

Tofacitinib Treatment Suppresses CD4+ T-Cell Activation and Th1 Response, Contributing to Protection against Staphylococcal Toxic Shock

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
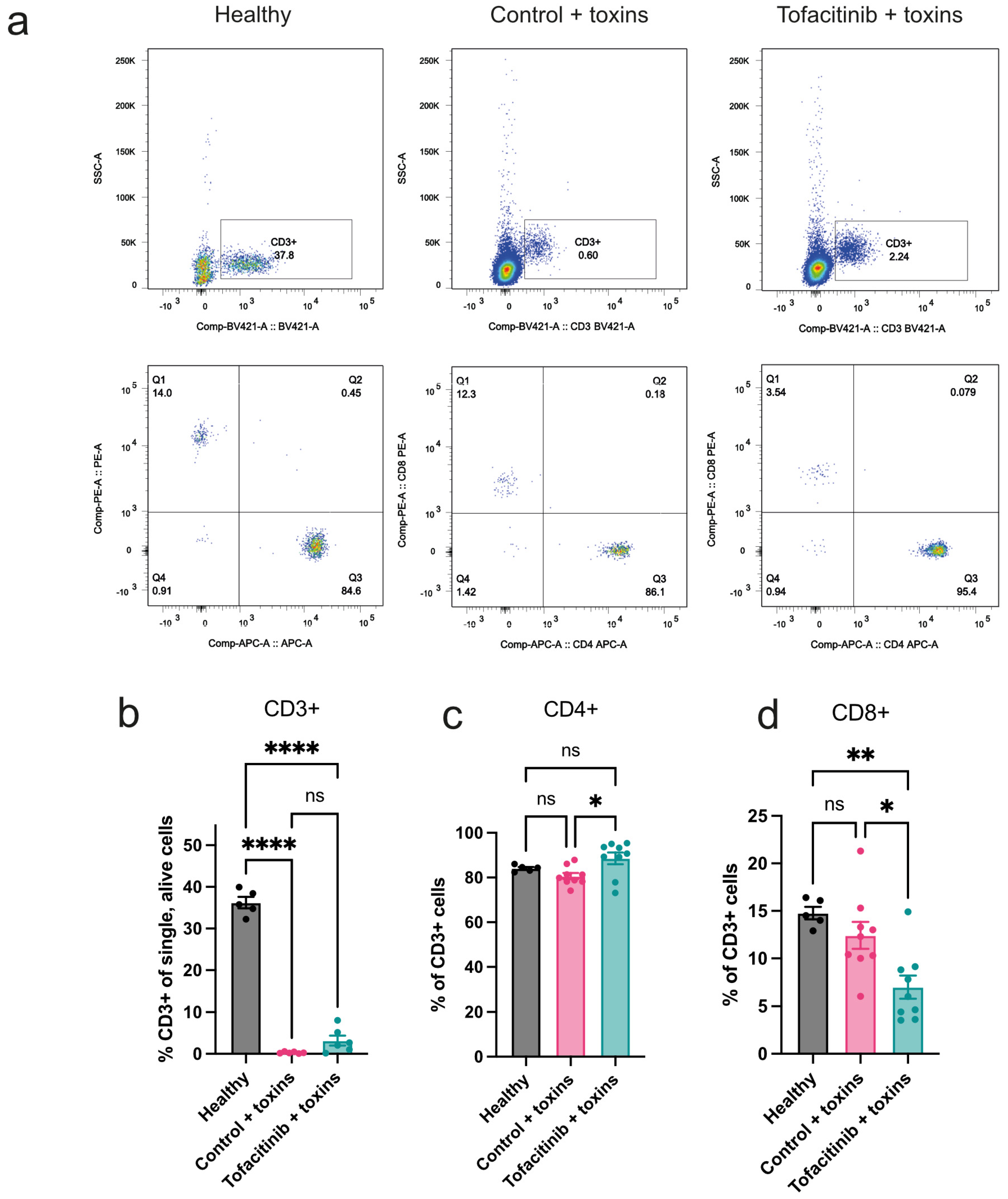
2.1. Tofacitinib Has a Limited Effect on Mitigating the Loss of Peripheral T Cells Caused by Toxic Shock
2.2. Tofacitinib Reduces Activation of T Cells in Peripheral Blood
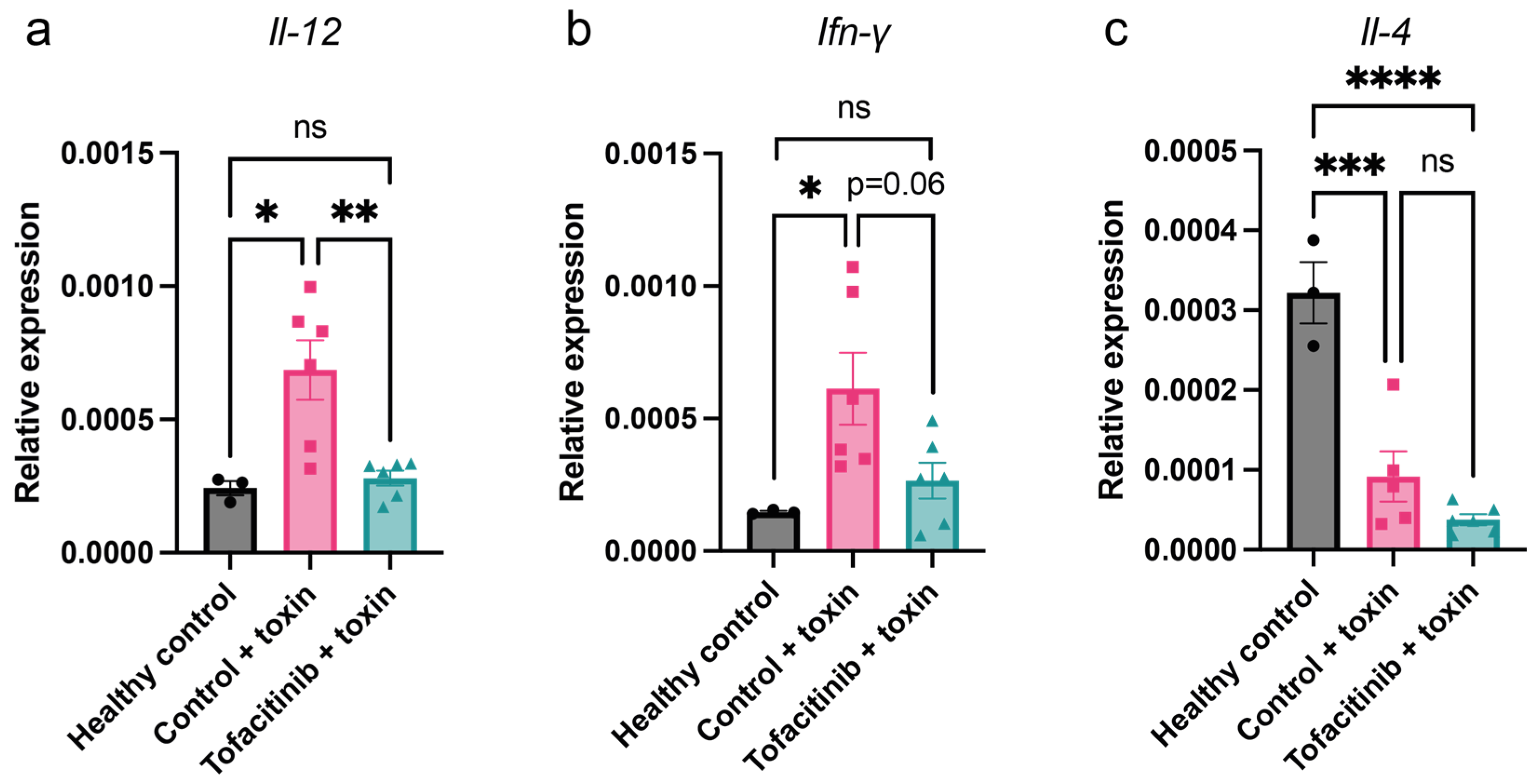
2.3. Tofacitinib Reduces Th1 Cytokine Expression in Mice with Toxic Shock
2.4. Tofacitinib Reduces Blood Levels of IL-12, IFN-γ, and IL-6 in Mice with Toxic Shock
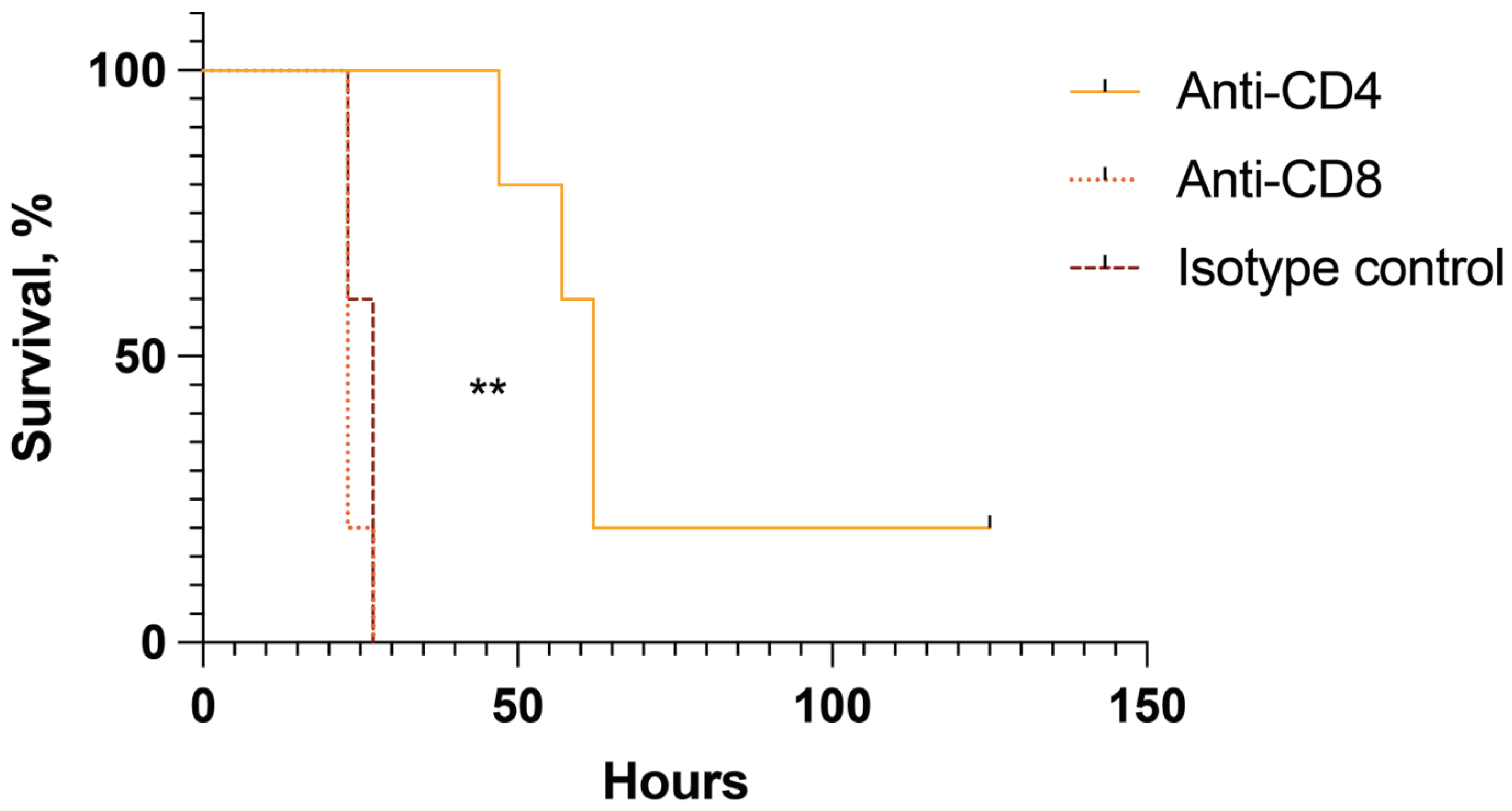
2.5. CD4+ but Not CD8+ Cells Are Pathogenic in Mice with Toxin-Induced Shock
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Mouse Model of Toxin-Induced Shock
4.3. Treatment with Tofacitinib
4.4. T-Cell Depletion
4.5. Flow Cytometry
4.6. RNA Extraction, cDNA Synthesis, and qRT-PCR
4.7. Enzyme-Linked Immunosorbent Assay
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Krakauer, T. Staphylococcal Superantigens: Pyrogenic Toxins Induce Toxic Shock. Toxins 2019, 11, 178. [Google Scholar] [CrossRef] [PubMed]
- Atchade, E.; De Tymowski, C.; Grall, N.; Tanaka, S.; Montravers, P. Toxic Shock Syndrome: A Literature Review. Antibiotics 2024, 13, 96. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.X.; McCormick, J.K. Staphylococcal superantigens in colonization and disease. Front. Cell Infect. Microbiol. 2012, 2, 52. [Google Scholar] [CrossRef] [PubMed]
- Tuffs, S.W.; Dufresne, K.; Rishi, A.; Walton, N.R.; McCormick, J.K. Novel insights into the immune response to bacterial T cell superantigens. Nat. Rev. Immunol. 2024, 24, 417–434. [Google Scholar] [CrossRef] [PubMed]
