, J.P.; Luckman, S.M.; Murphy, D.; Gainer, H. Gene regulation in the magnocellular hypothalamo-neurohypophysial system. Physiol. Rev. 2001, 81, 1197–1267. [Google Scholar] [CrossRef] [PubMed]
- Sparapani, S.; Millet-Boureima, C.; Oliver, J.; Mu, K.; Hadavi, P.; Kalostian, T.; Ali, N.; Avelar, C.M.; Bardies, M.; Barrow, B.; et al. The Biology of Vasopressin. Biomedicines 2021, 9, 89. [Google Scholar] [CrossRef] [PubMed]
- Morgenthaler, N.G.; Struck, J.; Alonso, C.; Bergmann, A. Assay for the measurement of copeptin, a stable peptide derived from the precursor of vasopressin. Clin. Chem. 2006, 52, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Rechlin, T.; Hochholzer, W.; Stelzi, C.; Laule, K.; Freidank, H.; Morgenthaler, N.G.; Bergmann, A.; Potocki, M.; Noveanu, M.; Breidthardt, T.; et al. Incremental value of Copeptin for rapid rule out of acute myocardial infarction. J. Am. Coll. Cardiol. 2009, 54, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Roffi, M.; Patrono, C. CardioPulse: ‘Ten Commandments’ of 2015 European Society of Cardiology Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation (NSTE-ACS). Eur. Heart J. 2016, 37, 208. [Google Scholar] [CrossRef] [PubMed]
- Aikins, A.O.; Nguyen, D.H.; Paundralingga, O.; Farmer, G.E.; Shimoura, C.G.; Brock, C.; Cunningham, J.T. Cardiovascular Neuroendocrinology: Emerging Role for Neurohypophyseal Hormones in Pathophysiology. Endocrinology 2021, 162, bqab082. [Google Scholar] [CrossRef] [PubMed]
- Costello, H.M.; Krilis, G.; Grenier, C.; Severs, D.; Czopek, A.; Ivy, J.R.; Nixon, M.; Holmes, M.C.; Livingstone, D.E.W.; Hoorn, E.J.; et al. High salt intake activates the hypothalamic-pituitary-adrenal axis, amplifies the stress response, and alters tissue glucocorticoid exposure in mice. Cardiovasc. Res. 2023, 119, 1740–1750. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, D.S. The extended autonomic system, dyshomeostasis, and COVID-19. Clin. Auton. Res. 2020, 30, 299–315. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, Y.; Oiso, Y.; Saito, H.; Majzoub, J.A. Positive and negative regulation of the rat vasopressin gene promoter. Endocrinology 1997, 138, 5266–5274. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Rivier, C. Hypophysiotropic role and hypothalamic gene expression of corticotropin-releasing factor and vasopressin in rats injected with interleukin-1 beta systemically or into the brain ventricles. J. Neuroendocr. 1994, 6, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Grinevich, V.; Ma, X.M.; Jirikowski, G.; Verbalis, J.; Aguilera, G. Lipopolysaccharide endotoxin potentiates the effect of osmotic stimulation on vasopressin synthesis and secretion in the rat hypothalamus. J. Neuroendocr. 2003, 15, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Pardy, K.; Murphy, D.; Carter, D.; Hui, K.M. The influence of interleukin-2 on vasopressin and oxytocin gene expression in the rodent hypothalamus. J. Neuroimmunol. 1993, 42, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Zelazowski, P.; Patchev, V.K.; Zelazowska, E.B.; Chrousos, G.P.; Gold, P.W.; Sternberg, E.M. Release of hypothalamic corticotropin-releasing hormone and arginine-vasopressin by interleukin 1 beta and alpha MSH: Studies in rats with different susceptibility to inflammatory disease. Brain Res. 1993, 631, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Antoni, F.A. Magnocellular Vasopressin and the Mechanism of “Glucocorticoid Escape”. Front. Endocrinol. 2019, 10, 422. [Google Scholar] [CrossRef] [PubMed]
- Luo X, Kiss A, Makara G, Lolait SJ, Aguilera G Stress-specific regulation of corticotropin releasing hormone receptor expression in the paraventricular and supraoptic nuclei of the hypothalamus in the rat. J. Neuroendocr. 1994, 6, 689–696. [CrossRef] [PubMed]
- Sawchenko, P.E. Adrenalectomy-induced enhancement of CRF and vasopressin immunoreactivity in parvocellular neurosecretory neurons: Anatomic, peptide, and steroid specificity. J. Neurosci. 1987, 7, 1093–1106. [Google Scholar] [CrossRef] [PubMed]
- Kovács, K.J.; Földes, A.; Sawchenko, P.E. Glucocorticoid negative feedback selectively targets vasopressin transcription in parvocellular neurosecretory neurons. J. Neurosci. 2000, 20, 3843–3852. [Google Scholar] [CrossRef] [PubMed]
- Kageyama, K.; Suda, T. Regulatory mechanisms underlying corticotropin-releasing factor gene expression in the hypothalamus. Endocr. J. 2009, 56, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M. Gene regulation system of vasopressin and corticotropin-releasing hormone. Gene Regul. Syst. Bio. 2008, 2, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Mills, N.J.; Sharma, K.; Haque, M.; Moore, M.; Teruyama, R. Aldosterone Mediated Regulation of Epithelial Sodium Channel (ENaC) Subunits in the Rat Hypothalamus. Neuroscience 2018, 390, 278–292. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, G.; Liu, Y. The molecular physiology of CRH neurons. Front. Neuroendocrinol. 2012, 33, 67–84. [Google Scholar] [CrossRef] [PubMed]
