
Pulmonary Vascular Responses to Chronic Intermittent Hypoxia in a Guinea Pig Model of Obstructive Sleep Apnea

 ,
,  , , and
, , and
Abstract
1. Introduction
2. Results
2.1. Animal’s General Status, Body Weight, and Hematocrit
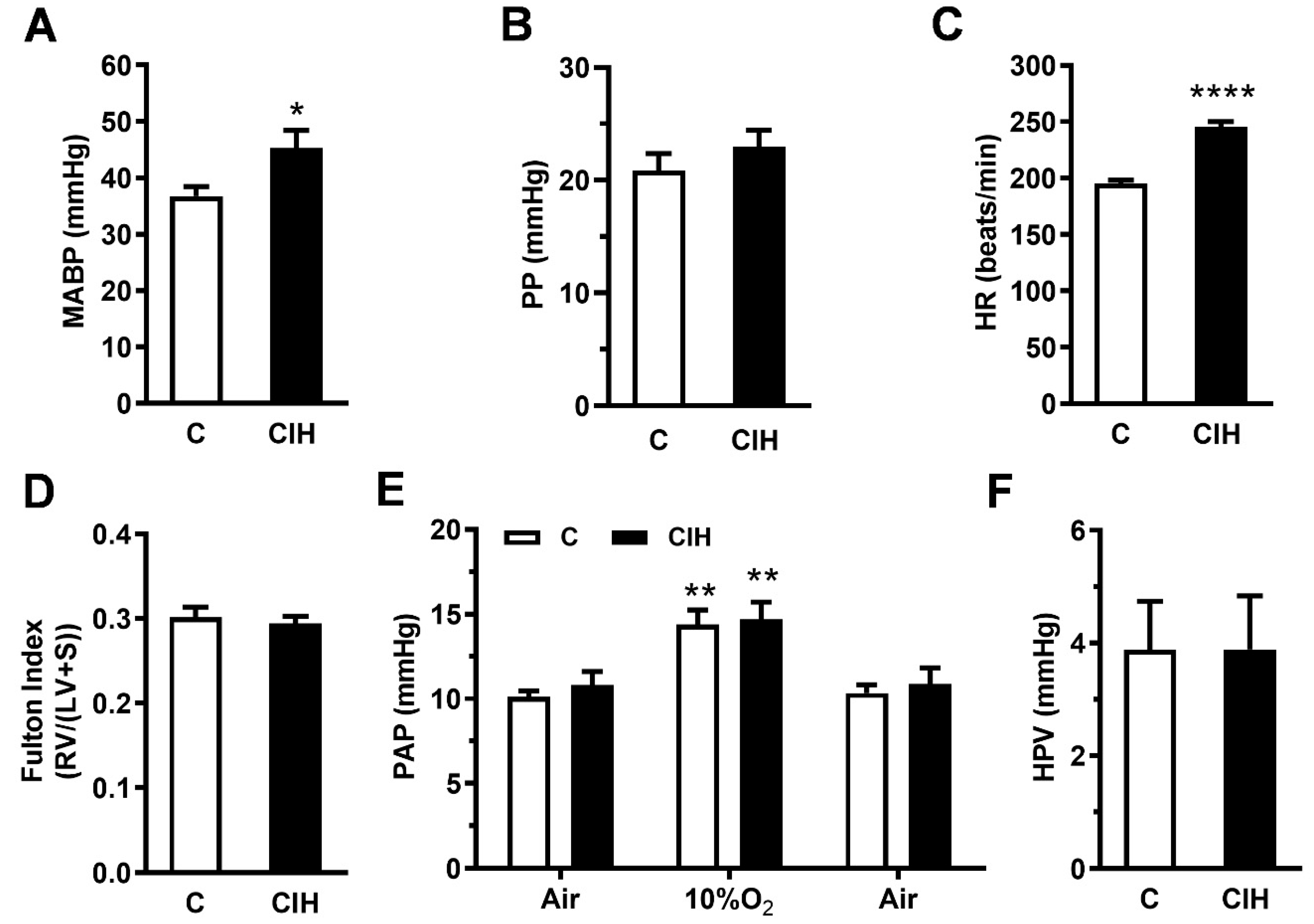
2.2. Pulmonary Hemodynamics and Right Ventricle Hypertrophy
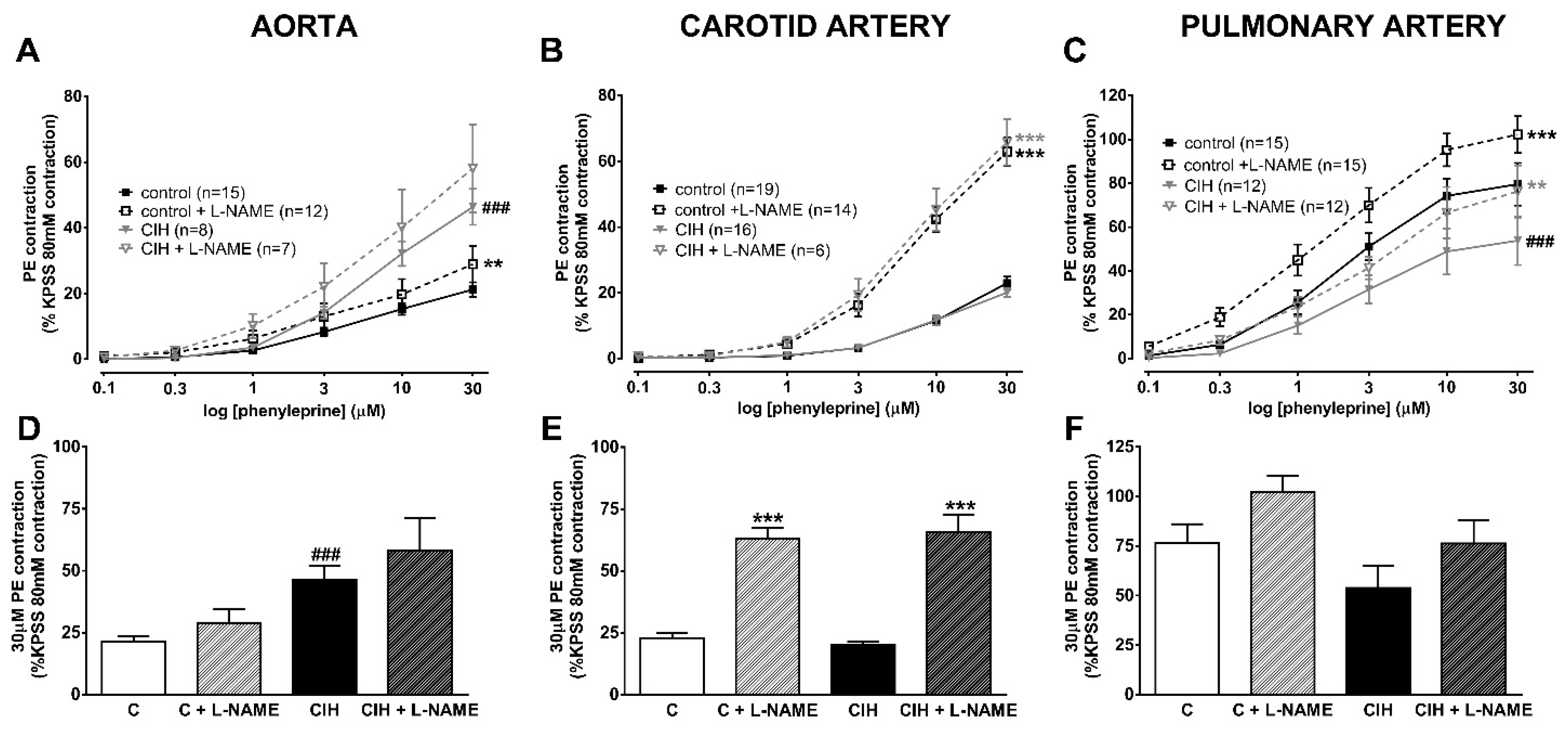
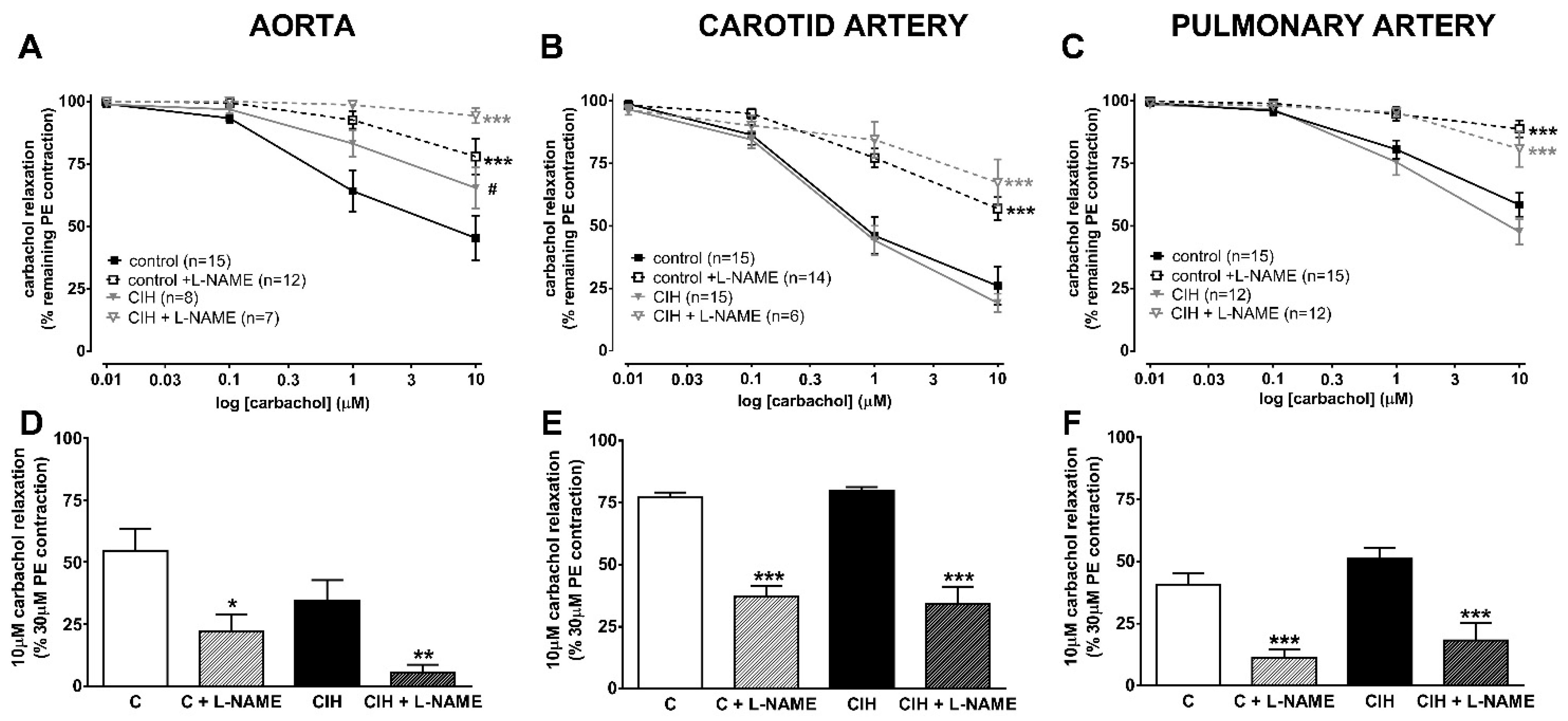
2.3. Vascular Contractility and Endothelial Function
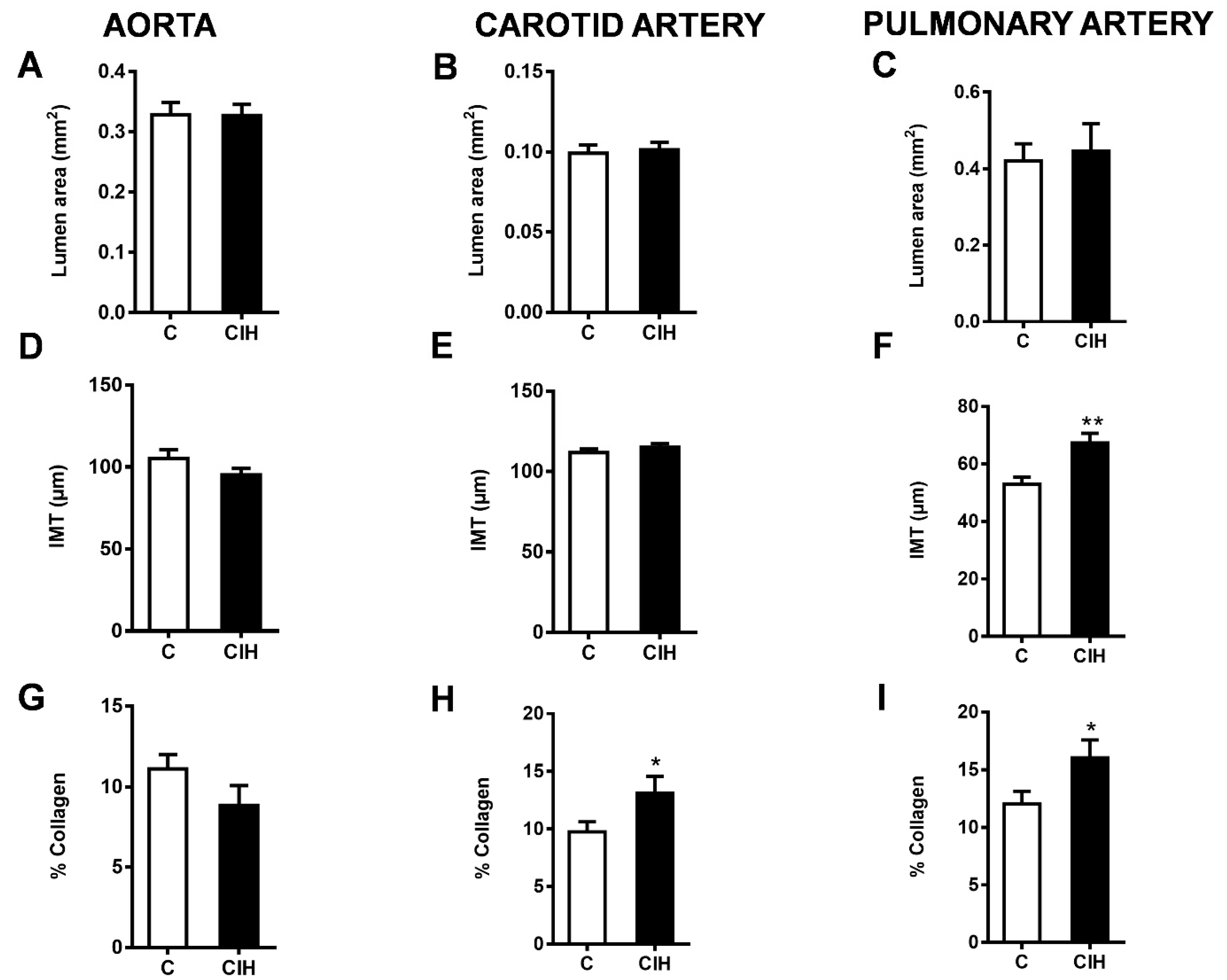
2.4. Morphometric and Histological Assessments of Vascular Remodeling
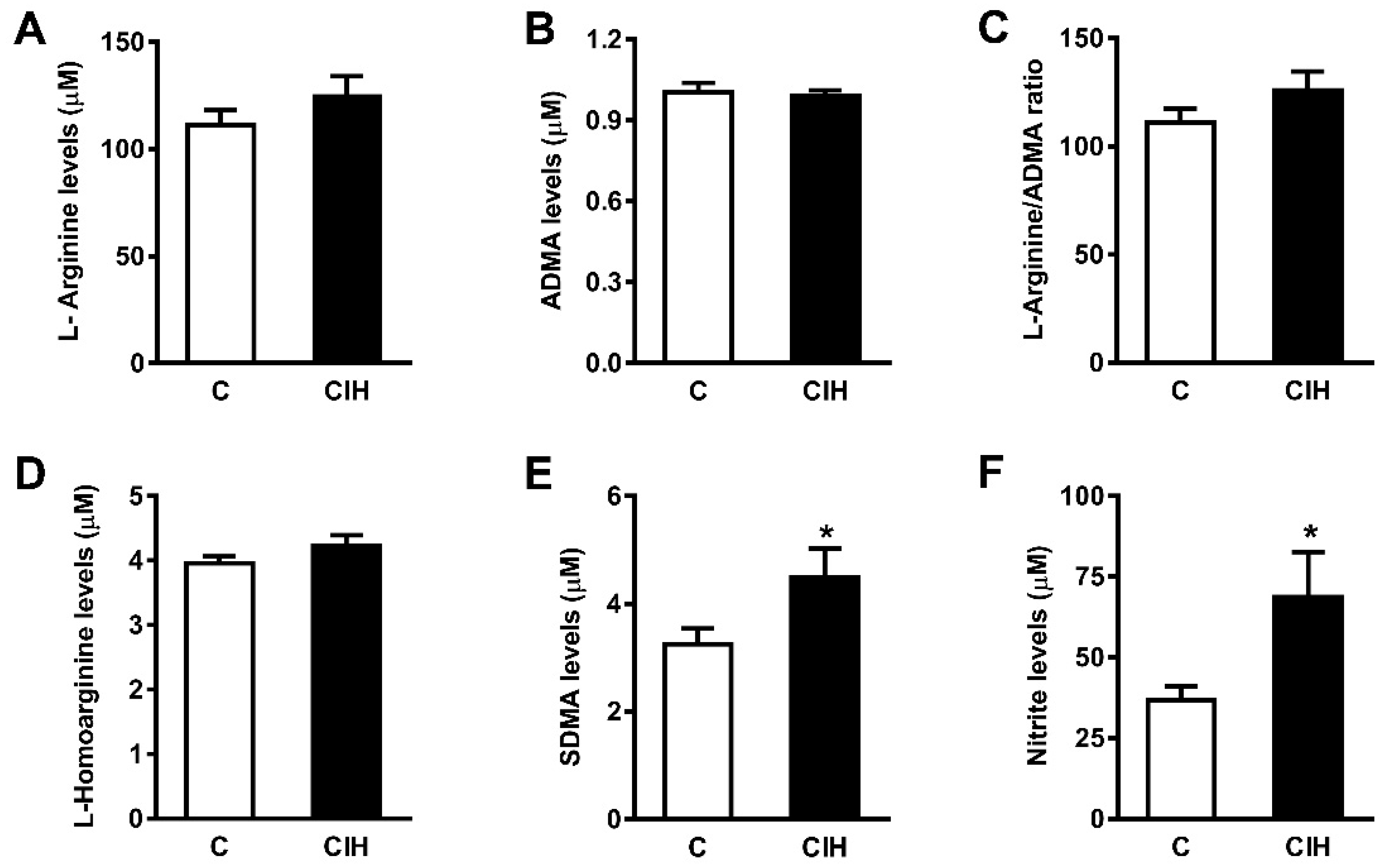
2.5. NO Bioavailability: Plasma Nitrites and Nitrates and L-arginine and Its Metabolites
2.6. Plasma Endothelin-1 (ET-1), Angotensin II (ANG II), Atrial Natriuretic Peptide (ANP), Vascular Endothelial Growth Factor (VEGF), and Catecholamines
2.7. Aconitase: Fumarase Activity Ratio and Nuclear Factor Kappa B (NF-kB)
3. Discussion
4. Materials and Methods
4.1. Chronic Intermittent Hypoxia Protocol
4.2. In Vivo Pulmonary and Systemic Blood Pressure Measurements
4.3. Vessel Reactivity and Endothelial Function
4.4. Fulton Index
4.5. Morphometric and Histological Assessments
4.6. L-arginine and Metabolites
4.7. Plasma Chemistry of Vasoactive Agents
4.8. Drugs
4.9. Data Presentation and Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Benjafield, A.V.; Ayas, N.T.; Eastwood, P.R.; Heinzer, R.; Ip, M.S.; Morrell, M.J.; Nuñez, C.M.; Patel, S.R.; Penzel, T.; Pepin, J.L.; et al. Estimation of the global prevalence and burden of obstructive sleep apnoea: A literature-based analysis. Lancet Respir. Med. 2019, 7, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Wolk, R.; Shamsuzzaman, A.S.; Somers, V.K. Obesity, Sleep Apnea, and Hypertension. Hypertension 2003, 42, 1067–1074. [Google Scholar] [CrossRef]
- Lavie, L. Oxidative stress in obstructive sleep apnea and intermittent hypoxia—Revisited—The bad ugly and good: Implications to the heart and brain. Sleep Med. Rev. 2015, 20, 27–45. [Google Scholar] [CrossRef] [PubMed]
- Lévy, P.; Kohler, M.; McNicholas, W.T.; Barbé, F.; McEvoy, R.D.; Somers, V.K.; Lavie, L.; Pepin, J.L. Obstructive sleep apnoea syndrome. Nat. Rev. Dis. Primers 2015, 1, 15015. [Google Scholar] [CrossRef] [PubMed]
- Dematteis, M.; Godin-Ribuot, D.; Arnaud, C.; Ribuot, C.; Stanke-Labesque, F.; Pépin, J.L.; Levy, P. Cardiovascular Consequences of Sleep-Disordered Breathing: Contribution of Animal Models to Understanding of the Human Disease. ILAR J. 2009, 50, 262–281. [Google Scholar] [CrossRef] [PubMed]
- Ryan, S.; Cummins, E.P.; Farre, R.; Gileles-Hillel, A.; Jun, J.C.; Oster, H.; Pepin, J.L.; Ray, D.W.; Reutrakul, S.; Sanchez-de-la-Torre, M.; et al. Understanding the pathophysiological mechanisms of cardiometabolic complications in obstructive sleep apnoea: Towards personalised treatment approaches. Eur. Respir. J. 2020, 56, 1902295. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, E.C.; Lesske, J.; Qian, W.; Miller, C.C.; Unger, T. Repetitive, episodic hypoxia causes diurnal elevation of blood pressure in rats. Hypertension 1992, 19, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, N.R. Carotid body chemoreflex: A driver of autonomic abnormalities in sleep apnoea. Exp. Physiol. 2016, 101, 975–985. [Google Scholar] [CrossRef]
