
Liver and Pancreatic Toxicity of Endocrine-Disruptive Chemicals: Focus on Mitochondrial Dysfunction and Oxidative Stress
 , ,
, , 
Abstract
1. Introduction
2. Data Sources
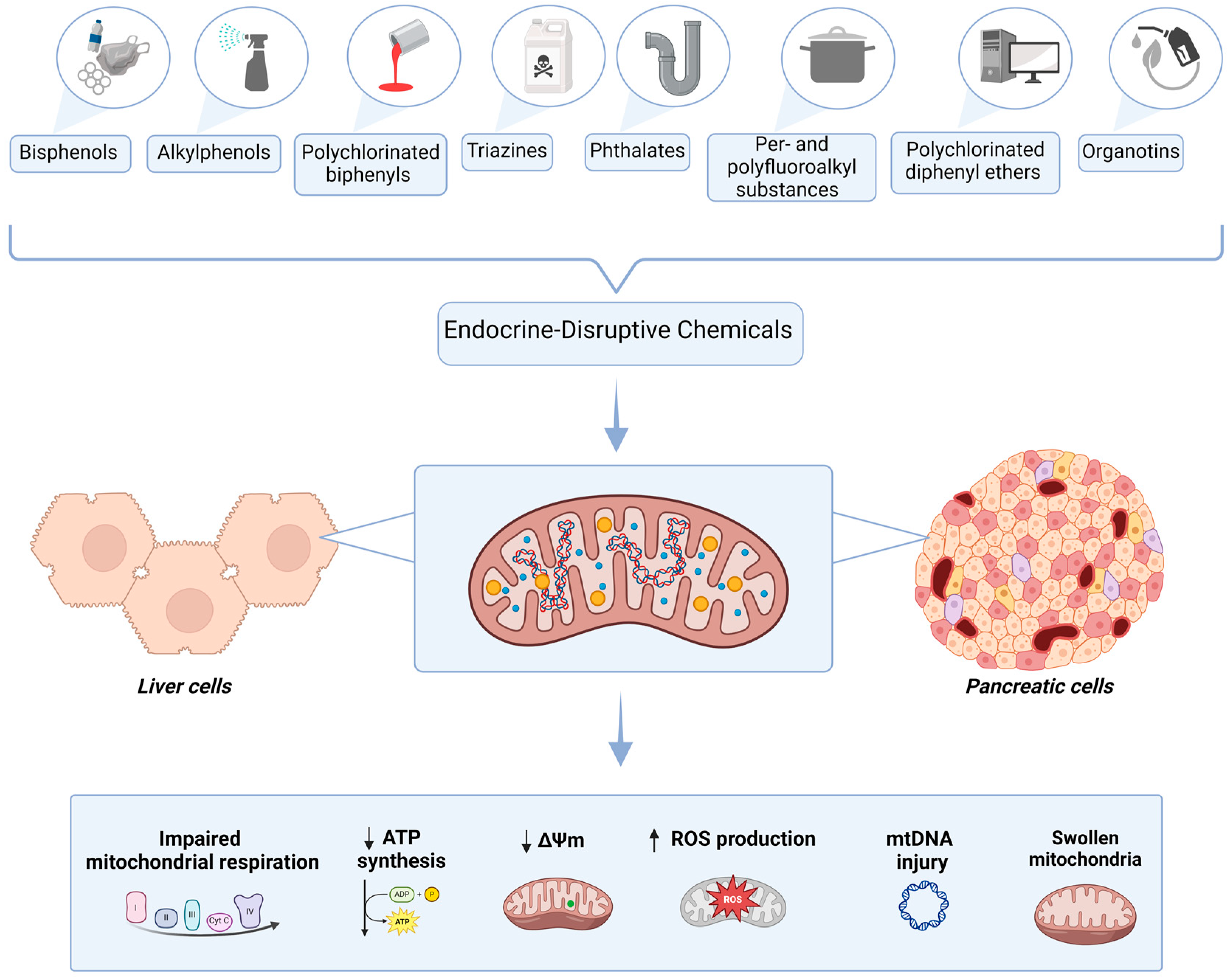
3. Mitochondrial Dysfunction and Oxidative Stress Are Common Pathomechanisms of EDC Toxicity and Metabolic Pathologies
4. EDCs and Liver Toxicity
4.1. Bisphenols (BPs)
4.2. Alkylphenols
4.3. Polychlorinated Biphenyls (PCBs)
4.4. Triazines
4.5. Phthalates
4.6. Per- and Polyfluoroalkyl Substances (PFASs)
4.7. Polychlorinated Diphenyl Ethers (PCDEs)
4.8. Mixtures of EDCs

{kind=link}
| Endocrine Disruptor | Experimental Model | Dose | Duration | Effects | Ref. |
|---|---|---|---|---|---|
| Bisphenol A (BPA) | HepG2 cells | to M | Acute: 24, 48 (and 72) h |
| [50] |
| Bisphenol A (BPA) | HepG2 cells | 10 or 100 nM | Acute: 2, 6, 12 or 24 h |
| [51] |
| Mouse liver | 1.2 mg/kg bw/day | Chronic: 5 days |
| ||
| Bisphenol A (BPA) | Mouse liver | BPA water 50 μg/kg/day | Chronic: 10 weeks |
| [56] |
| Bisphenol A (BPA) | Mouse liver | 50 µg/kg along with HFD (after 12 weeks of HFD feeding) | Chronic: 3 weeks |
| [58] |
| Bisphenol A | Mouse liver | 50 mg/kg/bw along with HFCCD bw | Chronic: 8 weeks |
| [59] |
| Bisphenol A (BPA) | Neonatal rat liver | 1.47 ng/mL | Acute: 1 h |
| [103] |
| Bisphenol A (BPA) | Rat liver | 30 mg/kg bw/day | Chronic: 6 weeks |
| [57] |
| Bisphenol A (BPA) | Rat liver | 50 mg/kg/day | Chronic: 4 weeks |
| [52] |
| Bisphenol A (BPA) | Rat liver | 150 mg/kg, 250 mg/kg and 500 mg/kg | Chronic: 14 days |
| [53] |
| Bisphenol A (BPA) | Rat liver | 10 and 50 mg/kg | Chronic: 30 days |
| [54] |
| Bisphenol A (BPA) | Rat liver | 0.5 mg/kg (low dose), 5 mg/kg (medium dose, NOAEL) or 50 mg/kg (high dose) | Chronic: 30 days |
| [55] |
| Bisphenol A (BPA) | Rat liver | 100 mg/kg | Chronic: 30 days |
| [66] |
| Bisphenol A (BPA) | Rat liver | 90 and 270 mg/kg bw | Chronic: 30 days |
| [60] |
| Bisphenol A (BPA) | Rat liver (L-NAME-induced hypertensive Wistar rats) | 50 µg/kg | Chronic: 30 days |
| [67] |
| Bisphenol A (BPA) | Rat offspring liver | 40 µg/kg/day | Chronic: during gestation and lactation |
| [61] |
| Bisphenol A | Rat liver (from pregnant rats and female postnatal day-6 offspring) | 0.036 mg/kg bw/day and 3.42 mg/kg bw/day | Chronic: during premating, mating, pregnancy, lactation |
| [62] |
| Bisphenol A (BPA) | Common carp (Cyprinus carpio) liver | 0.1, 1, 10, 100 and 1000 µg/L | Chronic: 30 days |
| [63] |
| Bisphenol A (BPA) | Common carp (Cyprinus carpio) liver | 4.5 and 6 mg/L | Chronic: 30 days |
| [64] |
| Bisphenol A (BPA) and S (BPS) | Rat liver | 50 or 500 μg/kg/day of BPA or BPS; 50 μg/kg/day of both BPA and BPS | Chronic: 20 weeks |
| [69] |
| Bisphenol S (BPS) | Mouse liver | 100 µg/kg/day in drinking water | Chronic: 10 weeks |
| [104] |
| Bisphenol S (BPS) | Mouse liver | 0.1–1 mM | Acute: 12 h |
| [70] |
| Bisphenol S (BPS) | Fish (Labeo rohita) liver | Groups II, III and IV were exposed to 80 mg BPS/L, Groups V, VI and VII were exposed to 100 mg BPS/L, and Groups VIII, IX and X to 120 mg BPS/L | Chronic: 7, 14 and 21 days |
| [72] |
| Bisphenols: bisphenol A (BPA), bisphenol F (BPF) and bisphenol AF (BPAF) | Mouse offspring liver | 100 ng/g bw/day | Chronic: from the 7th day of pregnancy to the 21st day after delivery |
| [68] |
| Bisphenol A (BPA) + nonylphenol (NP) | Rat liver | 100 µg/kg | Chronic: 56 consecutive days |
| [75] |
| Nonylphenol (NP) | Zebrafish (Danio rerio) liver | 50 and 100 μg/L | Chronic: 21 days |
| [76] |
| Polychlorinated biphenyls (PCBs) | Rat liver | Contaminated (two times the TDI) goat milk administered daily by gavage (6 µL/g bw) | Chronic: 8 weeks |
| [84] |
| Polychlorinated biphenyls (PCBs)—PCB126 | Mouse liver | 1.53 μmol/kg | Chronic: 10 weeks |
| [85] |
| Polychlorinated biphenyls (PCBs) and 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) | Rat liver | TCDD at 3, 10 or 100 ng/kg/day; PCB 126 at 10, 100 or 1000 ng/kg/day; PCB 153 at 10, 100 or 1000 μg/kg/day; Binary mixture of PCB 126 and PCB 153 (10 ng/kg/day + 10 μg/kg/day, 100 ng/kg/day + 100 μg/kg/day or 1000 ng/kg/day + 1000 μg/kg/day | Chronic: 13 and 52 weeks |
| [81] |
| Atrazine (ATZ) and its metabolite diaminochlorotriazine (DACT) | Mouse liver | ATZ and DACT (100 and 200 mg/kg/bw, respectively) | Chronic: 1 week |
| [86] |
| Atrazine (ATZ) | HepG2 cells | 0.05–2 mM | Acute: 3 h and 6 h |
| [87] |
| Di (2-ethylhexyl) phthalate (DEHP) | HSC-T6 cells | 50 and 100 µM | Chronic: 3.5 months |
| [93] |
| Di (2-ethylhexyl) phthalate (DEHP) | Rat liver | 600 mg/kg/day | Chronic: 12 weeks |
| [94] |
| Di (2-ethylhexyl) phthalate (DEHP) | Female quail (Coturnix japonica) liver | 250, 500 and 1000 mg/kg bw/day | Chronic: 45 days |
| [95] |
| Mono-(2-ethylhexyl) phthalate (MEHP) | Zebrafish liver | 31.25, 62.5, 125, 250, 500 or 1000 mg/L | Acute: 24 h |
| [96] |
| Perfluorooctane sulphonate (PFOS) | Rat liver | Single dose of 1 or 10 mg/kg body | Chronic: 28 consecutive days |
