
The Degradation of Botulinum Neurotoxin Light Chains Using PROTACs
 ,
,  , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
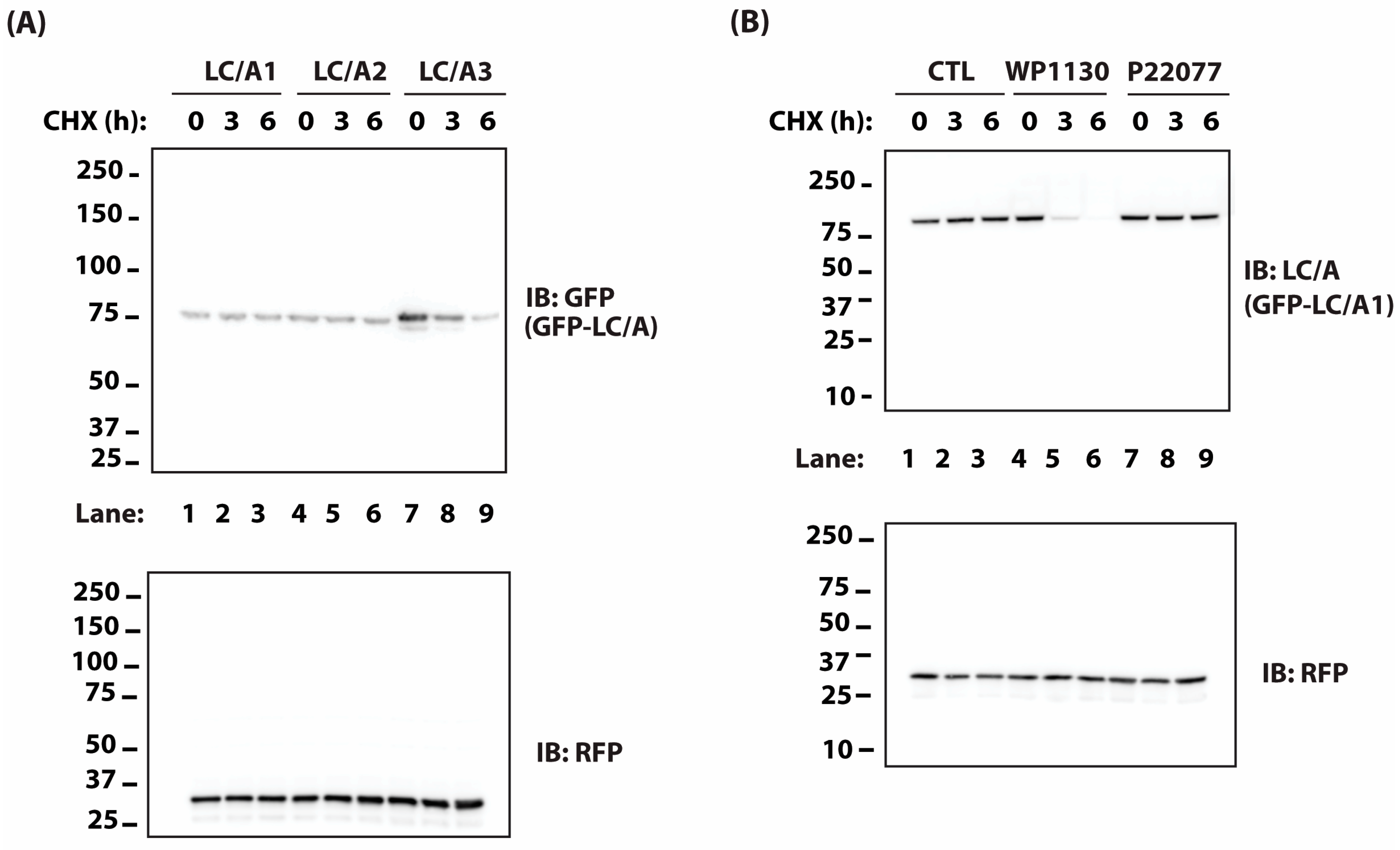
2.1. BoNT Persistence in Animals Correlates with the Rate of Protease Turnover in Transfected Cells
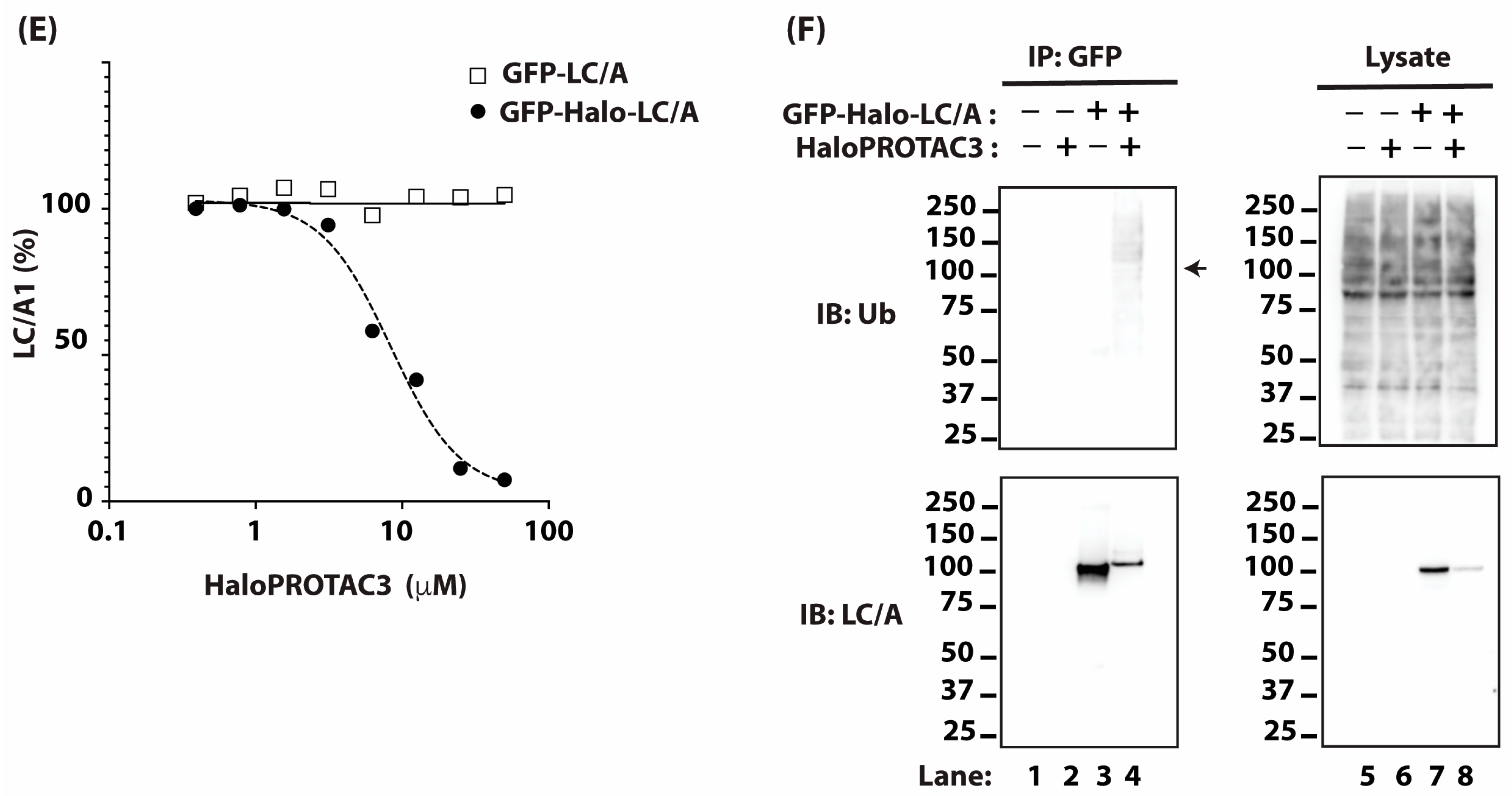
2.2. Targeting BoNT/A Light Chain with PROTAC
2.3. Targeting BoNT/A Light Chain Using a VHH and PROTAC
3. Methods
3.1. Plasmids
3.2. Antibodies
3.3. PROTACs
3.4. Cell Culture
3.5. Ubiquitination Experiments
3.6. Cell Culture Experiments
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Habermann, E.; Dreyer, F. Clostridial neurotoxins: Handling and action at the cellular and molecular level. Curr. Top Microbiol. Immunol. 1986, 129, 93–179. [Google Scholar]
- Jahn, R.; Niemann, H. Molecular mechanisms of clostridial neurotoxins. Ann. N. Y. Acad. Sci. 1994, 733, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.A.; Montecucco, C. Botulism. Handb. Clin. Neurol. 2008, 91, 333–368. [Google Scholar] [PubMed]
- Schiavo, G.; Matteoli, M.; Montecucco, C.; Kasai, H.; Takahashi, N.; Tokumaru, H.; Martin, J.H.; Engber, D.; Meng, Z.; Banerjee, A.; et al. Neurotoxins Affecting Neuroexocytosis. Physiol. Rev. 2000, 80, 717–766. [Google Scholar] [CrossRef] [PubMed]
- Dolly, J.O.; Black, J.; Williams, R.S.; Melling, J. Acceptors for botulinum neurotoxin reside on motor nerve terminals and mediate its internalization. Nature 1984, 307, 457–460. [Google Scholar] [CrossRef] [PubMed]
- Pantano, S.; Montecucco, C. The blockade of the neurotransmitter release apparatus by botulinum neurotoxins. Cell. Mol. Life Sci. 2013, 71, 793–811. [Google Scholar] [CrossRef]
- Montecucco, C.; Schiavo, G. Structure and function of tetanus and botulinum neurotoxins. Q. Rev. Biophys. 1995, 28, 423–472. [Google Scholar] [CrossRef] [PubMed]
- Eleopra, R.; Tugnoli, V.; Rossetto, O.; De Grandis, D.; Montecucco, C. Different time courses of recovery after poisoning with botulinum neurotoxin serotypes A and E in humans. Neurosci. Lett. 1998, 256, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Foran, P.G.; Mohammed, N.; Lisk, G.O.; Nagwaney, S.; Lawrence, G.W.; Johnson, E.; Smith, L.; Aoki, K.R.; Dolly, J.O. Evaluation of the therapeutic usefulness of botulinum neurotoxin B, C1, E, and F compared with the long lasting type A. Basis for distinct durations of inhibition of exocytosis in central neurons. J. Biol. Chem. 2003, 278, 1363–1371. [Google Scholar] [CrossRef]