- Abdelnour, A.; Bremell, T.; Tarkowski, A. Toxic shock syndrome toxin 1 contributes to the arthritogenicity of Staphylococcus aureus. J. Infect. Dis. 1994, 170, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, M.; Long, B.; Koyfman, A. The Evaluation and Management of Toxic Shock Syndrome in the Emergency Department: A Review of the Literature. J. Emerg. Med. 2018, 54, 807–814. [Google Scholar] [CrossRef] [PubMed]
- Giamarellos-Bourboulis, E.J.; Aschenbrenner, A.C.; Bauer, M.; Bock, C.; Calandra, T.; Gat-Viks, I.; Kyriazopoulou, E.; Lupse, M.; Monneret, G.; Pickkers, P.; et al. The pathophysiology of sepsis and precision-medicine-based immunotherapy. Nat. Immunol. 2024, 25, 19–28. [Google Scholar] [CrossRef] [PubMed]
- McLornan, D.P.; Pope, J.E.; Gotlib, J.; Harrison, C.N. Current and future status of JAK inhibitors. Lancet 2021, 398, 803–816. [Google Scholar] [CrossRef]
- Philips, R.L.; Wang, Y.; Cheon, H.; Kanno, Y.; Gadina, M.; Sartorelli, V.; Horvath, C.M.; Darnell, J.E., Jr.; Stark, G.R.; O’Shea, J.J. The JAK-STAT pathway at 30: Much learned, much more to do. Cell 2022, 185, 3857–3876. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Plenge, R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity 2012, 36, 542–550. [Google Scholar] [CrossRef]
- Peña, G.; Cai, B.; Deitch, E.A.; Ulloa, L. JAK2 inhibition prevents innate immune responses and rescues animals from sepsis. J. Mol. Med. 2010, 88, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Tsirigotis, P.; Papanikolaou, N.; Elefanti, A.; Konstantinou, P.; Gkirkas, K.; Rontogianni, D.; Siafakas, N.; Karakitsos, P.; Roilides, E.; Dimitriadis, G.; et al. Treatment of Experimental Candida Sepsis with a Janus Kinase Inhibitor Controls Inflammation and Prolongs Survival. Antimicrob. Agents Chemother. 