| [97] |
| Polychlorinated diphenyl ethers (PCDEs) | Zebrafish liver | 1, 10 and 50 μg/L | Chronic: 14 days |
| [99] |
| Endocrine disruptors mixture | Rabbit liver | 10 × ADI | Chronic: 12 months |
| [101] |
5. EDCs and Pancreatic Toxicity
5.1. Bisphenols (BPs)
5.2. Alkylphenols
5.3. Organotins
5.4. Mixture of EDCs
6. Discussion
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
List of Abbreviations
| 8-OHdG | 8-hydroxydeoxyguanosine |
| α-SMA | alpha-smooth muscle actin |
| ACE | angiotensin-converting enzyme |
| ADI | acceptable daily intake |
| ALP | alkaline phosphatase |
| ALT | alanine aminotransferase |
| AST | aspartate aminotransferase |
| ATP | adenosine triphosphate |
| ATZ | atrazine |
| Bax | Bcl-2-associated X |
| Bcl-2 | B-cell lymphoma-2 |
| BPA | bisphenol A |
| BPAF | bisphenol AF |
| BPF | bisphenol F |
| BPS | bisphenol S |
| BPs | bisphenols |
| BuPB | butylparaben |
| BW | body weight |
| CAT | catalase |
| CoA | coenzyme A |
| COX | cyclooxygenase |
| COX-2 | cyclooxygenase-2 |
| COX5B | cytochrome c oxidase subunit 5B mitochondrial |
| COQ9 | ubiquinone biosynthesis protein COQ9_mitochondrial |
| DACT | diaminochlorotriazine |
| DDE | dichlorodiphenyldichloroethylene |
| DEHP | di-(2-ethylhexyl) phthalate |
| DES | diethylstilbestrol |
| DNA | deoxyribonucleic acid |
| EDCs | endocrine-disruptive chemicals |
| ERK | extracellular signal-regulated kinase |
| ETS | electron transport system |
| F1 | first-generation adult mice offspring |
| F2 | second-generation adult mice offspring |
| GPx | glutathione peroxidase |
| GPx3 | glutathione peroxidase 3 |
| GR | glutathione reductase |
| GSH | reduced glutathione |
| GSHPx | glutathione peroxidase |
| GSIS | glucose-stimulated insulin secretion |
| GSSG | glutathione disulfide |
| GST | glutathione-S-transferase |
| H2O2 | hydrogen peroxide |
| HFCCD | high-fat/high-cholesterol/high-cholic acid diet |
| HFD | high-fat diet |
| HSC | hepatic stellate cells |
| IL-1β | interleukin-1β |
| IL-6 | interleukin-6 |
| iNOS | inducible nitric oxide synthase |
| JAK/STAT | Janus kinase/signal transduction and transcription activation |
| L-NAME | N-nitro-L-arginine methyl ester |
| LPO | lipid peroxidation |
| MAFLD | metabolic-associated fatty liver disease |
| MAPK | mitogen-activated protein kinase |
| MDA | malondialdehyde |
| MDC | metabolism-disrupting chemicals |
| MEHP | mono(2-ethylhexyl) phthalate |
| MePB | methylparaben |
| mtDNA | mitochondrial DNA |
| MMP | mitochondrial membrane potential |
| MRC | mitochondrial respiratory chain |
| NAC | N-acetylcysteine |
| NAFLD | non-alcoholic fatty liver disease |
| NF-κB | nuclear factor kappa-B |
| NO | nitric oxide |
| NOAEL | no observed adverse effect level |
| NP | nonylphenol |
| Nrf1 | nuclear respiratory factor 1 |
| Nrf2 | nuclear factor erythroid 2-related factor factor 2 |
| NTP | National Toxicology Program |
| OCR | oxygen consumption rate |
| Ogdh | oxoglutarate degydrogenase |
| OH-PCBs | hydroxylated PCBs |
| OP | octylphenol |
| OXPHOS | oxidative phosphorylation |
| PARP-1 | poly [ADP-ribose] polymerase 1 |
| PCBs | polychlorinated biphenyls |
| PCDEs | polychlorinated diphenyl ethers |
| PFOA | perfluorooctanoic acid |
| PFOS | perfluorooctane sulfonate |
| PGC-1α | peroxisome proliferator-activated receptor-gamma coactivator 1α |
| PKC | protein kinase C |
| PND6 | postnatal day 6 |
| POD | peroxidase |
| PrPB | propylparaben |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| Sdhb | succinate dehydrogenase iron-sulfur subunit |
| SOD | superoxide dismutase |
| SIRT1 | sirtuin-1 |
| SIRT3 | sirtuin-3 |
| T-AOC | total antioxidant capacity |
| TBARS | thiobarbituric acid reactive substances |
| TBT | trybutyltin |
| TCDD | 2,3,7,8-tetrachlorodibenzo-p-dioxin |
| TCS | triclosan |
| TFAM | mitochondrial transcription factor A |
| TPP | triphenylphosphate |
| TDI | tolerable daily intake |
| TNF-α | tumor necrosis factor-α |
| UCP2 | uncoupling protein 2 |
References
- Lopez-Jimenez, F.; Almahmeed, W.; Bays, H.; Cuevas, A.; Di Angelantonio, E.; le Roux, C.W.; Sattar, N.; Sun, M.C.; Wittert, G.; Pinto, F.J.; et al. Obesity and cardiovascular disease: Mechanistic insights and management strategies. A joint position paper by the World Heart Federation and World Obesity Federation. Eur. J. Prev. Cardiol. 2022, 29, 2218–2237. [Google Scholar] [CrossRef] [PubMed]
- Preda, A.; Carbone, F.; Tirandi, A.; Montecucco, F.; Liberale, L. Obesity phenotypes and cardiovascular risk: From pathophysiology to clinical management. Rev. Endocr. Metab. Disord. 2023, 24, 901–919. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.W.; Seeley, R.J.; Zeltser, L.M.; Drewnowski, A.; Ravussin, E.; Redman, L.M.; Leibel, R.L. Obesity Pathogenesis: An Endocrine Society Scientific Statement. Endocr. Rev. 2017, 38, 267–296. [Google Scholar] [CrossRef] [PubMed]