- Fonfria, E.; Maignel, J.; Lezmi, S.; Martin, V.; Splevins, A.; Shubber, S.; Kalinichev, M.; Foster, K.; Picaut, P.; Krupp, J. The Expanding Therapeutic Utility of Botulinum Neurotoxins. Toxins 2018, 10, 208. [Google Scholar] [CrossRef]
- Pirazzini, M.; Rossetto, O.; Eleopra, R.; Montecucco, C. Botulinum Neurotoxins: Biology, Pharmacology, and Toxicology. Pharmacol. Rev. 2017, 69, 200–235. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.L.; Schlessinger, J.; Cox, S.E.; Lin, X. Reloxin Investigational Group An Analysis of the Long-Term Safety Data of Repeat Administrations of Botulinum Neurotoxin Type A-ABO for the Treatment of Glabellar Lines. Aesthet. Surg. J. 2009, 29 (Suppl. 6), S43–S49. [Google Scholar] [CrossRef] [PubMed]
- Naumann, M.; Jankovic, J. Safety of botulinum toxin type A: A systematic review and meta-analysis. Curr. Med. Res. Opin. 2004, 20, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Bakheit, A.M.O.; Severa, S.; Cosgrove, A.; Morton, R.; Roussounis, S.H.; Doderlein, L.; Lin, J.-P. Safety profile and efficacy of botulinum toxin A (Dysport) in children with muscle spasticity. Dev. Med. Child Neurol. 2001, 43, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Dorner, M.B.; Wilking, H.; Skiba, M.; Wilk, L.; Steinberg, M.; Worbs, S.; Çeken, S.; Kaygusuz, S.; Simon, S.; Becher, F.; et al. A large travel-associated outbreak of iatrogenic botulism in four European countries following intragastric botulinum neurotoxin injections for weight reduction, Türkiye, February to March 2023. Eurosurveillance 2023, 28, 2300203. [Google Scholar] [CrossRef] [PubMed]
- Machamer, J.B.; Vazquez-Cintron, E.J.; O’brien, S.W.; Kelly, K.E.; Altvater, A.C.; Pagarigan, K.T.; Dubee, P.B.; Ondeck, C.A.; McNutt, P.M. Antidotal treatment of botulism in rats by continuous infusion with 3,4-diaminopyridine. Mol. Med. 2022, 28, 61. [Google Scholar] [CrossRef]