- Bous, J.; Fouillen, A.; Orcel, H.; Granier, S.; Bron, P.; Mouillac, B. Structures of the arginine-vasopressin and oxytocin receptor signaling complexes. Vitam. Horm. 2023, 123, 67–107. [Google Scholar] [CrossRef] [PubMed]
- Holmes, C.L.; Landry, D.W.; Granton, J.T. Science Review: Vasopressin and the cardiovascular system part 2—Clinical physiology. Crit. Care 2004, 8, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Bao, A.M.; Meynen, G.; Swaab, D.F. The stress system in depression and neurodegeneration: Focus on the human hypothalamus. Brain Res. Rev. 2008, 57, 531–553. [Google Scholar] [CrossRef] [PubMed]
- Viau, V.; Chu, A.; Soriano, L.; Dallman, M.F. Independent and overlapping effects of corticosterone and testosterone on corticotropin-releasing hormone and arginine vasopressin mRNA expression in the paraventricular nucleus of the hypothalamus and stress-induced adrenocorticotropic hormone release. J. Neurosci. 1999, 19, 6684–6693. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.P.; Tasker, J.G. Paraventricular Hypothalamic Mechanisms of Chronic Stress Adaptation. Front. Endocrinol. 2016, 7, 137. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.A.; Tan, S.M.L.; Hale, T.M.; Handa, R.J. Androgens and Their Role in Regulating Sex Differences in the Hypothalamic/Pituitary/Adrenal Axis Stress Response and Stress-Related Behaviors. Androg. Clin. Res. Ther. 2021, 2, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Thai, B.S.; Chia, L.Y.; Nguyen, A.T.N.; Qin, C.; Ritchie, R.H.; Hutchinson, D.S.; Kompa, A.; White, P.J.; May, L.T. Targeting G protein-coupled receptors for heart failure treatment. Br. J. Pharmacol. 2024, 181, 2270–2286. [Google Scholar] [CrossRef] [PubMed]
- Morello, J.P.; Bichet, D.G. Nephrogenic diabetes insipidus. Annu. Rev. Physiol. 2001, 63, 607–630. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Tian, D.; Cen, J.; Duan, L.; Xia, W. Novel AVPR2 mutations and clinical characteristics in 28 Chinese families with congenital nephrogenic diabetes insipidus. J. Endocrinol. Investig. 2021, 44, 2777–2783. [Google Scholar] [CrossRef] [PubMed]
- Dekan, Z.; Kremsmayr, T.; Keov, P.; Godin, M.; Teakle, N.; Dürrauer, L.; Xiang, H.; Gharib, D.; Bergmayr, C.; Hellinger, R.; et al. Nature-inspired dimerization as a strategy to modulate neuropeptide pharmacology exemplified with vasopressin and oxytocin. Chem. Sci. 2021, 12, 4057–4062. [Google Scholar] [CrossRef] [PubMed]
- Murat, B.; Devost, D.; Andrés, M.; Mion, J.; Boulay, V.; Corbani, M.; Zingg, H.H.; Guillon, G. V1b and CRHR1 receptor heterodimerization mediates synergistic biological actions of vasopressin and CRH. Mol. Endocrinol. 2012, 26, 502–520. [Google Scholar] [CrossRef] [PubMed]
- Patchev, V.K.; Almeida, O.F. Corticosteroid regulation of gene expression and binding characteristics of vasopressin receptors in the rat brain. Eur. J. Neurosci. 1995, 7, 1579–1583. [Google Scholar] [CrossRef] [PubMed]
- Wasilewski, M.A.; Grisanti, L.A.; Song, J.; Carter, R.L.; Repas, A.A.; Myers, V.D.; Gao, E.; Koch, W.J.; Cheung, J.Y.; Feldman, A.M.; et al. Vasopressin type 1A receptor deletion enhances cardiac contractility, β-adrenergic receptor sensitivity and acute cardiac injury-induced dysfunction. Clin. Sci. 2016, 130, 2017–2027. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, M.; Tsuchiya, K.; Maruyama, R.; Takemura, G.; Minatoguchi, S.; Fujiwara, H. Vasopressin inhibits sarcolemmal ATP-sensitive K+ channels via V1 receptors activation in the guinea pig heart. Circ. J. 2002, 66, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Hantash, B.M.; Thomas, A.P.; Reeves, J.P. Regulation of the cardiac L-type calcium channel in L6 cells by arginine-vasopressin. Biochem. J. 2006, 400, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Tilley, D.G.; Zhu, W.; Myers, V.D.; Barr, L.A.; Gao, E.; Li, X.; Song, J.; Carter, R.L.; Makarewich, C.A.; Yu, D.; et al. β-adrenergic receptor-mediated cardiac contractility is inhibited via vasopressin type 1A-receptor-dependent signaling. Circulation 2014, 130, 1800–1811. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Tilley, D.G.; Myers, V.D.; Coleman, R.C.; Feldman, A.M. Arginine vasopressin enhances cell survival via a G protein-coupled receptor kinase 2/β-arrestin1/extracellular-regulated kinase 1/2-dependent pathway in H9c2 cells. Mol. Pharmacol. 2013, 84, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Tilley, D.G.; Myers, V.D.; Tsai, E.J.; Feldman, A.M. Increased vasopressin 1A receptor expression in failing human hearts. J. Am. Coll. Cardiol. 2014, 63, 375–376. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Sun, S.; Wang, X.; Ni, E.; Zhao, L.; Zhu, W. GRK2 Mediates Arginine Vasopressin-Induced Interleukin-6 Production via Nuclear Factor-kappaB Signaling Neonatal Rat Cardiac Fibroblast. Mol. Pharmacol. 2017, 92, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Bucher, M.; Hobbhahn, J.; Taeger, K.; Kurtz, A. Cytokine-mediated downregulation of vasopressin V(1A) receptors during acute endotoxemia in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 282, R979–R984. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.; Innala, L.; Viau, V. Central vasopressin V1A receptor blockade impedes hypothalamic-pituitary-adrenal habituation to repeated restraint stress exposure in adult male rats. Neuropsychopharmacology 2012, 37, 2712–2719. [Google Scholar] [CrossRef] [PubMed]
- Roper, J.; O’Carroll, A.M.; Young, W., 3rd; Lolait, S. The vasopressin Avpr1b receptor: Molecular and pharmacological studies. Stress 2011, 14, 98–115. [Google Scholar] [CrossRef] [PubMed]
- Lolait, S.J.; O’Carroll, A.M.; Mahan, L.C.; Felder, C.C.; Button, D.C.; Young, W.S., 3rd; Mezey, E.; Brownstein, M.J. Extrapituitary expression of the rat V1b vasopressin receptor gene. Proc. Natl. Acad. Sci. USA 1995, 92, 6783–6787. [Google Scholar] [CrossRef] [PubMed]
- O’Carroll, A.M.; Howell, G.M.; Roberts, E.M.; Lolait, S.J. Vasopressin potentiates corticotropin-releasing hormone-induced insulin release from mouse pancreatic beta-cells. J. Endocrinol. 2008, 197, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Raghuram, V.; Chen, L.; Chou, C.L.; Yang, C.R.; Khundmiri, S.J.; Knepper, M.A. Vasopressin V2 receptor, tolvaptan, and ERK1/2 phosphorylation in the renal collecting duct. Am. J. Physiol. Ren. Physiol. 2024, 326, F57–F68. [Google Scholar] [CrossRef] [PubMed]
- Schrier, R.W. Molecular mechanisms of clinical concentrating and diluting disorders. Prog. Brain Res. 2008, 170, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Lightman, S.L.; Birnie, M.T.; Conway-Campbell, B.L. Dynamics of ACTH and Cortisol Secretion and Implications for Disease. Endocr. Rev. 2020, 41, bnaa002. [Google Scholar] [CrossRef]
- Yates, F.E.; Russell, S.M.; Dallman, M.F.; Hodge, G.A.; McCann, S.M.; Dhariwal, A.P. Potentiation by vasopressin of corticotropin release induced by corticotropin-releasing factor. Endocrinology 1971, 88, 3–15. [Google Scholar] [CrossRef]
- Joseph, D.N.; Whirledge, S. Stress and the HPA Axis: Balancing Homeostasis and Fertility. Int. J. Mol. Sci. 2017, 18, 2224. [Google Scholar] [CrossRef]
- Robertson-Dixon, I.; Murphy, M.J.; Crewther, S.G.; Riddell, N. The Influence of Light Wavelength on Human HPA Axis Rhythms: A Systematic Review. Life 2023, 13, 1968. [Google Scholar] [CrossRef]
- Otsuka, H.; Abe, M.; Kobayashi, H. The Effect of Aldosterone on Cardiorenal and Metabolic Systems. Int. J. Mol. Sci. 2023, 24, 5370. [Google Scholar] [CrossRef]
- Sztechman, D.; Czarzasta, K.; Cudnoch-Jedrzejewska, A.; Szczepanska-Sadowska, E.; Zera, T. Aldosterone and mineralocorticoid receptors in regulation of the cardiovascular system and pathological remodelling of the heart and arteries. J. Physiol. Pharmacol. 2018, 69, 829–845. [Google Scholar] [CrossRef]
- Albert, K.M.; Newhouse, P.A. Estrogen, Stress, and Depression: Cognitive and Biological Interactions. Annu. Rev. Clin. Psychol. 2019, 15, 399–423. [Google Scholar] [CrossRef]
- Patel, S.; Homaei, A.; Raju, A.B.; Meher, B.R. Estrogen: The necessary evil for human health, and ways to tame it. Biomed. Pharmacother. 2018, 102, 403–411. [Google Scholar] [CrossRef]
- Nelson, L.R.; Bulun, S.E. Estrogen production and action. J. Am. Acad. Dermatol. 2001, 45 (Suppl. S3), S116–S124. [Google Scholar] [CrossRef]
- Naamneh Elzenaty, R.; du Toit, T.; Flück, C.E. Basics of androgen synthesis and action. Best Pract. Res. Clin. Endocrinol. Metab. 2022, 36, 101665. [Google Scholar] [CrossRef]
- Fanelli, F.; Baronio, F.; Ortolano, R.; Mezzullo, M.; Cassio, A.; Pagotto, U.; Balsamo, A. Normative Basal Values of Hormones and Proteins of Gonadal and Adrenal Functions from Birth to Adulthood. Sex. Dev. 2018, 12, 50–94. [Google Scholar] [CrossRef]
- Kulle, A.E.; Riepe, F.G.; Melchior, D.; Hiort, O.; Holterhus, P.M. A novel ultrapressure liquid chromatography tandem mass spectrometry method for the simultaneous determination of androstenedione, testosterone, and dihydrotestosterone in pediatric blood samples: Age- and sex-specific reference data. J. Clin. Endocrinol. Metab. 2010, 95, 2399–2409. [Google Scholar] [CrossRef]
- Zirkin, B.R.; Papadopoulos, V. Leydig cells: Formation, function, and regulation. Biol. Reprod. 2018, 99, 101–111. [Google Scholar] [CrossRef]