2015, 59, 7367–7373. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, P.O.; Quirk, D.; Furtado, R.H.; Maia, L.N.; Saraiva, J.F.; Antunes, M.O.; Kalil Filho, R.; Junior, V.M.; Soeiro, A.M.; Tognon, A.P.; et al. Tofacitinib in Patients Hospitalized with Covid-19 Pneumonia. N. Engl. J. Med. 2021, 385, 406–415. [Google Scholar] [CrossRef] [PubMed]
- Jarneborn, A.; Mohammad, M.; Engdahl, C.; Hu, Z.; Na, M.; Ali, A.; Jin, T. Tofacitinib treatment aggravates Staphylococcus aureus septic arthritis, but attenuates sepsis and enterotoxin induced shock in mice. Sci. Rep. 2020, 10, 10891. [Google Scholar] [CrossRef] [PubMed]
- Marrack, P.; Kappler, J. The staphylococcal enterotoxins and their relatives. Science 1990, 248, 705–711. [Google Scholar] [CrossRef]
- Spellberg, B.; Edwards, J.E., Jr. Type 1/Type 2 immunity in infectious diseases. Clin. Infect. Dis. 2001, 32, 76–102. [Google Scholar] [CrossRef] [PubMed]
- Dinges, M.M.; Schlievert, P.M. Role of T cells and gamma interferon during induction of hypersensitivity to lipopolysaccharide by toxic shock syndrome toxin 1 in mice. Infect. Immun. 2001, 69, 1256–1264. [Google Scholar] [CrossRef]
- Saha, B.; Jaklic, B.; Harlan, D.M.; Gray, G.S.; June, C.H.; Abe, R. Toxic shock syndrome toxin-1-induced death is prevented by CTLA4Ig. J. Immunol. 1996, 157, 3869–3875. [Google Scholar] [CrossRef] [PubMed]