- McClintic, W.T.; Chandler, Z.D.; Karchalla, L.M.; Ondeck, C.A.; O’brien, S.W.; Campbell, C.J.; Jacobson, A.R.; McNutt, P.M. Aminopyridines Restore Ventilation and Reverse Respiratory Acidosis at Late Stages of Botulism in Mice. J. Pharmacol. Exp. Ther. 2023, 388, 637–646. [Google Scholar] [CrossRef] [PubMed]
- McNutt, P.M.; Vazquez-Cintron, E.J.; Tenezaca, L.; Ondeck, C.A.; Kelly, K.E.; Mangkhalakhili, M.; Machamer, J.B.; Angeles, C.A.; Glotfelty, E.J.; Cika, J.; et al. Neuronal delivery of antibodies has therapeutic effects in animal models of botulism. Sci. Transl. Med. 2021, 13, eabd7789. [Google Scholar] [CrossRef]
- Miyashita, S.-I.; Zhang, J.; Zhang, S.; Shoemaker, C.B.; Dong, M. Delivery of single-domain antibodies into neurons using a chimeric toxin–based platform is therapeutic in mouse models of botulism. Sci. Transl. Med. 2021, 13, eaaz4197. [Google Scholar] [CrossRef]
- Vazquez-Cintron, E.; Machamer, J.; Ondeck, C.; Pagarigan, K.; Winner, B.; Bodner, P.; Kelly, K.; Pennington, M.R.; McNutt, P. Symptomatic treatment of botulism with a clinically approved small molecule. J. Clin. Investig. 2020, 5, e132891. [Google Scholar] [CrossRef]
- Tsai, Y.C.; Kotiya, A.; Kiris, E.; Yang, M.; Bavari, S.; Tessarollo, L.; Oyler, G.A.; Weissman, A.M. Deubiquitinating enzyme VCIP135 dictates the duration of botulinum neurotoxin type A intoxication. Proc. Natl. Acad. Sci. USA 2017, 114, E5158–E5166. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.C.; Maditz, R.; Kuo, C.-L.; Fishman, P.S.; Shoemaker, C.B.; Oyler, G.A.; Weissman, A.M. Targeting botulinum neurotoxin persistence by the ubiquitin-proteasome system. Proc. Natl. Acad. Sci. USA 2010, 107, 16554–16559. [Google Scholar] [CrossRef]
- Keller, J.E.; Neale, E.A.; Oyler, G.; Adler, M. Persistence of botulinum neurotoxin action in cultured spinal cord cells. FEBS Lett. 1999, 456, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Vagin, O.; Tokhtaeva, E.; Garay, P.E.; Souda, P.; Bassilian, S.; Whitelegge, J.P.; Lewis, R.; Sachs, G.; Wheeler, L.; Aoki, R.; et al. Recruitment of septin cytoskeletal proteins by Botulinum toxin A protease determines its remarkable stability. J. Cell Sci. 2014, 127, 3294–3308. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.