- Midzak, A.; Akula, N.; Lecanu, L.; Papadopoulos, V. Novel androstenetriol interacts with the mitochondrial translocator protein and controls steroidogenesis. J. Biol. Chem. 2011, 286, 9875–9887. [Google Scholar] [CrossRef]
- Beattie, M.C.; Adekola, L.; Papadopoulos, V.; Chen, H.; Zirkin, B.R. Leydig cell aging and hypogonadism. Exp. Gerontol. 2015, 68, 87–91. [Google Scholar] [CrossRef]
- Payne, A.H.; Hales, D.B. Overview of steroidogenic enzymes in the pathway from cholesterol to active steroid hormones. Endocr. Rev. 2004, 25, 947–970. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, F.; Ye, L.; Zirkin, B.; Chen, H. Steroidogenesis in Leydig cells: Effects of aging and environmental factors. Reproduction 2017, 154, R111–R122. [Google Scholar] [CrossRef]
- Kuo, T.; McQueen, A.; Chen, T.C.; Wang, J.C. Regulation of Glucose Homeostasis by Glucocorticoids. Adv. Exp. Med. Biol. 2015, 872, 99–126. [Google Scholar] [CrossRef]
- Yoshimura, M.; Conway-Campbell, B.; Ueta, Y. Arginine vasopressin: Direct and indirect action on metabolism. Peptides 2021, 142, 170555. [Google Scholar] [CrossRef]
- Nakata, M.; Gantulga, D.; Santoso, P.; Zhang, B.; Masuda, C.; Mori, M.; Okada, T.; Yada, T. Paraventricular NUCB2/Nesfatin-1 Supports Oxytocin and Vasopressin Neurons to Control Feeding Behavior and Fluid Balance in Male Mice. Endocrinology 2016, 157, 2322–2332. [Google Scholar] [CrossRef]
- Mohan, S.; Flatt, P.R.; Irwin, N.; Moffett, R.C. Weight-reducing, lipid-lowering and antidiabetic activities of a novel arginine vasopressin analogue acting at the V1a and V1b receptors in high-fat-fed mice. Diabetes Obes. Metab. 2021, 23, 2215–2225. [Google Scholar] [CrossRef]
- Haam, J.; Halmos, K.C.; Di, S.; Tasker, J.G. Nutritional state-dependent ghrelin activation of vasopressin neurons via retrograde trans-neuronal-glial stimulation of excitatory GABA circuits. J. Neurosci. 2014, 34, 6201–6213. [Google Scholar] [CrossRef]
- Iwama, S.; Sugimura, Y.; Murase, T.; Hiroi, M.; Goto, M.; Hayashi, M.; Arima, H.; Oiso, Y. Central adiponectin functions to inhibit arginine vasopressin release in conscious rats. J. Neuroendocr. 2009, 21, 753–759. [Google Scholar] [CrossRef]
- Küchler, S.; Perwitz, N.; Schick, R.R.; Klein, J.; Westphal, S. Arginine-vasopressin directly promotes a thermogenic and pro-inflammatory adipokine expression profile in brown adipocytes. Regul. Pept. 2010, 164, 126–132. [Google Scholar] [CrossRef]
- Rofe, A.M.; Williamson, D.H. Metabolic effects of vasopressin infusion in the starved rat. Reversal of ketonaemia. Biochem. J. 1983, 212, 231–239. [Google Scholar] [CrossRef]
- Vaughan, M. Effect of pitressin on lipolysis and on phosphorylase activity in rat adipose tissue. Am. J. Physiol. 1964, 207, 1166–1168. [Google Scholar] [CrossRef]
- Hiroyama, M.; Aoyagi, T.; Fujiwara, Y.; Oshikawa, S.; Sanbe, A.; Endo, F.; Tanoue, A. Hyperammonaemia in V1a vasopressin receptor knockout mice caused by the promoted proteolysis and reduced intrahepatic blood volume. J. Physiol. 2007, 581 Pt. 3, 1183–1192. [Google Scholar] [CrossRef]
- Hiroyama, M.; Fujiwara, Y.; Nakamura, K.; Aoyagi, T.; Mizutani, R.; Sanbe, A.; Tasaki, R.; Tanoue, A. Altered lipid metabolism in vasopressin V1B receptor-deficient mice. Eur. J. Pharmacol. 2009, 602, 455–461. [Google Scholar] [CrossRef]
- Velho, G.; El Boustany, R.; Lefèvre, G.; Mohammedi, K.; Fumeron, F.; Potier, L.; Bankir, L.; Bouby, N.; Hadjadj, S.; Marre, M.; et al. Plasma Copeptin, Kidney Outcomes, Ischemic Heart Disease, and All-Cause Mortality in People With Long-standing Type 1 Diabetes. Diabetes Care 2016, 39, 2288–2295. [Google Scholar] [CrossRef]
- Vanhaecke, T.; Perrier, E.T.; Melander, O. A Journey through the Early Evidence Linking Hydration to Metabolic Health. Ann. Nutr. Metab. 2020, 76 (Suppl. S1), 4–9. [Google Scholar] [CrossRef]
- Roussel, R.; El Boustany, R.; Bouby, N.; Potier, L.; Fumeron, F.; Mohammedi, K.; Balkau, B.; Tichet, J.; Bankir, L.; Marre, M.; et al. Plasma Copeptin, AVP Gene Variants, and Incidence of Type 2 Diabetes in a Cohort From the Community. J. Clin. Endocrinol. Metab. 2016, 101, 2432–2439. [Google Scholar] [CrossRef]
- Enhörning, S.; Leosdottir, M.; Wallström, P.; Gullberg, B.; Berglund, G.; Wirfält, E.; Melander, O. Relation between human vasopressin 1a gene variance, fat intake, and diabetes. Am. J. Clin. Nutr. 2009, 89, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Enhörning, S.; Sjögren, M.; Hedblad, B.; Nilsson, P.M.; Struck, J.; Melander, O. Genetic vasopressin 1b receptor variance in overweight and diabetes mellitus. Eur. J. Endocrinol. 2016, 174, 69–75. [Google Scholar] [CrossRef]
- Hew-Butler, T.; Smith-Hale, V.; Pollard-McGrandy, A.; VanSumeren, M. Of Mice and Men-The Physiology, Psychology, and Pathology of Overhydration. Nutrients 2019, 11, 1539. [Google Scholar] [CrossRef]