- Papalou, O.; Kandaraki, E.A.; Papadakis, G.; Diamanti-Kandarakis, E. Endocrine Disrupting Chemicals: An Occult Mediator of Metabolic Disease. Front. Endocrinol. 2019, 10, 112. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.E.; Sharma, A.M.; Ardern, C.I.; Mirdamadi, P.; Mirdamadi, P.; Kuk, J.L. Secular differences in the association between caloric intake, macronutrient intake, and physical activity with obesity. Obes. Res. Clin. Pract. 2016, 10, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Neel, B.A.; Sargis, R.M. The Paradox of Progress: Environmental Disruption of Metabolism and the Diabetes Epidemic. Diabetes 2011, 60, 1838–1848. [Google Scholar] [CrossRef] [PubMed]
- Baillie-Hamilton, P.F. Chemical Toxins: A Hypothesis to Explain the Global Obesity Epidemic. J. Altern. Complement. Med. 2002, 8, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Schnegelberger, R.D.; Lang, A.L.; Arteel, G.E.; Beier, J.I. Environmental toxicant-induced maladaptive mitochondrial changes: A potential unifying mechanism in fatty liver disease? Acta Pharm. Sin. B 2021, 11, 3756–3767. [Google Scholar] [CrossRef] [PubMed]
- Colborn, T.; Saal, F.S.V.; Soto, A.M. Developmental effects of endocrine-disrupting chemicals in wildlife and humans. Environ. Health Perspect. 1993, 101, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Grun, F.; Blumberg, B. Environmental Obesogens: Organotins and Endocrine Disruption via Nuclear Receptor Signaling. Endocrinology 2006, 147, s50–s55. [Google Scholar] [CrossRef] [PubMed]
- Heindel, J.J.; Blumberg, B. Environmental Obesogens: Mechanisms and Controversies. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 89–106. [Google Scholar] [CrossRef] [PubMed]
- La Merrill, M.; Birnbaum, L.S. Childhood Obesity and Environmental Chemicals. Mt. Sinai J. Med. 2011, 78, 22–48. [Google Scholar] [CrossRef] [PubMed]
- Heindel, J.J.; Blumberg, B.; Cave, M.; Machtinger, R.; Mantovani, A.; Mendez, M.A.; Nadal, A.; Palanza, P.; Panzica, G.; Sargis, R.; et al. Metabolism disrupting chemicals and metabolic disorders. Reprod. Toxicol. 2017, 68, 3–33. [Google Scholar] [CrossRef] [PubMed]
- Mosca, A.; Manco, M.; Braghini, M.R.; Cianfarani, S.; Maggiore, G.; Alisi, A.; Vania, A. Environment, Endocrine Disruptors, and Fatty Liver Disease Associated with Metabolic Dysfunction (MASLD). Metabolites 2024, 14, 71. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, K.; Ziková-Kloas, A.; Marx-Stoelting, P.; Braeuning, A. Metabolism-Disrupting Chemicals Affecting the Liver: Screening, Testing, and Molecular Pathway Identification. Int. J. Mol. Sci. 2023, 24, 2686. [Google Scholar] [CrossRef] [PubMed]
- Amato, A.A.; Wheeler, H.B.; Blumberg, B. Obesity and endocrine-disrupting chemicals. Endocr. Connect. 2021, 10, R87–R105. [Google Scholar] [CrossRef] [PubMed]
- Hinault, C.; Caroli-Bosc, P.; Bost, F.; Chevalier, N. Critical Overview on Endocrine Disruptors in Diabetes Mellitus. Int. J. Mol. Sci. 2023, 24, 4537. [Google Scholar] [CrossRef] [PubMed]
- Predieri, B.; Bruzzi, P.; Bigi, E.; Ciancia, S.; Madeo, S.F.; Lucaccioni, L.; Iughetti, L. Endocrine Disrupting Chemicals and Type 1 Diabetes. Int. J. Mol. Sci. 2020, 21, 2937. [Google Scholar] [CrossRef] [PubMed]
- Massart, J.; Begriche, K.; Corlu, A.; Fromenty, B. Xenobiotic-Induced Aggravation of Metabolic-Associated Fatty Liver Disease. Int. J. Mol. Sci. 2022, 23, 1062. [Google Scholar] [CrossRef] [PubMed]
- Reddam, A.; McLarnan, S.; Kupsco, A. Environmental Chemical Exposures and Mitochondrial Dysfunction: A Review of Recent Literature. Curr. Environ. Health Rep. 2022, 9, 631–649. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.N.; Chan, S.S. Sources, mechanisms, and consequences of chemical-induced mitochondrial toxicity. Toxicology 2017, 391, 2–4. [Google Scholar] [CrossRef] [PubMed]
- Blajszczak, C.; Bonini, M.G. Mitochondria targeting by environmental stressors: Implications for redox cellular signaling. Toxicology 2017, 391, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Leung, M.C.; Meyer, J.N. Mitochondria as a target of organophosphate and carbamate pesticides: Revisiting common mechanisms of action with new approach methodologies. Reprod. Toxicol. 2019, 89, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, S.; Nottola, S.A.; Torge, D.; Palmerini, M.G.; Necozione, S.; Macchiarelli, G. Association between Female Reproductive Health and Mancozeb: Systematic Review of Experimental Models. Int. J. Environ. Res. Public Health 2020, 17, 2580. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Tan, J.; Wan, Z.; Zou, Y.; Afewerky, H.K.; Zhang, Z.; Zhang, T. Effects of Commonly Used Pesticides in China on the Mitochondria and Ubiquitin-Proteasome System in Parkinson’s Disease. Int. J. Mol. Sci. 2017, 18, 2507. [Google Scholar] [CrossRef]
- Monzel, A.S.; Enríquez, J.A.; Picard, M. Multifaceted mitochondria: Moving mitochondrial science beyond function and dysfunction. Nat. Metab. 2023, 5, 546–562. [Google Scholar] [CrossRef] [PubMed]
- Picard, M.; Shirihai, O.S. Mitochondrial signal transduction. Cell Metab. 2022, 34, 1620–1653. [Google Scholar] [CrossRef] [PubMed]