- Semenza, G.L.; Nanduri, X.; Prabhakar, R. Neural regulation of hypoxia-inducible factors and redox state drives the pathogenesis of hypertension in a rodent model of sleep apnea. J. Appl. Physiol. 2015, 119, 1152–1156. [Google Scholar] [CrossRef]
- Lesske, J.; Fletcher, E.C.; Bao, G.; Unger, T. Hypertension caused by chronic intermittent hypoxia—Influence of chemoreceptors and sympathetic nervous system. J. Hypertens. 1997, 15, 1593–1603. [Google Scholar]
- Del Rio, R.; Andrade, D.C.; Lucero, C.; Arias, P.; Iturriaga, R. Carotid Body Ablation Abrogates Hypertension and Autonomic Alterations Induced by Intermittent Hypoxia in Rats. Hypertension 2016, 68, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, J.A.; Veasey, S.C.; Morgan, B.J.; O’Donnell, C.P. Pathophysiology of Sleep Apnea. Physiol. Rev. 2010, 90, 47–112. [Google Scholar] [CrossRef] [PubMed]
- Iturriaga, R.; Castillo-Galan, S. The Beneficial Effect of the Blockade of Stim-Activated TRPC-ORAI Channels on Vascular Remodeling and Pulmonary Hypertension Induced by Intermittent Hypoxia Is Independent of Oxidative Stress. Adv. Exp. Med. Biol. 2023, 1427, 53–60. [Google Scholar] [PubMed]
- Prieto-Lloret, J.; Olea, E.; Gordillo-Cano, A.; Docio, I.; Obeso, A.; Gomez-Niño, A.; Aaronson, P.I.; Rocher, A. Maladaptive pulmonary vascular responses to chronic sustained and chronic intermittent hypoxia in rat. Antioxidants 2022, 11, 54. [Google Scholar] [CrossRef]
- Fagan, K.A. Selected Contribution: Pulmonary hypertension in mice following intermittent hypoxia. J. Appl. Physiol. 2001, 90, 2502–2507. [Google Scholar] [CrossRef] [PubMed]
- Docio, I.; Olea, E.; Prieto-Lloret, J.; Gallego-Martin, T.; Obeso, A.; Gomez-Niño, A.; Rocher, A. Guinea Pig as a Model to Study the Carotid Body Mediated Chronic Intermittent Hypoxia Effects. Front. Physiol. 2018, 9, 694. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, E.C.; Lesske, J.; Behm, R.; Miller, C.C.; Stauss, H.; Unger, T. Carotid chemoreceptors, systemic blood pressure, and chronic episodic hypoxia mimicking sleep apnea. J. Appl. Physiol. 1992, 72, 1978–1984. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, C.; Beguin, P.C.; Lantuejoul, S.; Pepin, J.L.; Guillermet, C.; Pelli, G.; Burger, F.; Buatois, V.; Ribuot, C.; Baguet, J.P.; et al. The Inflammatory Preatherosclerotic Remodeling Induced by Intermittent Hypoxia Is Attenuated by RANTES/CCL5 Inhibition. Am. J. Respir. Crit. Care Med. 2011, 184, 724–731. [Google Scholar] [CrossRef] [PubMed]
- Gras, E.; Belaidi, E.; Briançon-Marjollet, A.; Pépin, J.L.; Arnaud, C.; Godin-Ribuot, D. Endothelin-1 mediates intermittent hypoxia-induced inflammatory vascular remodeling through HIF-1 activation. J. Appl. Physiol. 2016, 120, 437–443. [Google Scholar] [CrossRef]