-L.; Oyler, G.A.; Shoemaker, C.B. Accelerated neuronal cell recovery from botulinum neurotoxin intoxication by targeted ubiquitination. PLoS ONE 2011, 6, e20352. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, J.M.; Kuo, C.-L.; Abeijon, C.; Sepulveda, J.; Oyler, G.; Hu, X.; Jin, M.M.; Shoemaker, C.B. Camelid single domain antibodies (VHHs) as neuronal cell intrabody binding agents and inhibitors of Clostridium botulinum neurotoxin (BoNT) proteases. Toxicon 2010, 56, 990–998. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef] [PubMed]
- Buckley, D.L. Small-molecule inhibitors of the interaction between the E3 ligase VHL and HIF1alpha. Angew. Chem. Int. Ed. Engl. 2012, 51, 11463–11467. [Google Scholar] [CrossRef]
- Buckley, D.L.; Van Molle, I.; Gareiss, P.C.; Tae, H.S.; Michel, J.; Noblin, D.J.; Jorgensen, W.L.; Ciulli, A.; Crews, C.M. Targeting the von Hippel–Lindau E3 ubiquitin ligase using small molecules to disrupt the VHL/HIF-1α interaction. J. Am. Chem. Soc. 2012, 134, 4465–4468. [Google Scholar] [CrossRef]
- Ottis, P.; Toure, M.; Cromm, P.M.; Ko, E.; Gustafson, J.L.; Crews, C.M. Assessing Different E3 Ligases for Small Molecule Induced Protein Ubiquitination and Degradation. ACS Chem. Biol. 2017, 12, 2570–2578. [Google Scholar] [CrossRef]
- Lu, J.; Qian, Y.; Altieri, M.; Dong, H.; Wang, J.; Raina, K.; Hines, J.; Winkler, J.D.; Crew, A.P.; Coleman, K.; et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol. 2015, 22, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. [Google Scholar] [CrossRef] [PubMed]
- Buckley, D.L.; Raina, K.; Darricarrere, N.; Hines, J.; Gustafson, J.L.; Smith, I.E.; Miah, A.H.; Harling, J.D.; Crews, C.M. HaloPROTACS: Use of Small Molecule PROTACs to Induce Degradation of HaloTag Fusion Proteins. ACS Chem. Biol. 2015, 10, 1831–1837. [Google Scholar] [CrossRef] [PubMed]
- Nabet, B.; Ferguson, F.M.; Seong, B.K.A.; Kuljanin, M.; Leggett, A.L.; Mohardt, M.L.; Robichaud, A.; Conway, A.S.; Buckley, D.L.; Mancias, J.D.; et al. Rapid and direct control of target protein levels with VHL-recruiting dTAG molecules. Nat. Commun. 2020, 11, 4687. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Khoo, R.; Peh, K.M.; Teo, J.; Chang, S.C.; Ng, S.; Beilhartz, G.L.; Melnyk, R.A.; Johannes, C.W.; Brown, C.J.; et al. bioPROTACs as versatile modulators of intracellular therapeutic targets including proliferating cell nuclear antigen (PCNA). Proc. Natl. Acad. Sci. USA 2020, 117, 5791–5800. [Google Scholar] [CrossRef] [PubMed]