- Mourokh, L.; Friedman, J. Mitochondria at the Nanoscale: Physics Meets Biology—What Does It Mean for Medicine? Int. J. Mol. Sci. 2024, 25, 2835. [Google Scholar] [CrossRef] [PubMed]
- Bleier, L.; Wittig, I.; Heide, H.; Steger, M.; Brandt, U.; Dröse, S. Generator-specific targets of mitochondrial reactive oxygen species. Free Radic. Biol. Med. 2015, 78, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Muntean, D.M.; Sturza, A.; Dănilă, M.D.; Borza, C.; Duicu, O.M.; Mornoș, C. The Role of Mitochondrial Reactive Oxygen Species in Cardiovascular Injury and Protective Strategies. Oxidative Med. Cell. Longev. 2016, 2016, 8254942. [Google Scholar] [CrossRef] [PubMed]
- Hartsoe, P.; Holguin, F.; Chu, H.W. Mitochondrial Dysfunction and Metabolic Reprogramming in Obesity and Asthma. Int. J. Mol. Sci. 2024, 25, 2944. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-L.; Liu, J.-F.; Liu, S.-F. Mitochondrial Dysfunction in Chronic Obstructive Pulmonary Disease: Unraveling the Molecular Nexus. Biomedicines 2024, 12, 814. [Google Scholar] [CrossRef] [PubMed]
- Marroqui, L.; Tudurí, E.; Alonso-Magdalena, P.; Quesada, I.; Nadal, Á.; dos Santos, R.S. Mitochondria as target of endocrine-disrupting chemicals: Implications for type 2 diabetes. J. Endocrinol. 2018, 239, R27–R45. [Google Scholar] [CrossRef]
- Chen, J.; Stimpson, S.E.; Fernandez-Bueno, G.A.; Mathews, C.E. Mitochondrial Reactive Oxygen Species and Type 1 Diabetes. Antioxid. Redox Signal. 2018, 29, 1361–1372. [Google Scholar] [CrossRef] [PubMed]
- Avram, V.F.; Merce, A.P.; Hâncu, I.M.; Bătrân, A.D.; Kennedy, G.; Rosca, M.G.; Muntean, D.M. Impairment of Mitochondrial Respiration in Metabolic Diseases: An Overview. Int. J. Mol. Sci. 2022, 23, 8852. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, S.; Di Lisa, F.; Kaludercic, N. Mitochondrial reactive oxygen species in physiology and disease. Cell Calcium 2021, 94, 102344. [Google Scholar] [CrossRef] [PubMed]
- Nickel, A.; Kohlhaas, M.; Maack, C. Mitochondrial reactive oxygen species production and elimination. J. Mol. Cell. Cardiol. 2014, 73, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Chen, Y.; Zhang, Y. Mitochondrial reactive oxygen species cause major oxidative mitochondrial DNA damages and repair pathways. J. Biosci. 2020, 45, 84. [Google Scholar] [CrossRef]
- Corkey, B.E. Reactive oxygen species: Role in obesity and mitochondrial energy efficiency. Philos. Trans. R. Soc. B Biol. Sci. 2023, 378, 20220210. [Google Scholar] [CrossRef]
- Heindel, J.J.; Lustig, R.H.; Howard, S.; Corkey, B.E. Obesogens: A unifying theory for the global rise in obesity. Int. J. Obes. 2024, 48, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Ge, Y.; Liu, X.; Deng, S.; Li, J.; Tan, P.; Yang, Y.; Wu, Z. Exposure to the environmental pollutant chlorpyrifos induces hepatic toxicity through activation of the JAK/STAT and MAPK pathways. Sci. Total. Environ. 2024, 928, 171711. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Sheng, J.; Pei, H.; Sheng, Y.; Wang, J.; Zhou, X.; Li, W.; Cao, C.; Yang, Y. Environmental toxin chlorpyrifos induces liver injury by activating P53-mediated ferroptosis via GSDMD-mtROS. Ecotoxicol. Environ. Saf. 2023, 257, 114938. [Google Scholar] [CrossRef] [PubMed]
- Ahn, C.; Jeung, E.-B. Endocrine-Disrupting Chemicals and Disease Endpoints. Int. J. Mol. Sci. 2023, 24, 5342. [Google Scholar] [CrossRef] [PubMed]
- Bergman, Å.; Heindel, J.J.; Kasten, T.; Kidd, K.A.; Jobling, S.; Neira, M.; Zoeller, R.T.; Becher, G.; Bjerregaard, P.; Bornman, R.; et al. The Impact of Endocrine Disruption: A Consensus Statement on the State of the Science. Environ. Health Perspect. 2013, 121, A104–A106. [Google Scholar] [CrossRef] [PubMed]
- Vandenberg, L.N.; Maffini, M.V.; Sonnenschein, C.; Rubin, B.S.; Soto, A.M. Bisphenol-A and the Great Divide: A Review of Controversies in the Field of Endocrine Disruption. Endocr. Rev. 2009, 30, 75–95. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.G.; Tungekar, B.; Adiga, D.; Chakrabarty, S.; Rai, P.S.; Kabekkodu, S.P. Alterations induced by Bisphenol A on cellular organelles and potential relevance on human health. Biochim. Biophys. Acta Mol. Cell Res. 2023, 1870, 119505. [Google Scholar] [CrossRef] [PubMed]
- Bindhumol, V.; Chitra, K.; Mathur, P. Bisphenol A induces reactive oxygen species generation in the liver of male rats. Toxicology 2003, 188, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Lang, I.A.; Galloway, T.S.; Scarlett, A.; Henley, W.E.; Depledge, M.; Wallace, R.B.; Melzer, D. Association of Urinary Bisphenol A Concentration with Medical Disorders and Laboratory Abnormalities in Adults. JAMA 2008, 300, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Rancière, F.; Lyons, J.G.; Loh, V.H.; Botton, J.; Galloway, T.; Wang, T.; Shaw, J.E.; Magliano, D.J. Bisphenol A and the risk of cardiometabolic disorders: A systematic review with meta-analysis of the epidemiological evidence. Environ. Health 2015, 14, 46. [Google Scholar] [CrossRef] [PubMed]