- Phillips, S.A.; Olson, E.B.; Morgan, B.J.; Lombard, J.H. Chronic intermittent hypoxia impairs endothelium-dependent dilation in rat cerebral and skeletal muscle resistance arteries. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H388–H393. [Google Scholar] [CrossRef]
- Arnaud, C.; Poulain, L.; Lévy, P.; Dematteis, M. Inflammation contributes to the atherogenic role of intermittent hypoxia in apolipoprotein-E knock out mice. Atherosclerosis 2011, 219, 425–431. [Google Scholar] [CrossRef]
- Drager, L.F.; Yao, Q.; Hernandez, K.L.; Shin, M.K.; Bevans-Fonti, S.; Gay, J.; Sussan, T.E.; Jun, J.C.; Myers, A.C.; Olivecrona, G.; et al. Chronic intermittent hypoxia induces atherosclerosis via activation of adipose angiopoietin-like 4. Am. J. Respir. Crit. Care Med. 2013, 188, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.J.; Yang, C.C.; Hsu, Y.Y.; Lin, Y.N.; Kuo, T.B. Enhanced sympathetic outflow and decreased baroreflex sensitivity are associated with intermittent hypoxia-induced systemic hypertension in conscious rats. J. Appl. Physiol. 2006, 100, 1974–1982. [Google Scholar] [CrossRef] [PubMed]
- Lucking, E.F.; O’Halloran, K.D.; Jones, J.F. Increased cardiac output contributes to the development of chronic intermittent hypoxia-induced hypertension. Exp. Physiol. 2014, 99, 1312–1324. [Google Scholar] [CrossRef] [PubMed]
- Iturriaga, R.; Oyarce, M.P.; Dias, A.C. Role of Carotid Body in Intermittent Hypoxia-Related Hypertension. Curr. Hypertens. Rep. 2017, 19, 38. [Google Scholar] [CrossRef] [PubMed]
- Peppard, P.E.; Young, T.; Palta, M.; Skatrud, J. Prospective study of the association between sleep-disordered breathing and hypertension. N. Engl. J. Med. 2000, 342, 1378–1384. [Google Scholar] [CrossRef]
- Fletcher, E.C. Physiological consequences of intermittent hypoxia: Systemic blood pressure. J. Appl. Physiol. 2001, 90, 1600–1605. [Google Scholar] [CrossRef]
- Lucking, E.F.; O’Connor, K.M.; Strain, C.R.; Fouhy, F.; Bastiaanssen, T.F.; Burns, D.P.; Golubeva, A.V.; Stanton, C.; Clarke, G.; Cryan, J.F.; et al. Chronic intermittent hypoxia disrupts cardiorespiratory homeostasis and gut microbiota composition in adult male guinea-pigs. EBioMedicine 2018, 38, 191–205. [Google Scholar] [CrossRef]
- Sun, M.K.; Reis, D.J. Hypoxia selectively excites vasomotor neurons of rostral ventrolateral medulla in rats. Am. J. Physiol. 1994, 266, R245–R256. [Google Scholar] [CrossRef]
- Marina, N.; Tang, F.; Figueiredo, M.; Mastitskaya, S.; Kasimov, V.; Mohamed-Ali, V.; Roloff, E.; Teschemacher, A.G.; Gourine, A.V.; Kasparov, S. Purinergic signalling in the rostral ventro-lateral medulla controls sympathetic drive and contributes to the progression of heart failure following myocardial infarction in rats. Basic Res. Cardiol. 2013, 108, 317. [Google Scholar] [CrossRef]
- Machado, B.H.; Zoccal, D.B.; Moraes, D.J. Neurogenic hypertension and the secrets of respiration. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2017, 312, R864–R872. [Google Scholar] [CrossRef]
- Gonzalez-Obeso, E.; Docio, I.; Olea, E.; Cogolludo, A.; Obeso, A.; Rocher, A.; Gomez-Niño, A. Guinea Pig Oxygen-Sensing and Carotid Body Functional Properties. Front. Physiol. 2017, 8, 285. [Google Scholar] [CrossRef]