- Szczepanska-Sadowska, E.; Wsol, A.; Cudnoch-Jedrzejewska, A.; Żera, T. Complementary Role of Oxytocin and Vasopressin in Cardiovascular Regulation. Int. J. Mol. Sci. 2021, 22, 11465. [Google Scholar] [CrossRef]
- Bichet, D.G. Regulation of Thirst and Vasopressin Release. Annu. Rev. Physiol. 2019, 81, 359–373. [Google Scholar] [CrossRef]
- Szczepanska-Sadowska, E. Neuromodulation of Cardiac Ischemic Pain: Role of the Autonomic Nervous System and Vasopressin. J. Integr. Neurosci. 2024, 23, 49. [Google Scholar] [CrossRef] [PubMed]
- Danziger, J.; Zeidel, M.L. Osmotic homeostasis. Clin. J. Am. Soc. Nephrol. 2015, 10, 852–862. [Google Scholar] [CrossRef]
- Zimmerman, C.A.; Lin, Y.C.; Leib, D.E.; Guo, L.; Huey, E.L.; Daly, G.E.; Chen, Y.; Knight, Z.A. Thirst neurons anticipate the homeostatic consequences of eating and drinking. Nature 2016, 537, 680–684. [Google Scholar] [CrossRef]
- Zaelzer, C.; Hua, P.; Prager-Khoutorsky, M.; Ciura, S.; Voisin, D.L.; Liedtke, W.; Bourque, C.W. ΔN-TRPV1: A Molecular Co-detector of Body Temperature and Osmotic Stress. Cell Rep. 2015, 13, 23–30. [Google Scholar] [CrossRef]
- Saker, P.; Farrell, M.J.; Adib, F.R.; Egan, G.F.; McKinley, M.J.; Denton, D.A. Regional brain responses associated with drinking water during thirst and after its satiation. Proc. Natl. Acad. Sci. USA 2014, 111, 5379–5384. [Google Scholar] [CrossRef]
- Cheung, P.W.; Bouley, R.; Brown, D. Targeting the Trafficking of Kidney Water Channels for Therapeutic Benefit. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 175–194. [Google Scholar] [CrossRef]
- Bankir, L.; Bichet, D.G.; Bouby, N. Vasopressin V2 receptors, ENaC, and sodium reabsorption: A risk factor for hypertension? Am. J. Physiol. Ren. Physiol. 2010, 299, F917–F928. [Google Scholar] [CrossRef] [PubMed]
- Fenton, R.A. Essential role of vasopressin-regulated urea transport processes in the mammalian kidney. Pflug. Arch. 2009, 458, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Bankir, L. Antidiuretic action of vasopressin: Quantitative aspects and interaction between V1a and V2 receptor-mediated effects. Cardiovasc. Res. 2001, 51, 372–390. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, C.; Summer, S.N.; Falk, S.; Cadnapaphornchai, M.A.; Chen, Y.C.; Schrier, R.W. Molecular analysis of impaired urinary diluting capacity in glucocorticoid deficiency. Am. J. Physiol. Ren. Physiol. 2006, 290, F1135–F1142. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Huang, Y.; Li, S.; Ge, N.; Li, T.; Wang, Y.; Liu, K.; Liu, C. Glucocorticoids Reverse Diluted Hyponatremia Through Inhibiting Arginine Vasopressin Pathway in Heart Failure Rats. J. Am. Heart Assoc. 2020, 9, e014950. [Google Scholar] [CrossRef] [PubMed]
- Frenkel, A.; Abuhasira, R.; Bichovsky, Y.; Bukhin, A.; Novack, V.; Brotfain, E.; Zlotnik, A.; Klein, M. Examination of the association of steroids with fluid accumulation in critically ill patients, considering the possibility of biases. Sci. Rep. 2021, 11, 5557. [Google Scholar] [CrossRef] [PubMed]
- Kenyon, C.J.; Saccoccio, N.A.; Morris, D.J. Aldosterone effects on water and electrolyte metabolism. J. Endocrinol. 1984, 100, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.G.; Welch, A.K.; Cain, B.D.; Sayeski, P.P.; Gumz, M.L.; Wingo, C.S. Aldosterone: Renal Action and Physiological Effects. Compr. Physiol. 2023, 13, 4409–4491. [Google Scholar] [CrossRef]
- Sladek, C.D.; Somponpun, S.J. Estrogen receptors: Their roles in regulation of vasopressin release for maintenance of fluid and electrolyte homeostasis. Front. Neuroendocr. 2008, 29, 114–127. [Google Scholar] [CrossRef]
- Wang, Y.X.; Crofton, J.T.; Liu, H.; Sato, K.; Brooks, D.P.; Share, L. Estradiol attenuates the antidiuretic action of vasopressin in ovariectomized rats. Am. J. Physiol. 1995, 268 Pt. 2, R951–R957. [Google Scholar] [CrossRef]
- Somponpun, S.J. Neuroendocrine regulation of fluid and electrolyte balance by ovarian steroids: Contributions from central oestrogen receptors. J. Neuroendocr. 2007, 19, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Somponpun, S.J.; Johnson, A.K.; Beltz, T.; Sladek, C.D. Estrogen receptor-alpha expression in osmosensitive elements of the lamina terminalis: Regulation by hypertonicity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 287, R661–R669. [Google Scholar] [CrossRef] [PubMed]
- Voisin, D.L.; Simonian, S.X.; Herbison, A.E. Identification of estrogen receptor-containing neurons projecting to the rat supraoptic nucleus. Neuroscience 1997, 78, 215–228. [Google Scholar] [CrossRef]
- Kuiper, G.G.; Carlsson, B.; Grandien, K.; Enmark, E.; Häggblad, J.; Nilsson, S.; Gustafsson, J.A. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology 1997, 138, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Kuang, W.; Qiu, Z.; Zhou, Z. G protein-coupled estrogen receptor: A promising therapeutic target for aldosterone-induced hypertension. Front. Endocrinol. 2023, 14, 1226458. [Google Scholar] [CrossRef]