- Huc, L.; Lemarié, A.; Guéraud, F.; Héliès-Toussaint, C. Low concentrations of bisphenol A induce lipid accumulation mediated by the production of reactive oxygen species in the mitochondria of HepG2 cells. Toxicol. In Vitro 2012, 26, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Moon, M.K.; Kim, M.J.; Jung, I.K.; Koo, Y.D.; Ann, H.Y.; Lee, K.J.; Kim, S.H.; Yoon, Y.C.; Cho, B.-J.; Park, K.S.; et al. Bisphenol A Impairs Mitochondrial Function in the Liver at Doses below the No Observed Adverse Effect Level. J. Korean Med. Sci. 2012, 27, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Hassan, Z.K.; Elobeid, M.A.; Virk, P.; Omer, S.A.; ElAmin, M.; Daghestani, M.H.; AlOlayan, E.M. Bisphenol A Induces Hepatotoxicity through Oxidative Stress in Rat Model. Oxidative Med. Cell. Longev. 2012, 2012, 194829. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Beigh, S.; Chaudhari, B.P.; Sharma, S.; Abdi, S.A.H.; Ahmad, S.; Ahmad, F.; Parvez, S.; Raisuddin, S. Mitochondrial dysfunction induced by Bisphenol A is a factor of its hepatotoxicity in rats. Environ. Toxicol. 2015, 31, 1922–1934. [Google Scholar] [CrossRef] [PubMed]
- Kourouma, A.; Quan, C.; Duan, P.; Qi, S.; Yu, T.; Wang, Y.; Yang, K. Bisphenol A Induces Apoptosis in Liver Cells through Induction of ROS. Adv. Toxicol. 2015, 2015, 901983. [Google Scholar] [CrossRef]
- Hassani, F.V.; Abnous, K.; Mehri, S.; Jafarian, A.; Birner-Gruenberger, R.; Robati, R.Y.; Hosseinzadeh, H. Proteomics and phosphoproteomics analysis of liver in male rats exposed to bisphenol A: Mechanism of hepatotoxicity and biomarker discovery. Food Chem. Toxicol. 2018, 112, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhao, Z.; Ji, W. Bisphenol A induces apoptosis, oxidative stress and inflammatory response in colon and liver of mice in a mitochondria-dependent manner. Biomed. Pharmacother. 2019, 117, 109182. [Google Scholar] [CrossRef] [PubMed]
- Eweda, S.M.; Newairy, A.S.A.; Abdou, H.M.; Gaber, A.S. Bisphenol A-induced oxidative damage in the hepatic and cardiac tissues of rats: The modulatory role of sesame lignans. Exp. Ther. Med. 2020, 19, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Pirozzi, C.; Lama, A.; Annunziata, C.; Cavaliere, G.; Ruiz-Fernandez, C.; Monnolo, A.; Comella, F.; Gualillo, O.; Stornaiuolo, M.; Mollica, M.P.; et al. Oral Bisphenol A Worsens Liver Immune-Metabolic and Mitochondrial Dysfunction Induced by High-Fat Diet in Adult Mice: Cross-Talk between Oxidative Stress and Inflammasome Pathway. Antioxidants 2020, 9, 1201. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-L.; Wang, Y.-C.; Hsu, Y.-A.; Chen, C.-S.; Weng, R.-C.; Lu, Y.-P.; Chuang, C.-Y.; Wan, L. Bisphenol A Coupled with a High-Fat Diet Promotes Hepatosteatosis through Reactive-Oxygen-Species-Induced CD36 Overexpression. Toxics 2022, 10, 208. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Liu, B.; Tian, L.; Jiang, X.; Li, X.; Cai, D.; Sun, J.; Bai, W.; Jin, Y. Exposure to Bisphenol A Caused Hepatoxicity and Intestinal Flora Disorder in Rats. Int. J. Mol. Sci. 2022, 23, 8042. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Xia, W.; Zhu, Y.; Li, X.; Wang, D.; Liu, J.; Chang, H.; Li, G.; Xu, B.; Chen, X.; et al. Mitochondrial dysfunction in early life resulted from perinatal bisphenol A exposure contributes to hepatic steatosis in rat offspring. Toxicol. Lett. 2014, 228, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Linillos-Pradillo, B.; Rancan, L.; Paredes, S.D.; Schlumpf, M.; Lichtensteiger, W.; Vara, E.; Tresguerres, J.F. Low Dose of BPA Induces Liver Injury through Oxidative Stress, Inflammation and Apoptosis in Long–Evans Lactating Rats and Its Perinatal Effect on Female PND6 Offspring. Int. J. Mol. Sci. 2023, 24, 4585. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.; Chen, J.; Li, Y.; Chen, Z.; Jiang, L.; Yang, M.; Wu, M. Oxidative stress and immune disturbance after long-term exposure to bisphenol A in juvenile common carp (Cyprinus carpio). Ecotoxicol. Environ. Saf. 2016, 130, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Afzal, G.; Ahmad, H.I.; Hussain, R.; Jamal, A.; Kiran, S.; Hussain, T.; Saeed, S.; Nisa, M.U. Bisphenol A Induces Histopathological, Hematobiochemical Alterations, Oxidative Stress, and Genotoxicity in Common Carp (Cyprinus carpio L.). Oxidative Med. Cell. Longev. 2022, 2022, 5450421. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yang, R.; Shi, W.; Zhou, X.; Sun, S. The association between bisphenol A exposure and oxidative damage in rats/mice: A systematic review and meta-analysis. Environ. Pollut. 2021, 292, 118444. [Google Scholar] [CrossRef] [PubMed]