- Greenberg, H.E.; Sica, A.; Batson, D.; Scharf, S.M. Chronic intermittent hypoxia increases sympathetic responsiveness to hypoxia and hypercapnia. J. Appl. Physiol. 1999, 86, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Kraiczi, H.; Magga, J.; Sun, X.Y.; Ruskoaho, H.; Zhao, X.; Hedner, J. Hypoxic pressor response, cardiac size, and natriuretic peptides are modified by long-term intermittent hypoxia. J. Appl. Physiol. 1999, 87, 2025–2031. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.H.; Tang, Y.; Niu, X.; Sun, H.Y. The role of nitric oxide (NO) levels in patients with obstructive sleep apnea-hypopnea syndrome: A meta-analysis. Sleep Breath. 2021, 25, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Moller, D.S.; Lind, P.; Strunge, B.; Pedersen, E.B. Abnormal vasoactive hormones and 24-hour blood pressure in obstructive sleep apnea. Am. J. Hypertens. 2003, 16, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Phillips, B.G.; Narkiewicz, K.; Pesek, C.A.; Haynes, W.G.; Dyken, M.E.; Somers, V.K. Effects of obstructive sleep apnea on endothelin-1 and blood pressure. J. Hypertens. 1999, 17, 61–66. [Google Scholar] [CrossRef]
- Lemmey, H.A.; Garland, C.J.; Dora, K.A. Intrinsic regulation of microvascular tone by myoendothelial feedback circuits. Curr. Top. Membr. 2020, 85, 327–355. [Google Scholar]
- Castillo, S.; Arenas, G.A.; Reyes, R.V.; Krause, B.J.; Iturriaga, R. Stim-activated TRPC-ORAI channels in pulmonary hypertension induced by chronic intermittent hypoxia. Pulm. Circ. 2020, 10, 13–22. [Google Scholar] [CrossRef]
- Snow, J.B.; Norton, C.E.; Sands, M.A.; Weise-Cross, L.; Yan, S.; Herbert, L.M.; Sheak, J.R.; Gonzalez Bosc, L.V.; Walker, B.R.; Kanagy, N.L.; et al. Intermittent Hypoxia Augments Pulmonary Vasoconstrictor Reactivity through PKCbeta/Mitochondrial Oxidant Signaling. Am. J. Respir. Cell Mol. Biol. 2020, 62, 732–746. [Google Scholar] [CrossRef]
- Tozzi, C.A.; Poiani, G.J.; Edelman, N.H.; Riley, D.J. Vascular collagen affects reactivity of hypertensive pulmonary arteries of the rat. J. Appl. Physiol. 1989, 66, 1730–1735. [Google Scholar] [CrossRef]
- Sandow, S.L.; Gzik, D.J.; Lee, R.M. Arterial internal elastic lamina holes: Relationship to function? J. Anat. 2009, 214, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, B.; Godin-Ribuot, D.; Joyeux-Faure, M.; Caron, F.; Bessard, G.; Lévy, P.; Stanke-Labesque, F. Functional assessment of vascular reactivity after chronic intermittent hypoxia in the rat. Respir. Physiol. Neurobiol. 2005, 150, 278–286. [Google Scholar] [CrossRef]
- Allahdadi, K.J.; Walker, B.R.; Kanagy, N.L. Augmented Endothelin Vasoconstriction in Intermittent Hypoxia-Induced Hypertension. Hypertension 2005, 45, 705–709. [Google Scholar] [CrossRef] [PubMed]
- Daly, C.J.; Ross, R.A.; Whyte, J.; Henstridge, C.M.; Irving, A.J.; McGrath, J.C. Fluorescent ligand binding reveals heterogeneous distribution of adrenoceptors and “cannabinoid-like” receptors in small arteries. Br. J. Pharmacol. 2010, 159, 787–796. [Google Scholar] [CrossRef]