- Venet, F.; Davin, F.; Guignant, C.; Larue, A.; Cazalis, M.A.; Darbon, R.; Allombert, C.; Mougin, B.; Malcus, C.; Poitevin-Later, F.; et al. Early assessment of leukocyte alterations at diagnosis of septic shock. Shock 2010, 34, 358–363. [Google Scholar] [CrossRef]
- Ghoreschi, K.; Jesson, M.I.; Li, X.; Lee, J.L.; Ghosh, S.; Alsup, J.W.; Warner, J.D.; Tanaka, M.; Steward-Tharp, S.M.; Gadina, M.; et al. Modulation of innate and adaptive immune responses by tofacitinib (CP-690,550). J. Immunol. 2011, 186, 4234–4243. [Google Scholar] [CrossRef]
- De Wilde, V.; Benghiat, F.S.; Novalrivas, M.; Lebrun, J.F.; Kubjak, C.; Oldenhove, G.; Verdebout, J.M.; Goldman, M.; Le Moine, A. Endotoxin hyperresponsiveness upon CD4+ T cell reconstitution in lymphopenic mice: Control by natural regulatory T cells. Eur. J. Immunol. 2008, 38, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.; Luz, A.; Bendigs, S.; Erdmann, A.; Wagner, H.; Heeg, K. Superantigen and endotoxin synergize in the induction of lethal shock. Eur. J. Immunol. 1997, 27, 825–833. [Google Scholar] [CrossRef] [PubMed]
- Arad, G.; Hillman, D.; Levy, R.; Kaempfer, R. Superantigen antagonist blocks Th1 cytokine gene induction and lethal shock. J. Leukoc. Biol. 2001, 69, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Tuffs, S.W.; Haeryfar, S.M.M.; McCormick, J.K. Manipulation of Innate and Adaptive Immunity by Staphylococcal Superantigens. Pathogens 2018, 7, 53. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B. IFNγ: Signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Fei, Y.; Wang, W.; Kwiecinski, J.; Josefsson, E.; Pullerits, R.; Jonsson, I.M.; Magnusson, M.; Jin, T. The combination of a tumor