- Ijaz, M.U.; Shahab, M.S.; Samad, A.; Ashraf, A.; Al-Ghanim, K.; Mruthinti, S.S.; Mahboob, S. Tangeretin ameliorates bisphenol induced hepatocyte injury by inhibiting inflammation and oxidative stress. Saudi J. Biol. Sci. 2022, 29, 1375–1379. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, M.; Maadurshni, G.B.; Manivannan, J. Bisphenol A (BPA) exposure aggravates hepatic oxidative stress and inflammatory response under hypertensive milieu—Impact of low dose on hepatocytes and influence of MAPK and ER stress pathways. Food Chem. Toxicol. 2024, 183, 114197. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Tian, S.; Yan, J.; Jia, M.; Yan, S.; Li, R.; Zhang, R.; Zhu, W.; Zhou, Z. Effects of perinatal exposure to BPA, BPF and BPAF on liver function in male mouse offspring involving in oxidative damage and metabolic disorder. Environ. Pollut. 2019, 247, 935–943. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, L.F.; Carneiro, M.F.H.; Dechandt, C.R.P.; Cassoli, J.S.; Alberici, L.C.; Barbosa, F., Jr. Global liver proteomic analysis of Wistar rats chronically exposed to low-levels of bisphenol A and S. Environ. Res. 2020, 182, 109080. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Liu, R.; Zong, W. Bisphenol S Interacts with Catalase and Induces Oxidative Stress in Mouse Liver and Renal Cells. J. Agric. Food Chem. 2016, 64, 6630–6640. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Kadannagari, S.; Deruiter, J.; Pathak, S.; Abbott, K.L.; Salamat, J.M.; Pondugula, S.R.; Akingbemi, B.T.; Dhanasekaran, M. Effects of developmental exposures to Bisphenol-A and Bisphenol-S on hepatocellular function in male Long-Evans rats. Life Sci. 2023, 326, 121752. [Google Scholar] [CrossRef] [PubMed]
- Mahim, S.S.; Anjali, V.R.; Remya, V.S.; Reshmi, S.; Devi, C.A. Oxidative stress responses of a freshwater fish, Labeo rohita, to a xenobiotic, bisphenol S. J. Biochem. Mol. Toxicol. 2021, 35, e22820. [Google Scholar] [CrossRef]
- Alharbi, H.F.; Algonaiman, R.; Alduwayghiri, R.; Aljutaily, T.; Algheshairy, R.M.; Almutairi, A.S.; Alharbi, R.M.; Alfurayh, L.A.; Alshahwan, A.A.; Alsadun, A.F.; et al. Exposure to Bisphenol A Substitutes, Bisphenol S and Bisphenol F, and Its Association with Developing Obesity and Diabetes Mellitus: A Narrative Review. Int. J. Environ. Res. Public Health 2022, 19, 15918. [Google Scholar] [CrossRef] [PubMed]
- Štefunková, N.; Greifová, H.; Jambor, T.; Tokárová, K.; Zuščíková, L.; Bažány, D.; Massányi, P.; Capcarová, M.; Lukáč, N. Comparison of the Effect of BPA and Related Bisphenols on Membrane Integrity, Mitochondrial Activity, and Steroidogenesis of H295R Cells In Vitro. Life 2023, 14, 3. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Liu, Z.; Liu, T. The antagonistic effect of bisphenol A and nonylphenol on liver and kidney injury in rats. Immunopharmacol. Immunotoxicol. 2021, 43, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, U.; Samanta, A.; Biswas, S.; Ghosh, S.; Das, S.; Banerjee, S.; Maitra, S. Chronic exposure to nonylphenol induces oxidative stress and liver damage in male zebrafish (Danio rerio): Mechanistic insight into cellular energy sensors, lipid accumulation and immune modulation. Chem. Interact. 2022, 351, 109762. [Google Scholar] [CrossRef] [PubMed]
- Espina, C.; Straif, K.; Friis, S.; Kogevinas, M.; Saracci, R.; Vainio, H.; Schüz, J. European Code against Cancer 4th Edition: Environment, occupation and cancer. Cancer Epidemiol. 2015, 39 (Suppl. 1), S84–S92. [Google Scholar] [CrossRef] [PubMed]
- McCann, M.S.; Fernandez, H.R.; Flowers, S.A.; Maguire-Zeiss, K.A. Polychlorinated biphenyls induce oxidative stress and metabolic responses in astrocytes. Neurotoxicology 2021, 86, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Thompson, B.L.; Wahlang, B.; Jordan, C.T.; Hilt, J.Z.; Hennig, B.; Dziubla, T. The environmental pollutant, polychlorinated biphenyls, and cardiovascular disease: A potential target for antioxidant nanotherapeutics. Drug Deliv. Transl. Res. 2017, 8, 740–759. [Google Scholar] [CrossRef] [PubMed]
- Dhakal, K.; Gadupudi, G.S.; Lehmler, H.-J.; Ludewig, G.; Duffel, M.W.; Robertson, L.W. Sources and toxicities of phenolic polychlorinated biphenyls (OH-PCBs). Environ. Sci. Pollut. Res. 2018, 25, 16277–16290. [Google Scholar] [CrossRef] [PubMed]
- VanEtten, S.L.; Bonner, M.R.; Ren, X.; Birnbaum, L.S.; Kostyniak, P.J.; Wang, J.; Olson, J.R. Effect of exposure to 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) and polychlorinated biphenyls (PCBs) on mitochondrial DNA (mtDNA) copy number in rats. Toxicology 2021, 454, 152744. [Google Scholar] [CrossRef] [PubMed]
- Van Den Berg, M.; Birnbaum, L.S.; Denison, M.; De Vito, M.; Farland, W.; Feeley, M.; Fiedler, H.; Håkansson, H.; Hanberg, A.; Haws, L.; et al. The 2005 World Health Organization Reevaluation of Human and Mammalian Toxic Equivalency Factors for Dioxins and Dioxin-Like Compounds. Toxicol. Sci. 2006, 93, 223–241. [Google Scholar] [CrossRef] [PubMed]
- National Toxicology Program. Toxicology and carcinogenesis studies of a binary mixture of 3,3′,4,4′,5-pentachlorobiphenyl (PCB 126) (Cas No. 57465-28-8) and 2,3′,4,4′,5-pentachlorobiphenyl (PCB 118) (Cas No. 31508-00-6) in female Harlan Sprague-Dawley rats (gavage studies). Natl. Toxicol. Program Tech. Rep. Ser. 2006, 1–258.