- Minoguchi, K.; Yokoe, T.; Tazaki, T.; Minoguchi, H.; Tanaka, A.; Oda, N.; Okada, S.; Ohta, S.; Naito, H.; Adachi, M. Increased Carotid Intima-Media Thickness and Serum Inflammatory Markers in Obstructive Sleep Apnea. Am. J. Respir. Crit. Care Med. 2005, 172, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Hodis, H.N.; Mack, W.J.; LaBree, L.; Selzer, R.H.; Liu, C.R.; Liu, C.H.; Liu, C.H.; Azen, S.P. The role of carotid arterial intima—Media thickness in predicting clinical coronary events. Ann. Intern. Med. 1998, 128, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Gileles-Hillel, A.; Almendros, I.; Khalyfa, A.; Zhang, S.X.; Wang, Y.; Gozal, D. Early Intermittent Hypoxia Induces Proatherogenic Changes in Aortic Wall Macrophages in a Murine Model of Obstructive Sleep Apnea. Am. J. Respir. Crit. Care Med. 2014, 190, 958–961. [Google Scholar] [CrossRef]
- Dewan, N.A.; Nieto, F.J.; Somers, V.K. Intermittent Hypoxemia and OSA: Implications for Comorbidities. Chest 2015, 147, 266–274. [Google Scholar] [CrossRef]
- Quintero, M.; Gonzalez-Martin, M.C.; Vega-Agapito, V.; Gonzalez, C.; Obeso, A.; Farré, R.; Gonzalez, C. The effects of intermittent hypoxia on redox status, NF-κB activation, and plasma lipid levels are dependent on the lowest oxygen saturation. Free Radic. Biol. Med. 2013, 65, 1143–1154. [Google Scholar] [CrossRef]
- Wagenseil, J.E.; Mecham, R.P. Elastin in Large Artery Stiffness and Hypertension. J. Cardiovasc. Transl. Res. 2012, 5, 264–273. [Google Scholar] [CrossRef]
- Campen, M.J.; Shimoda, L.A.; O’Donnell, C.P. Acute and chronic cardiovascular effects of intermittent hypoxia in C57BL/6J mice. J. Appl. Physiol. 2005, 99, 2028–2035. [Google Scholar] [CrossRef] [PubMed]
- Nisbet, R.E.; Graves, A.S.; Kleinhenz, D.J.; Rupnow, H.L.; Reed, A.L.; Fan, T.H.; Mitchell, P.O.; Sutliff, R.L.; Hart, C.M. The Role of NADPH Oxidase in Chronic Intermittent Hypoxia-Induced Pulmonary Hypertension in Mice. Am. J. Respir. Cell Mol. Biol. 2009, 40, 601–609. [Google Scholar] [CrossRef]
- Castillo-Galan, S.; Riquelme, B.; Iturriaga, R. Crucial Role of Stromal Interaction Molecule-Activated TRPC-ORAI Channels in Vascular Remodeling and Pulmonary Hypertension Induced by Intermittent Hypoxia. Front. Physiol. 2022, 13, 841828. [Google Scholar] [CrossRef] [PubMed]
- Chaouat, A.; Weitzenblum, E.; Krieger, J.; Oswald, M.; Kessler, R. Pulmonary Hemodynamics in the Obstructive Sleep Apnea Syndrome: Results in 220 Consecutive Patients. Chest 1996, 109, 380–386. [Google Scholar] [CrossRef]
- Yang, J.Z.; Mokhlesi, B.; Mesarwi, O.A. Obstructive sleep apnea and pulmonary hypertension: The pendulum swings again. J. Clin. Sleep Med. 2023, 19, 209–211. [Google Scholar] [CrossRef]
- Khosla, U.M.; Zharikov, S.; Finch, J.L.; Nakagawa, T.; Roncal, C.; Mu, W.; Krotova, K.; Block, E.R.; Prabhakar, S.; Johnson, R.J. Hyperuricemia induces endothelial dysfunction. Kidney Int. 2005, 67, 1739–1742. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, L.C.; Tanus-Santos, J.E.; Castro, M.M. The potential of stimulating nitric oxide formation in the treatment of hypertension. Expert Opin. Ther. Targets 2017, 21, 543–556. [Google Scholar] [CrossRef]