- Rodriguez-Giustiniani, P.; Rodriguez-Sanchez, N.; Galloway, S.D.R. Fluid and electrolyte balance considerations for female athletes. Eur. J. Sport. Sci. 2022, 22, 697–708. [Google Scholar] [CrossRef]
- Swenson, K.L.; Sladek, C.D. Gonadal steroid modulation of vasopressin secretion in response to osmotic stimulation. Endocrinology 1997, 138, 2089–2097. [Google Scholar] [CrossRef]
- Siegenthaler, J.; Walti, C.; Urwyler, S.A.; Schuetz, P.; Christ-Crain, M. Copeptin concentrations during psychological stress: The PsyCo study. Eur. J. Endocrinol. 2014, 171, 737–742. [Google Scholar] [CrossRef]
- Swaab, D.F.; Bao, A.M.; Lucassen, P.J. The stress system in the human brain in depression and neurodegeneration. Aging Res. Rev. 2005, 4, 141–194. [Google Scholar] [CrossRef]
- Milutinović-Smiljanić, S.; Šarenac, O.; Lozić-Djurić, M.; Murphy, D.; Japundžić-Žigon, N. Evidence for involvement of central vasopressin V1b and V2 receptors in stress-induced baroreflex desensitization. Br. J. Pharmacol. 2013, 169, 900–908. [Google Scholar] [CrossRef]
- Grassi, D.; Lagunas, N.; Calmarza-Font, I.; Diz-Chaves, Y.; Garcia-Segura, L.M.; Panzica, G.C. Chronic unpredictable stress and long-term ovariectomy affect arginine-vasopressin expression in the paraventricular nucleus of adult female mice. Brain Res. 2014, 1588, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Russell, G.; Lightman, S. The human stress response. Nat. Rev. Endocrinol. 2019, 15, 525–534. [Google Scholar] [CrossRef]
- Borrow, A.P.; Bales, N.J.; Stover, S.A.; Handa, R.J. Chronic Variable Stress Induces Sex-Specific Alterations in Social Behavior and Neuropeptide Expression in the Mouse. Endocrinology 2018, 159, 2803–2814. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Komnenov, D.; Newhouse, L.; Rishi, A.K.; Rossi, N.F. Paraventricular Nucleus V1a Receptor Knockdown Blunts Neurocardiovascular Responses to Acute Stress in Male Rats after Chronic Mild Unpredictable Stress. Physiol. Behav. 2022, 253, 113867. [Google Scholar] [CrossRef] [PubMed]
- Komnenov, D.; Quaal, H.; Rossi, N.F. V1a and V1b vasopressin receptors within the paraventricular nucleus contribute to hypertension in male rats exposed to chronic mild unpredictable stress. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2021, 320, R213–R225. [Google Scholar] [CrossRef] [PubMed]
- Powell-Roach, K.L.; Yao, Y.; Jhun, E.H.; He, Y.; Suarez, M.L.; Ezenwa, M.O.; Molokie, R.E.; Wang, Z.J.; Wilkie, D.J. Vasopressin SNP pain factors and stress in sickle cell disease. PLoS ONE 2019, 14, e0224886. [Google Scholar] [CrossRef] [PubMed]
- Pavlidi, P.; Kokras, N.; Dalla, C. Sex Differences in Depression and Anxiety. Curr. Top. Behav. Neurosci. 2023, 62, 103–132. [Google Scholar] [CrossRef] [PubMed]
- Woodward, E.; Rangel-Barajas, C.; Ringland, A.; Logrip, M.L.; Coutellier, L. Sex-Specific Timelines for Adaptations of Prefrontal Parvalbumin Neurons in Response to Stress and Changes in Anxiety- and Depressive-Like Behaviors. eNeuro 2023, 10, ENEURO.0300-22.2023. [Google Scholar] [CrossRef] [PubMed]
- Rivier, C. Gender, sex steroids, corticotropin-releasing factor, nitric oxide, and the HPA response to stress. Pharmacol. Biochem. Behav. 1999, 64, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Viau, V. Functional cross-talk between the hypothalamic-pituitary-gonadal and -adrenal axes. J. Neuroendocr. 2002, 14, 506–513. [Google Scholar] [CrossRef]
- Teo, C.H.; Wong, A.C.H.; Sivakumaran, R.N.; Parhar, I.; Soga, T. Gender Differences in Cortisol and Cortisol Receptors in Depression: A Narrative Review. Int. J. Mol. Sci. 2023, 24, 7129. [Google Scholar] [CrossRef] [PubMed]
- Rosinger, Z.J.; Jacobskind, J.S.; De Guzman, R.M.; Justice, N.J.; Zuloaga, D.G. A sexually dimorphic distribution of corticotropin-releasing factor receptor 1 in the paraventricular hypothalamus. Neuroscience 2019, 409, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Cox, K.H.; Quinnies, K.M.; Eschendroeder, A.; Didrick, P.M.; Eugster, E.A.; Rissman, E.F. Number of X-chromosome genes influences social behavior and vasopressin gene expression in mice. Psychoneuroendocrinology 2015, 51, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.E.; Holsen, L.M.; Ironside, M.; Moser, A.D.; Duda, J.M.; Null, K.E.; Perlo, S.; Richards, C.E.; Nascimento, N.F.; Du, F.; et al. Neural response to stress differs by sex in young adulthood. Psychiatry Res. Neuroimaging 2023, 332, 111646. [Google Scholar] [CrossRef] [PubMed]