necrosis factor inhibitor and antibiotic alleviates staphylococcal arthritis and sepsis in mice. J. Infect. Dis. 2011, 204, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Tracey, K.J.; Fong, Y.; Hesse, D.G.; Manogue, K.R.; Lee, A.T.; Kuo, G.C.; Lowry, S.F.; Cerami, A. Anti-cachectin/TNF monoclonal antibodies prevent septic shock during lethal bacteraemia. Nature 1987, 330, 662–664. [Google Scholar] [CrossRef] [PubMed]
- Abraham, E.; Anzueto, A.; Gutierrez, G.; Tessler, S.; San Pedro, G.; Wunderink, R.; Dal Nogare, A.; Nasraway, S.; Berman, S.; Cooney, R.; et al. Double-blind randomised controlled trial of monoclonal antibody to human tumour necrosis factor in treatment of septic shock. NORASEPT II Study Group. Lancet 1998, 351, 929–933. [Google Scholar] [CrossRef] [PubMed]
- Steinhagen, F.; Schmidt, S.V.; Schewe, J.C.; Peukert, K.; Klinman, D.M.; Bode, C. Immunotherapy in sepsis—Brake or accelerate? Pharmacol. Ther. 2020, 208, 107476. [Google Scholar] [CrossRef]
- RECOVERY_Collaborative_Group. Tocilizumab in patients admitted to hospital with COVID-19 (RECOVERY): A randomised, controlled, open-label, platform trial. Lancet 2021, 397, 1637–1645. [Google Scholar] [CrossRef]
- Horby, P.; Lim, W.S.; Emberson, J.R.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Brightling, C.; Ustianowski, A.; Elmahi, E.; et al. Dexamethasone in Hospitalized Patients with COVID-19. N. Engl. J. Med. 2021, 384, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Kłak, M.; Anäkkälä, N.; Wang, W.; Lange, S.; Jonsson, I.M.; Tarkowski, A.; Jin, T. Tranexamic acid, an inhibitor of plasminogen activation, aggravates staphylococcal septic