- Böger, R.H. The emerging role of asymmetric dimethylarginine as a novel cardiovascular risk factor. Cardiovasc. Res. 2003, 59, 824–833. [Google Scholar] [CrossRef]
- Teerlink, T.; Luo, Z.; Palm, F.; Wilcox, C.S. Cellular ADMA: Regulation and action. Pharmacol. Res. 2009, 60, 448–460. [Google Scholar] [CrossRef]
- Cooke, J.P. Does ADMA Cause Endothelial Dysfunction? Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2032–2037. [Google Scholar] [CrossRef]
- Surdacki, A.; Nowicki, M.; Sandmann, J.; Tsikas, D.; Boeger, R.H.; Bode-Boeger, S.M.; Kruszelnicka-Kwiatkowska, O.; Kokot, F.; Dubiel, J.S.; Froelich, J.C. Reduced Urinary Excretion of Nitric Oxide Metabolites and Increased Plasma Levels of Asymmetric Dimethylarginine in Men with Essential Hypertension. J. Cardiovasc. Pharmacol. 1999, 33, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Böger, R.H.; Maas, R.; Schulze, F.; Schwedhelm, E. Asymmetric dimethylarginine (ADMA) as a prospective marker of cardiovascular disease and mortality—An update on patient populations with a wide range of cardiovascular risk. Pharmacol. Res. 2009, 60, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Böger, R.H.; Hannemann, J. Dual role of the L-arginine–ADMA–NO pathway in systemic hypoxic vasodilation and pulmonary hypoxic vasoconstriction. Pulm. Circ. 2020, 10, 2045894020918850. [Google Scholar] [CrossRef]
- Hannemann, J.; Siques, P.; Schmidt-Hutten, L.; Zummack, J.; Brito, J.; Boger, R. Association of Genes of the NO Pathway with Altitude Disease and Hypoxic Pulmonary Hypertension. J. Clin. Med. 2021, 10, 5761. [Google Scholar] [CrossRef] [PubMed]
- Kanagy, N.L.; Walker, B.R.; Nelin, L.D. Role of Endothelin in Intermittent Hypoxia-Induced Hypertension. Hypertension 2001, 37, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Rey, S.; Del Rio, R.; Iturriaga, R. Contribution of Endothelin-1 and Endothelin A and B Receptors to the Enhanced Carotid Body Chemosensory Responses Induced by Chronic Intermittent Hypoxia. Adv. Exp. Med. Biol. 2008, 605, 228–232. [Google Scholar] [PubMed]
- de Frutos, S.; Caldwell, E.; Nitta, C.H.; Kanagy, N.L.; Wang, J.; Wang, W.; Walker, M.K.; Gonzalez Bosc, L.V. NFATc3 contributes to intermittent hypoxia-induced arterial remodeling in mice. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H356–H363. [Google Scholar] [CrossRef] [PubMed]
- Chaumais, M.C.; Djessas, M.R.A.; Thuillet, R.; Cumont, A.; Tu, L.; Hebert, G.; Gaignard, P.; Huertas, A.; Savale, L.; Humbert, M.; et al. Additive protective effects of sacubitril/valsartan and bosentan on vascular remodelling in experimental pulmonary hypertension. Cardiovasc. Res. 2021, 117, 1391–1401. [Google Scholar] [CrossRef]
- Gjørup, P.H.; Sadauskiene, L.; Wessels, J.; Nyvad, O.; Strunge, B.; Pedersen, E.B. Abnormally Increased Endothelin-1 in Plasma During the Night in Obstructive Sleep Apnea: Relation to Blood Pressure and Severity of Disease. Am. J. Hypertens. 2007, 20, 44–52. [Google Scholar] [CrossRef]