- Pietranera, L.; Saravia, F.; Roig, P.; Lima, A.; De Nicola, A.F. Mineralocorticoid treatment upregulates the hypothalamic vasopressinergic system of spontaneously hypertensive rats. Neuroendocrinology 2004, 80, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Matsuguchi, H.; Schmid, P.G. Acute interaction of vasopressin and neurogenic mechanisms in DOC-salt hypertension. Am. J. Physiol. 1982, 242, H37–H43. [Google Scholar] [CrossRef] [PubMed]
- Ferrario, C.M.; Mohara, O.; Ueno, Y.; Brosnihan, K.B. Hemodynamic and neurohormonal changes in the development of DOC hypertension in the dog. Am. J. Med. Sci. 1988, 295, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Rademaker, M.T.; Charles, C.J.; Nicholls, M.G.; Richards, A.M. Interactions of enhanced urocortin 2 and mineralocorticoid receptor antagonism in experimental heart failure. Circ. Heart Fail. 2013, 6, 825–832. [Google Scholar] [CrossRef] [PubMed]
- Pasquali, R.; Gagliardi, L.; Vicennati, V.; Gambineri, A.; Colitta, D.; Ceroni, L.; Casimirri, F. ACTH and cortisol response to combined corticotropin releasing hormone-arginine vasopressin stimulation in obese males and its relationship to body weight, fat distribution and parameters of the metabolic syndrome. Int. J. Obes. Relat. Metab. Disord. 1999, 23, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Schinke, C.; Hesse, S.; Rullmann, M.; Becker, G.A.; Luthardt, J.; Zientek, F.; Patt, M.; Stoppe, M.; Schmidt, E.; Meyer, K.; et al. Central noradrenaline transporter availability is linked with HPA axis responsiveness and copeptin in human obesity and non-obese controls. Stress 2019, 22, 93–102. [Google Scholar] [CrossRef]
- Canivell, S.; Mohaupt, M.; Ackermann, D.; Pruijm, M.; Guessous, I.; Ehret, G.; Escher, G.; Pechère-Bertschi, A.; Vogt, B.; Devuyst, O.; et al. Copeptin and insulin resistance: Effect modification by age and 11 β-HSD2 activity in a population-based study. J. Endocrinol. Investig. 2018, 41, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Nye, E.J.; Bornstein, S.R.; Grice, J.E.; Tauchnitz, R.; Hockings, G.I.; Strakosch, C.R.; Jackson, R.V.; Torpy, D.J. Interactions between the stimulated hypothalamic-pituitary-adrenal axis and leptin in humans. J. Neuroendocr. 2000, 12, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Kacheva, S.; Kolk, K.; Morgenthaler, N.G.; Brabant, G.; Karges, W. Gender-specific co-activation of arginine vasopressin and the hypothalamic-pituitary-adrenal axis during stress. Clin. Endocrinol. 2015, 82, 570–576. [Google Scholar] [CrossRef] [PubMed]
- Zelena, D.; Mergl, Z.; Makara, G.B. The role of vasopressin in diabetes mellitus-induced hypothalamo-pituitary-adrenal axis activation: Studies in Brattleboro rats. Brain Res. Bull. 2006, 69, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Balapattabi, K.; Little, J.T.; Bachelor, M.E.; Cunningham, R.L.; Cunningham, J.T. Sex Differences in the Regulation of Vasopressin and Oxytocin Secretion in Bile Duct-Ligated Rats. Neuroendocrinology 2021, 111, 237–248. [Google Scholar] [CrossRef]
- Coiro, V.; Volpi, R.; Capretti, L.; Bacchi-Modena, A.; Cigarini, C.; Bianconi, L.; Rossi, G.; Gramellini, D.; Chiodera, P. Arginine vasopressin secretion in non-obese women with polycystic ovary syndrome. Acta Endocrinol. 1989, 121, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Vicennati, V.; Ceroni, L.; Gagliardi, L.; Pagotto, U.; Gambineri, A.; Genghini, S.; Pasquali, R. Response of the hypothalamic-pituitary-adrenal axis to small dose arginine-vasopressin and daily urinary free cortisol before and after alprazolam pre-treatment differs in obesity. J. Endocrinol. Investig. 2004, 27, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Zoorob, R.J.; Cender, D. A different look at corticosteroids. Am. Fam. Physician 1998, 58, 443–450. [Google Scholar] [PubMed]
- Erkut, Z.A.; Pool, C.; Swaab, D.F. Glucocorticoids suppress corticotropin-releasing hormone and vasopressin expression in human hypothalamic neurons. J. Clin. Endocrinol. Metab. 1998, 83, 2066–2073. [Google Scholar] [CrossRef] [PubMed]
- Zanardo, V.; Golin, R.; Chiozza, M.L.; Faggian, D. Dexamethasone does not affect vasopressin release in bronchopulmonary dysplasia. Pediatr. Nephrol. 2000, 15, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Gordijn, M.S.; Gemke, R.J.; van Dalen, E.C.; Rotteveel, J.; Kaspers, G.J. Hypothalamic-pituitary-adrenal (HPA) axis suppression after treatment with glucocorticoid therapy for childhood acute lymphoblastic leukaemia. Cochrane Database Syst. Rev. 2012, CD008727, Updated in Cochrane Database Syst. Rev. 2015, CD008727. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.Y.; Unmehopa, U.A.; Zhou, J.N.; Swaab, D.F. Glucocorticoids suppress vasopressin gene expression in human suprachiasmatic nucleus. J. Steroid Biochem. Mol. Biol. 2006, 98, 248–253. [Google Scholar] [CrossRef] [PubMed]