- Ounnas, F.; Privé, F.; Lamarche, F.; Salen, P.; Favier-Hininger, I.; Marchand, P.; Le Bizec, B.; Venisseau, A.; Batandier, C.; Fontaine, E.; et al. A relevant exposure to a food matrix contaminated environmentally by polychlorinated biphenyls induces liver and brain disruption in rats. Chemosphere 2016, 161, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Deng, P.; Barney, J.; Petriello, M.C.; Morris, A.J.; Wahlang, B.; Hennig, B. Hepatic metabolomics reveals that liver injury increases PCB 126-induced oxidative stress and metabolic dysfunction. Chemosphere 2019, 217, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Wang, L.; Chen, G.; Lin, X.; Miao, W.; Fu, Z. Exposure of mice to atrazine and its metabolite diaminochlorotriazine elicits oxidative stress and endocrine disruption. Environ. Toxicol. Pharmacol. 2014, 37, 782–790. [Google Scholar] [CrossRef] [PubMed]
- Sagarkar, S.; Gandhi, D.; Devi, S.S.; Sakharkar, A.; Kapley, A. Atrazine exposure causes mitochondrial toxicity in liver and muscle cell lines. Indian J. Pharmacol. 2016, 48, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Halden, R.U. Plastics and Health Risks. Annu. Rev. Public Health 2010, 31, 179–194. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-I.; Chiang, C.-W.; Lin, H.-C.; Zhao, J.-F.; Li, C.-T.; Shyue, S.-K.; Lee, T.-S. Maternal exposure to di-(2-ethylhexyl) phthalate exposure deregulates blood pressure, adiposity, cholesterol metabolism and social interaction in mouse offspring. Arch. Toxicol. 2016, 90, 1211–1224. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.-F.; Hsiao, S.-H.; Hsu, M.-H.; Pao, K.-C.; Kou, Y.R.; Shyue, S.-K.; Lee, T.-S. Di-(2-ethylhexyl) phthalate accelerates atherosclerosis in apolipoprotein E-deficient mice. Arch. Toxicol. 2016, 90, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhang, W.; Rui, B.-B.; Yang, S.-M.; Xu, W.-P.; Wei, W. Di(2-ethylhexyl) phthalate exacerbates non-alcoholic fatty liver in rats and its potential mechanisms. Environ. Toxicol. Pharmacol. 2016, 42, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Tickner, J.A.; Schettler, T.; Guidotti, T.; McCally, M.; Rossi, M. Health risks posed by use of Di-2-ethylhexyl phthalate (DEHP) in PVC medical devices: A critical review. Am. J. Ind. Med. 2001, 39, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-Y.; Suk, F.-M.; Twu, Y.-C.; Liao, Y.-J. Long-Term Exposure to Low-Dose Di-(2-ethylhexyl) Phthalate Impairs Cholesterol Metabolism in Hepatic Stellate Cells and Exacerbates Liver Fibrosis. Int. J. Environ. Res. Public Health 2020, 17, 3802. [Google Scholar] [CrossRef]
- Li, G.; Zhao, C.-Y.; Wu, Q.; Guan, S.-Y.; Jin, H.-W.; Na, X.-L.; Zhang, Y.-B. Integrated metabolomics and transcriptomics reveal di(2-ethylhexyl) phthalate-induced mitochondrial dysfunction and glucose metabolism disorder through oxidative stress in rat liver. Ecotoxicol. Environ. Saf. 2021, 228, 112988. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhao, Y.; Talukder, M.; Han, Y.; Zhang, C.; Li, X.-N.; Li, J.-L. Di(2-ethylhexyl) phthalate induced hepatotoxicity in quail (Coturnix japonica) via modulating the mitochondrial unfolded protein response and NRF2 mediated antioxidant defense. Sci. Total. Environ. 2019, 651, 885–894. [Google Scholar] [CrossRef] [PubMed]
- Park, C.G.; Sung, B.; Ryu, C.S.; Kim, Y.J. Mono-(2-ethylhexyl) phthalate induces oxidative stress and lipid accumulation in zebrafish liver cells. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2020, 230, 108704. [Google Scholar] [CrossRef] [PubMed]
- Han, R.; Hu, M.; Zhong, Q.; Wan, C.; Liu, L.; Li, F.; Zhang, F.; Ding, W. Perfluorooctane sulphonate induces oxidative hepatic damage via mitochondria-dependent and NF-kappa B/TNF-alpha-mediated pathway. Chemosphere 2018, 191, 1056–1064. [Google Scholar] [CrossRef]
- Domingo, J.L. Polychlorinated diphenyl ethers (PCDEs): Environmental levels, toxicity and human exposure: A review of the published literature. Environ. Int. 2006, 32, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Xiong, W.; Shi, S.; Shi, J.; Yang, W.; Zhang, X. Biomarker Responses, Gene Expression Alterations, and Histological Changes in Zebrafish (Danio rerio) after in vivo Exposure to Polychlorinated Diphenyl Ethers. Front. Physiol. 2022, 13, 907906. [Google Scholar] [CrossRef] [PubMed]
- Margina, D.; Nițulescu, G.M.; Ungurianu, A.; Mesnage, R.; Goumenou, M.; Sarigiannis, D.A.; Aschner, M.; Spandidos, D.A.; Renieri, E.A.; Tsatsakis, A.; et al. Overview of the effects of chemical mixtures with endocrine disrupting activity in the context of real-life risk simulation: An integrative approach (Review). World Acad. Sci. J. 2019, 1, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Vardakas, P.; Veskoukis, A.S.; Rossiou, D.; Gournikis, C.; Kapetanopoulou, T.; Karzi, V.; Docea, A.O.; Tsatsakis, A.; Kouretas, D. A Mixture of Endocrine Disruptors and the Pesticide Roundup® Induce Oxidative Stress in Rabbit Liver When Administered under the Long-Term Low-Dose Regimen: Reinforcing the Notion of Real-Life Risk Simulation. Toxics 2022, 10, 190. [Google Scholar] [CrossRef] [PubMed]
- Tsatsakis, A.; Goumenou, M.; Liesivuori, J.; Dekant, W.; Hernández, A.F. Toxicology for real-life risk simulation—Editorial preface to this special issue. Toxicol. Lett. 2019, 309, 33–34. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Jiang, Y.; Li, Y.; Wan, Y.; Liu, J.; Ma, Y.; Mao, Z.; Chang, H.; Li, G.; Xu, B.; et al. Early-Life Exposure to Bisphenol A Induces Liver Injury in Rats Involvement of Mitochondria-Mediated Apoptosis. PLoS ONE 2014, 9, e90443. [Google Scholar] [CrossRef] [PubMed]
- Mornagui, B.; Rezg, R.; Repond, C.; Pellerin, L. Bisphenol S favors hepatic steatosis development via an upregulation of liver MCT1 expression and an impairment of the mitochondrial respiratory system. J. Cell. Physiol. 2022, 237, 3057–3068. [Google Scholar] [CrossRef] [PubMed]
- Kim, K. The Role of Endocrine Disruption Chemical-Regulated Aryl Hydrocarbon Receptor Activity in the Pathogenesis of Pancreatic Diseases and Cancer. Int. J. Mol. Sci. 2024, 25, 3818. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Lin, Y.; Li, Y.; Ying, C.; Chen, J.; Song, L.; Zhou, Z.; Lv, Z.; Xia, W.; Chen, X.; et al. Perinatal Exposure to Bisphenol A at Reference Dose Predisposes Offspring to Metabolic Syndrome in Adult Rats on a High-Fat Diet. Endocrinology 2011, 152, 3049–3061. [Google Scholar] [CrossRef] [PubMed]
- Carchia, E.; Porreca, I.; Almeida, P.J.; D’Angelo, F.; Cuomo, D.; Ceccarelli, M.; De Felice, M.; Mallardo, M.; Ambrosino, C. Evaluation of low doses BPA-induced perturbation of glycemia by toxicogenomics points to a primary role of pancreatic islets and to the mechanism of toxicity. Cell Death Dis. 2015, 6, e1959. [Google Scholar] [CrossRef] [PubMed]
- Susiarjo, M.; Xin, F.; Bansal, A.; Stefaniak, M.; Li, C.; Simmons, R.A.; Bartolomei, M.S. Bisphenol A Exposure Disrupts Metabolic Health across Multiple Generations in the Mouse. Endocrinology 2015, 156, 2049–2058. [Google Scholar] [CrossRef] [PubMed]
- Bansal, A.; Rashid, C.; Xin, F.; Li, C.; Polyak, E.; Duemler, A.; van der Meer, T.; Stefaniak, M.; Wajid, S.; Doliba, N.; et al. Sex- and Dose-Specific Effects of Maternal Bisphenol A Exposure on Pancreatic Islets of First- and Second-Generation Adult Mice Offspring. Environ. Health Perspect. 2017, 125, 097022. [Google Scholar] [CrossRef] [PubMed]
- Moon, M.K.; Jeong, I.-K.; Oh, T.J.; Ahn, H.Y.; Kim, H.H.; Park, Y.J.; Jang, H.C.; Park, K.S. Long-term oral exposure to bisphenol A induces glucose intolerance and insulin resistance. J. Endocrinol. 2015, 226, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Xia, W.; Zhou, Z.; Li, Y.; Lin, Y.; Wei, J.; Wei, Z.; Xu, B.; Shen, J.; Li, W.; et al. Low-level phenolic estrogen pollutants impair islet morphology and β-cell function in isolated rat islets. J. Endocrinol. 2012, 215, 303–311. [Google Scholar] [CrossRef]
- Li, X.; Zhou, L.; Ni, Y.; Wang, A.; Hu, M.; Lin, Y.; Hong, C.; Wan, J.; Chen, B.; Fang, L.; et al. Nonylphenol induces pancreatic damage in rats through mitochondrial dysfunction and oxidative stress. Toxicol. Res. 2017, 6, 353–360. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 3032732, Tributyl tin. 2024. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Tributyl-tin (accessed on 1 July 2024).