- Atkeson, A.; Yeh, S.Y.; Malhotra, A.; Jelic, S. Endothelial Function in Obstructive Sleep Apnea. Prog. Cardiovasc. Dis. 2009, 51, 351–362. [Google Scholar] [CrossRef]
- Limberg, J.K.; Baker, S.E.; Ott, E.P.; Jacob, D.W.; Scruggs, Z.M.; Harper, J.L.; Manrique-Acevedo, C.M. Endothelin-1 receptor blockade does not alter the sympathetic and hemodynamic response to acute intermittent hypoxia in men. J. Appl. Physiol. 2022, 133, 867–875. [Google Scholar] [CrossRef] [PubMed]
- Marcus, N.J.; Li, Y.L.; Bird, C.E.; Schultz, H.D.; Morgan, B.J. Chronic intermittent hypoxia augments chemoreflex control of sympathetic activity: Role of the angiotensin II type 1 receptor. Respir. Physiol. Neurobiol. 2010, 171, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Saxena, A.; Little, J.T.; Nedungadi, T.P.; Cunningham, J.T. Angiotensin II type 1a receptors in subfornical organ contribute towards chronic intermittent hypoxia-associated sustained increase in mean arterial pressure. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H435–H446. [Google Scholar] [CrossRef] [PubMed]
- Mifflin, S.; Cunningham, J.T.; Toney, G.M.; Mifflin, S. Neurogenic mechanisms underlying the rapid onset of sympathetic responses to intermittent hypoxia. J. Appl. Physiol. 2015, 119, 1441–1448. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Wei, F.; Ning, S.; Hu, J.; Shi, H.; Yao, Z.; Tang, M.; Zhang, Y.; Gong, J.; Ge, J.; et al. PPARgamma Agonist Rosiglitazone and Antagonist GW9662: Antihypertensive Effects on Chronic Intermittent Hypoxia-Induced Hypertension in Rats. J. Cardiovasc. Transl. Res. 2024. ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Iturriaga, R.; Andrade, D.C.; Del Rio, R. Enhanced carotid body chemosensory activity and the cardiovascular alterations induced by intermittent hypoxia. Front. Physiol. 2014, 5, 468. [Google Scholar] [CrossRef]
- Pialoux, V.; Foster, G.E.; Ahmed, S.B.; Beaudin, A.E.; Hanly, P.J.; Poulin, M.J. Losartan abolishes oxidative stress induced by intermittent hypoxia in humans. J. Physiol. 2011, 589, 5529–5537. [Google Scholar] [CrossRef]
- Lam, S.Y.; Liu, Y.; Ng, K.M.; Liong, E.C.; Tipoe, G.L.; Leung, P.S.; Fung, L.M. Upregulation of a local renin–angiotensin system in the rat carotid body during chronic intermittent hypoxia. Exp. Physiol. 2014, 99, 220–231. [Google Scholar] [CrossRef]
- Morrell, N.W.; Morris, K.G.; Stenmark, K.R. Role of angiotensin-converting enzyme and angiotensin II in development of hypoxic pulmonary hypertension. Am. J. Physiol. 1995, 269, H1186–H1194. [Google Scholar] [CrossRef]
- Marciante, A.B.; Shell, B.; Farmer, G.E.; Cunningham, J.T. Role of angiotensin II in chronic intermittent hypoxia-induced hypertension and cognitive decline. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2021, 320, R519–R525. [Google Scholar] [CrossRef]
- Goldstein, D.S.; Eisenhofer, G.; Kopin, I.J. Sources and Significance of Plasma Levels of Catechols and Their Metabolites in Humans. J. Pharmacol. Exp. Ther. 2003, 305, 800–811. [Google Scholar] [CrossRef]