- von Bardeleben, U.; Holsboer, F.; Stalla, G.K.; Müller, O.A. Combined administration of human corticotropin-releasing factor and lysine vasopressin induces cortisol escape from dexamethasone suppression in healthy subjects. Life Sci. 1985, 37, 1613–1618. [Google Scholar] [CrossRef]
- Escudero, D.S.; Fantinelli, J.C.; Martínez, V.R.; González Arbeláez, L.F.; Amarillo, M.E.; Pérez, N.G.; Díaz, R.G. Hydrocortisone cardioprotection in ischaemia/reperfusion injury involves antioxidant mechanisms. Eur. J. Clin. Investig. 2024, 54, e14172. [Google Scholar] [CrossRef] [PubMed]
- Giugliano, G.R.; Giugliano, R.P.; Gibson, C.M.; Kuntz, R.E. Meta-analysis of corticosteroid treatment in acute myocardial infarction. Am. J. Cardiol. 2003, 91, 1055–1059. [Google Scholar] [CrossRef] [PubMed]
- Tol, M.M.; Shekar, K.; Barnett, A.G.; McGree, J.; McWhinney, B.C.; Ziegenfuss, M.; Ungerer, J.P.; Fraser, J.F. A preliminary investigation into adrenal responsiveness and outcomes in patients with cardiogenic shock after acute myocardial infarction. J. Crit. Care 2014, 29, 470.e1–470.e6. [Google Scholar] [CrossRef] [PubMed]
- Torgersen, C.; Luckner, G.; Schröder, D.C.; Schmittinger, C.A.; Rex, C.; Ulmer, H.; Dünser, M.W. Concomitant arginine-vasopressin and hydrocortisone therapy in severe septic shock: Association with mortality. Intensive Care Med. 2011, 37, 1432–1437. [Google Scholar] [CrossRef] [PubMed]
- Penn, J.; Douglas, W.; Curran, J.; Chaudhuri, D.; Dionne, J.C.; Fernando, S.M.; Granton, D.; Mathew, R.; Rochwerg, B. Efficacy and safety of corticosteroids in cardiac arrest: A systematic review, meta-analysis and trial sequential analysis of randomized control trials. Crit. Care 2023, 27, 12. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Andersen, L.W.; Isbye, D.; Kjærgaard, J.; Kristensen, C.M.; Darling, S.; Zwisler, S.T.; Fisker, S.; Schmidt, J.C.; Kirkegaard, H.; Grejs, A.M.; et al. Effect of Vasopressin and Methylprednisolone vs Placebo on Return of Spontaneous Circulation in Patients With In-Hospital Cardiac Arrest: A Randomized Clinical Trial. JAMA 2021, 326, 1586–1594. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Scott, L.V.; Dinan, T.G. Vasopressin as a target for antidepressant development: An assessment of the available evidence. J. Affect. Disord. 2002, 72, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Simon, N.G.; Guillon, C.; Fabio, K.; Heindel, N.D.; Lu, S.F.; Miller, M.; Ferris, C.F.; Brownstein, M.J.; Garripa, C.; Koppel, G.A. Vasopressin antagonists as anxiolytics and antidepressants: Recent developments. Recent. Pat. CNS Drug Discov. 2008, 3, 77–93. [Google Scholar] [CrossRef] [PubMed]
- Poretti, M.B.; Sawant, R.S.; Rask-Andersen, M.; de Cuneo, M.F.; Schiöth, H.B.; Perez, M.F.; Carlini, V.P. Reduced vasopressin receptors activation mediates the anti-depressant effects of fluoxetine and venlafaxine in bulbectomy model of depression. Psychopharmacology 2016, 233, 1077–1086. [Google Scholar] [CrossRef] [PubMed]
- Stewart, L.Q.; Roper, J.A.; Young, W.S., 3rd; O’Carroll, A.M.; Lolait, S.J. The role of the arginine vasopressin Avp1b receptor in the acute neuroendocrine action of antidepressants. Psychoneuroendocrinology 2008, 33, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Kiss, A.; Bundzikova, J.; Pirnik, Z.; Mikkelsen, J.D. Different antipsychotics elicit different effects on magnocellular oxytocinergic and vasopressinergic neurons as revealed by Fos immunohistochemistry. J. Neurosci. Res. 2010, 88, 677–685. [Google Scholar] [CrossRef]
- Florkowski, C.M.; Crozier, I.G.; Nightingale, S.; Evans, M.J.; Ellis, M.J.; Joyce, P.; Donald, R.A. Plasma cortisol, PRL, ACTH, AVP and corticotrophin releasing hormone responses to direct current cardioversion and electroconvulsive therapy. Clin. Endocrinol. 1996, 44, 163–168. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szczepanska-Sadowska, E.; Czarzasta, K.; Bogacki-Rychlik, W.; Kowara, M. The Interaction of Vasopressin with Hormones of the Hypothalamo–Pituitary–Adrenal Axis: The Significance for Therapeutic Strategies in Cardiovascular and Metabolic Diseases. Int. J. Mol. Sci. 2024, 25, 7394. https://doi.org/10.3390/ijms25137394
Szczepanska-Sadowska E, Czarzasta K, Bogacki-Rychlik W, Kowara M. The Interaction of Vasopressin with Hormones of the Hypothalamo–Pituitary–Adrenal Axis: The Significance for Therapeutic Strategies in Cardiovascular and Metabolic Diseases. International Journal of Molecular Sciences. 2024; 25(13):7394. https://doi.org/10.3390/ijms25137394
Chicago/Turabian StyleSzczepanska-Sadowska, Ewa, Katarzyna Czarzasta, Wiktor Bogacki-Rychlik, and Michał Kowara. 2024. "The Interaction of Vasopressin with Hormones of the Hypothalamo–Pituitary–Adrenal Axis: The Significance for Therapeutic Strategies in Cardiovascular and Metabolic Diseases" International Journal of Molecular Sciences 25, no. 13: 7394. https://doi.org/10.3390/ijms25137394