arthritis and sepsis. Scand. J. Infect. Dis. 2010, 42, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, M.; Nguyen, M.T.; Engdahl, C.; Na, M.; Jarneborn, A.; Hu, Z.; Karlsson, A.; Pullerits, R.; Ali, A.; Götz, F.; et al. The YIN and YANG of lipoproteins in developing and preventing infectious arthritis by Staphylococcus aureus. PLoS Pathog. 2019, 15, e1007877. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jarneborn, A.; Hu, Z.; Deshmukh, M.; Kopparapu, P.K.; Jin, T. Tofacitinib Treatment Suppresses CD4+ T-Cell Activation and Th1 Response, Contributing to Protection against Staphylococcal Toxic Shock. Int. J. Mol. Sci. 2024, 25, 7456. https://doi.org/10.3390/ijms25137456
Jarneborn A, Hu Z, Deshmukh M, Kopparapu PK, Jin T. Tofacitinib Treatment Suppresses CD4+ T-Cell Activation and Th1 Response, Contributing to Protection against Staphylococcal Toxic Shock. International Journal of Molecular Sciences. 2024; 25(13):7456. https://doi.org/10.3390/ijms25137456
Chicago/Turabian StyleJarneborn, Anders, Zhicheng Hu, Meghshree Deshmukh, Pradeep Kumar Kopparapu, and Tao Jin. 2024. "Tofacitinib Treatment Suppresses CD4+ T-Cell Activation and Th1 Response, Contributing to Protection against Staphylococcal Toxic Shock" International Journal of Molecular Sciences 25, no. 13: 7456. https://doi.org/10.3390/ijms25137456
APA StyleJarneborn, A., Hu, Z., Deshmukh, M., Kopparapu, P. K., & Jin, T. (2024). Tofacitinib Treatment Suppresses CD4+ T-Cell Activation and Th1 Response, Contributing to Protection against Staphylococcal Toxic Shock. International Journal of Molecular Sciences, 25(13), 7456. https://doi.org/10.3390/ijms25137456