- Kjær, M. Adrenal Gland: Fight or Flight Implications for Exercise and Sports. In The Endocrine System in Sports and Exercise; William, J., Kraemer, W.J., Rogol, A.D., Eds.; Wiley: Hoboken, NJ, USA, 2005; pp. 194–199. [Google Scholar]
- Schwenke, D.O.; Bolter, C.P.; Cragg, P.A. Are the carotid bodies of the guinea-pig functional? Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2007, 146, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Monge, C.; Leon-Velarde, F. Physiological adaptation to high altitude: Oxygen transport in mammals and birds. Physiol. Rev. 1991, 71, 1135–1172. [Google Scholar] [CrossRef]
- Zoccal, D.B.; Vieira, B.N.; Mendes, L.R.; Evangelista, A.B.; Leirão, I.P. Hypoxia sensing in the body: An update on the peripheral and central mechanisms. Exp. Physiol. 2024, 109, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Ricart-Jané, D.; Llobera, M.; López-Tejero, M.D. Anticoagulants and Other Preanalytical Factors Interfere in Plasma Nitrate/Nitrite Quantification by the Griess Method. Nitric Oxide 2002, 6, 178–185. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C | CIH | |
|---|---|---|
| Body weight (g) | 791 ± 19 | 715 ± 14 ** |
| Hematocrit (%) | 41.5 ± 0.8 | 41.4 ± 0.8 |
| EPO (mU/mL) | 141 ± 17 | 214 ± 52 * |
| pO2 (mm Hg) | 65 ± 6 | 64 ± 6 |
| pCO2 (mm Hg) | 36 ± 2 | 38 ± 3 |
| Aorta | Carotid Artery | Pulmonary Artery | ||||
|---|---|---|---|---|---|---|
| C | CIH | C | CIH | C | CIH | |
| Mean (mN) | 9.7 ± 1.4 | 6.5 ± 1.8 | 16.0 ± 1.7 | 16.9 ± 2.4 | 10.0 ± 1.4 | 10.3 ± 0.7 |
| n | 15 | 8 | 19 | 16 | 15 | 12 |
| Guinea Pig | C | CIH |
|---|---|---|
| ET-1 (pg/mL) | 19.9 ± 1.9 | 21.5 ± 1.8 |
| ANG II (pg/mL) | 355 ± 49 | 412 ± 67 |
| ANP (pg/mL) | 234 ± 15 | 209 ± 6 |
| VEGF (pg/mL) | 2.5 ± 0.2 | 2.6 ± 0.2 |
| NE (pmol/mL) | 7.2 ± 1.7 | 80.7 ± 24.0 * |
| E (pmol/mL) | 11 ± 0.4 | 64 ± 20 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olea, E.; Valverde-Pérez, E.; Docio, I.; Prieto-Lloret, J.; Aaronson, P.I.; Rocher, A. Pulmonary Vascular Responses to Chronic Intermittent Hypoxia in a Guinea Pig Model of Obstructive Sleep Apnea. Int. J. Mol. Sci. 2024, 25, 7484. https://doi.org/10.3390/ijms25137484
Olea E, Valverde-Pérez E, Docio I, Prieto-Lloret J, Aaronson PI, Rocher A. Pulmonary Vascular Responses to Chronic Intermittent Hypoxia in a Guinea Pig Model of Obstructive Sleep Apnea. International Journal of Molecular Sciences. 2024; 25(13):7484. https://doi.org/10.3390/ijms25137484
Chicago/Turabian StyleOlea, Elena, Esther Valverde-Pérez, Inmaculada Docio, Jesus Prieto-Lloret, Philip I. Aaronson, and Asunción Rocher. 2024. "Pulmonary Vascular Responses to Chronic Intermittent Hypoxia in a Guinea Pig Model of Obstructive Sleep Apnea" International Journal of Molecular Sciences 25, no. 13: 7484. https://doi.org/10.3390/ijms25137484
APA StyleOlea, E., Valverde-Pérez, E., Docio, I., Prieto-Lloret, J., Aaronson, P. I., & Rocher, A. (2024). Pulmonary Vascular Responses to Chronic Intermittent Hypoxia in a Guinea Pig Model of Obstructive Sleep Apnea. International Journal of Molecular Sciences, 25(13), 7484. https://doi.org/10.3390/ijms25137484