- Chen, Y.-W.; Lan, K.-C.; Tsai, J.-R.; Weng, T.-I.; Yang, C.-Y.; Liu, S.-H. Tributyltin exposure at noncytotoxic doses dysregulates pancreatic β-cell function in vitro and in vivo. Arch. Toxicol. 2017, 91, 3135–3144. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, R.S.; Babiloni-Chust, I.; Marroqui, L.; Nadal, A. Screening of Metabolism-Disrupting Chemicals on Pancreatic α-Cells Using In Vitro Methods. Int. J. Mol. Sci. 2022, 24, 231. [Google Scholar] [CrossRef] [PubMed]
- Makaji, E.; Raha, S.; Wade, M.G.; Holloway, A.C. Effect of Environmental Contaminants on Beta Cell Function. Int. J. Toxicol. 2011, 30, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Jaacks, L.M.; Vandevijvere, S.; Pan, A.; McGowan, C.J.; Wallace, C.; Imamura, F.; Mozaffarian, D.; Swinburn, B.; Ezzati, M. The obesity transition: Stages of the global epidemic. Lancet Diabetes Endocrinol. 2019, 7, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Küblbeck, J.; Vuorio, T.; Niskanen, J.; Fortino, V.; Braeuning, A.; Abass, K.; Rautio, A.; Hakkola, J.; Honkakoski, P.; Levonen, A.-L. The EDCMET Project: Metabolic Effects of Endocrine Disruptors. Int. J. Mol. Sci. 2020, 21, 3021. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, E.; Ladeira, C.; Viegas, S. EDCs Mixtures: A Stealthy Hazard for Human Health? Toxics 2017, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Duh-Leong, C.; Maffini, M.V.; Kassotis, C.D.; Vandenberg, L.N.; Trasande, L. The regulation of endocrine-disrupting chemicals to minimize their impact on health. Nat. Rev. Endocrinol. 2023, 19, 600–614. [Google Scholar] [CrossRef] [PubMed]
| Endocrine Disruptor | Experimental Model | Dose | Duration | Effects | Ref. |
|---|---|---|---|---|---|
| Bisphenol A (BPA) | β-cells of rat offspring | 50, 250 or 1250 µg/kg/day | Chronic: during gestation and lactation |
| [106] |
| Bisphenol A (BPA) | Primary murine pancreatic islets | M | Acute: 24 or 48 h |
| [107] |
| Bisphenol A (BPA) | Pancreatic islets of mice offspring | 10 µg/kg/d (low dose) and 10 mg/kg/d (high dose) | Chronic: during gestation and lactation |
| [109] |
| Bisphenol A (BPA) | αTC1-9 murine cell line | From 0.1 pM to 1 µM | Acute: 24 h |
| [115] |
| Tributyltin (TBT) | αTC1-9 murine cell line | From 0.1 pM to 1 µM | Acute: 24 h |
| [115] |
| Tributyltin (TBT) | β-cell-derived RIN-m5F rat cell line; pancreatic islets of mice and humans | 0.05–0.2 μM | Acute: 0.5–4 h |
| [114] |
| Octylphenol (OP) | Pancreatic islets of rats | 25 µg/L | Acute: 24 h |
| [111] |
| Nonylphenol (NP) | |||||
| Bisphenol A (BPA) | |||||
| 4-Nonylphenol (NP) | Pancreatic islets of rats | 60 mg/kg and 180 mg/kg | Chronic: 90 consecutive days |
| [112] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lința, A.V.; Lolescu, B.M.; Ilie, C.A.; Vlad, M.; Blidișel, A.; Sturza, A.; Borza, C.; Muntean, D.M.; Crețu, O.M. Liver and Pancreatic Toxicity of Endocrine-Disruptive Chemicals: Focus on Mitochondrial Dysfunction and Oxidative Stress. Int. J. Mol. Sci. 2024, 25, 7420. https://doi.org/10.3390/ijms25137420
Lința AV, Lolescu BM, Ilie CA, Vlad M, Blidișel A, Sturza A, Borza C, Muntean DM, Crețu OM. Liver and Pancreatic Toxicity of Endocrine-Disruptive Chemicals: Focus on Mitochondrial Dysfunction and Oxidative Stress. International Journal of Molecular Sciences. 2024; 25(13):7420. https://doi.org/10.3390/ijms25137420
Chicago/Turabian StyleLința, Adina V., Bogdan M. Lolescu, Cosmin A. Ilie, Mihaela Vlad, Alexandru Blidișel, Adrian Sturza, Claudia Borza, Danina M. Muntean, and Octavian M. Crețu. 2024. "Liver and Pancreatic Toxicity of Endocrine-Disruptive Chemicals: Focus on Mitochondrial Dysfunction and Oxidative Stress" International Journal of Molecular Sciences 25, no. 13: 7420. https://doi.org/10.3390/ijms25137420
APA StyleLința, A. V., Lolescu, B. M., Ilie, C. A., Vlad, M., Blidișel, A., Sturza, A., Borza, C., Muntean, D. M., & Crețu, O. M. (2024). Liver and Pancreatic Toxicity of Endocrine-Disruptive Chemicals: Focus on Mitochondrial Dysfunction and Oxidative Stress. International Journal of Molecular Sciences, 25(13), 7420. https://doi.org/10.3390/ijms25137420