- Pellett, S.; Tepp, W.H.; Whitemarsh, R.C.M.; Bradshaw, M.; Johnson, E.A. In vivo onset and duration of action varies for botulinum neurotoxin A subtypes 1-5. Toxicon 2015, 107, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Whitemarsh, R.C.M.; Tepp, W.H.; Johnson, E.A.; Pellett, S. Persistence of botulinum neurotoxin a subtypes 1-5 in primary rat spinal cord cells. PLoS ONE 2014, 9, e90252. [Google Scholar] [CrossRef] [PubMed]
- Sen, E.; Kota, K.P.; Panchal, R.G.; Bavari, S.; Kiris, E. Screening of a Focused Ubiquitin-Proteasome Pathway Inhibitor Library Identifies Small Molecules as Novel Modulators of Botulinum Neurotoxin Type A Toxicity. Front. Pharmacol. 2021, 12, 763950. [Google Scholar] [CrossRef] [PubMed]
- Schwickart, M.; Huang, X.; Lill, J.R.; Liu, J.; Ferrando, R.; French, D.M.; Maecker, H.; O’rourke, K.; Bazan, F.; Eastham-Anderson, J.; et al. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature 2009, 463, 103–107. [Google Scholar] [CrossRef]
- Los, G.V.; Encell, L.P.; McDougall, M.G.; Hartzell, D.D.; Karassina, N.; Zimprich, C.; Wood, M.G.; Learish, R.; Ohana, R.F.; Urh, M.; et al. HaloTag: A novel protein labeling technology for cell imaging and protein analysis. ACS Chem. Biol. 2008, 3, 373–382. [Google Scholar] [CrossRef]
- Tovell, H.; Testa, A.; Maniaci, C.; Zhou, H.; Prescott, A.R.; Macartney, T.; Ciulli, A.; Alessi, D.R. Rapid and Reversible Knockdown of Endogenously Tagged Endosomal Proteins via an Optimized HaloPROTAC Degrader. ACS Chem. Biol. 2019, 14, 882–892. [Google Scholar] [CrossRef] [PubMed]
- Bondeson, D.P.; Mares, A.; Smith, I.E.D.; Ko, E.; Campos, S.; Miah, A.H.; Mulholland, K.E.; Routly, N.; Buckley, D.L.; Gustafson, J.L.; et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015, 11, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Semenova, E.; Guerriero, M.L.; Zhang, B.; Hock, A.; Hopcroft, P.; Kadamur, G.; Afzal, A.M.; Lazic, S.E. Flexible Fitting of PROTAC Concentration–Response Curves with Changepoint Gaussian Processes. SLAS Discov. Adv. Sci. Drug Discov. 2021, 26, 1212–1224. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Cintron, E.J.; Beske, P.H.; Tenezaca, L.; Tran, B.Q.; Oyler, J.M.; Glotfelty, E.J.; Angeles, C.A.; Syngkon, A.; Mukherjee, J.; Kalb, S.R.; et al. Engineering Botulinum Neurotoxin C1 as a Molecular Vehicle for Intra-Neuronal Drug Delivery. Sci. Rep. 2017, 7, srep42923. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, J.M.; Vazquez-Cintron, E.; Lam, K.-H.; Mukherjee, J.; Bedenice, D.; Ondeck, C.A.; Conroy, M.T.; Bodt, S.M.L.; Winner, B.M.; Webb, R.P.; et al. Camelid VHH Antibodies that Neutralize Botulinum Neurotoxin Serotype E Intoxication or Protease Function. Toxins 2020, 12, 611. [Google Scholar] [CrossRef] [PubMed]
- Lam, K.-H.; Perry, K.; Shoemaker, C.B.; Jin, R. Two VHH Antibodies Neutralize Botulinum Neurotoxin E1 by Blocking Its Membrane Translocation in Host Cells. Toxins 2020, 12, 616. [Google Scholar] [CrossRef]
- Lam, K.-H.; Tremblay, J.M.; Vazquez-Cintron, E.; Perry, K.; Ondeck, C.; Webb, R.P.; McNutt, P.M.; Shoemaker, C.B.; Jin, R. Structural Insights into Rational Design of Single-Domain Antibody-Based Antitoxins against Botulinum Neurotoxins. Cell Rep. 2020, 30, 2526–2539.e6. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsai, Y.C.; Kozar, L.; Mawi, Z.P.; Ichtchenko, K.; Shoemaker, C.B.; McNutt, P.M.; Weissman, A.M. The Degradation of Botulinum Neurotoxin Light Chains Using PROTACs. Int. J. Mol. Sci. 2024, 25, 7472. https://doi.org/10.3390/ijms25137472
Tsai YC, Kozar L, Mawi ZP, Ichtchenko K, Shoemaker CB, McNutt PM, Weissman AM. The Degradation of Botulinum Neurotoxin Light Chains Using PROTACs. International Journal of Molecular Sciences. 2024; 25(13):7472. https://doi.org/10.3390/ijms25137472
Chicago/Turabian StyleTsai, Yien Che, Loren Kozar, Zo P. Mawi, Konstantin Ichtchenko, Charles B. Shoemaker, Patrick M. McNutt, and Allan M. Weissman. 2024. "The Degradation of Botulinum Neurotoxin Light Chains Using PROTACs" International Journal of Molecular Sciences 25, no. 13: 7472. https://doi.org/10.3390/ijms25137472