
scL-2PAM: A Novel Countermeasure That Ameliorates Neuroinflammation and Neuronal Losses in Mice Exposed to an Anticholinesterase Organophosphate

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
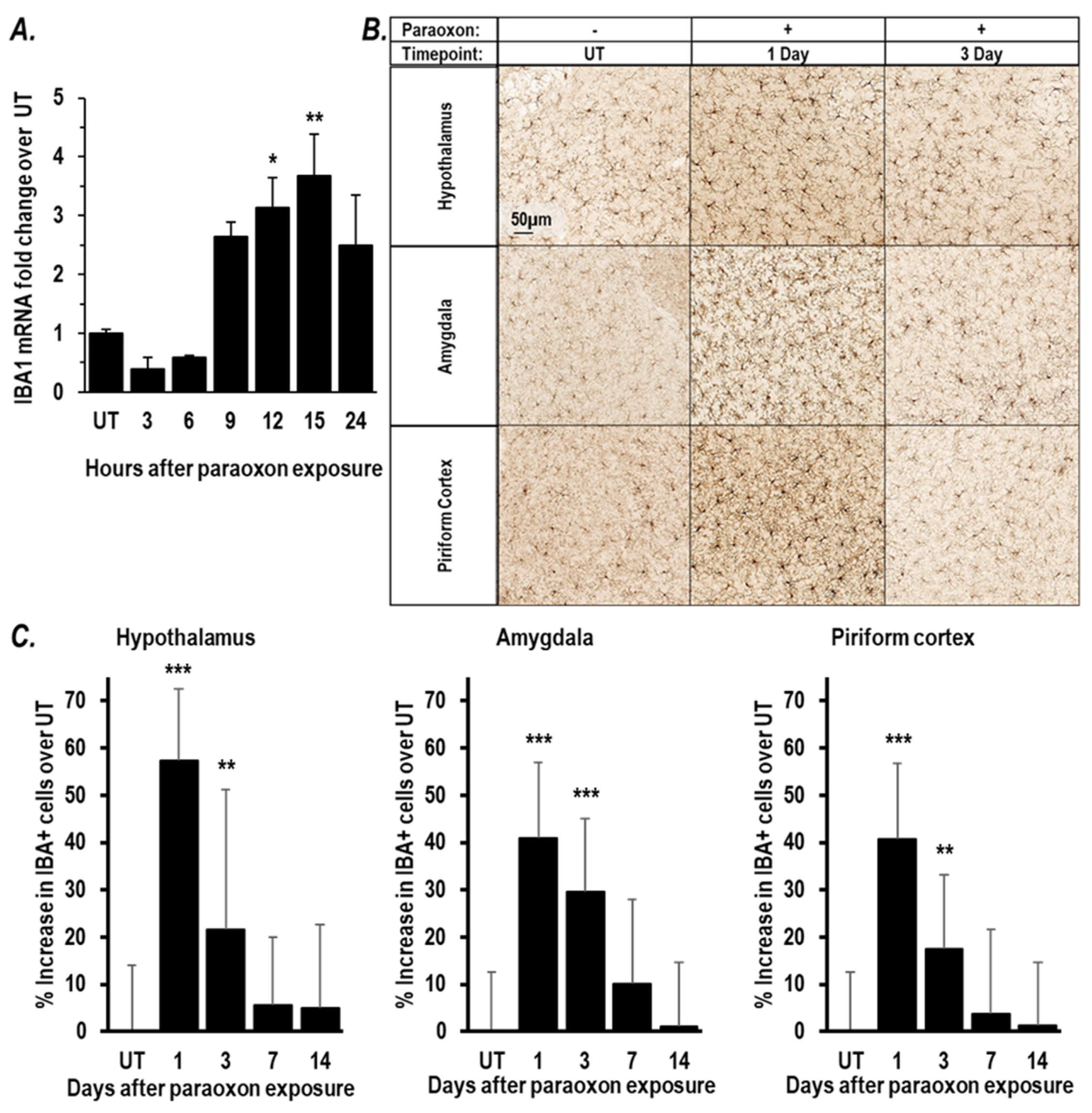
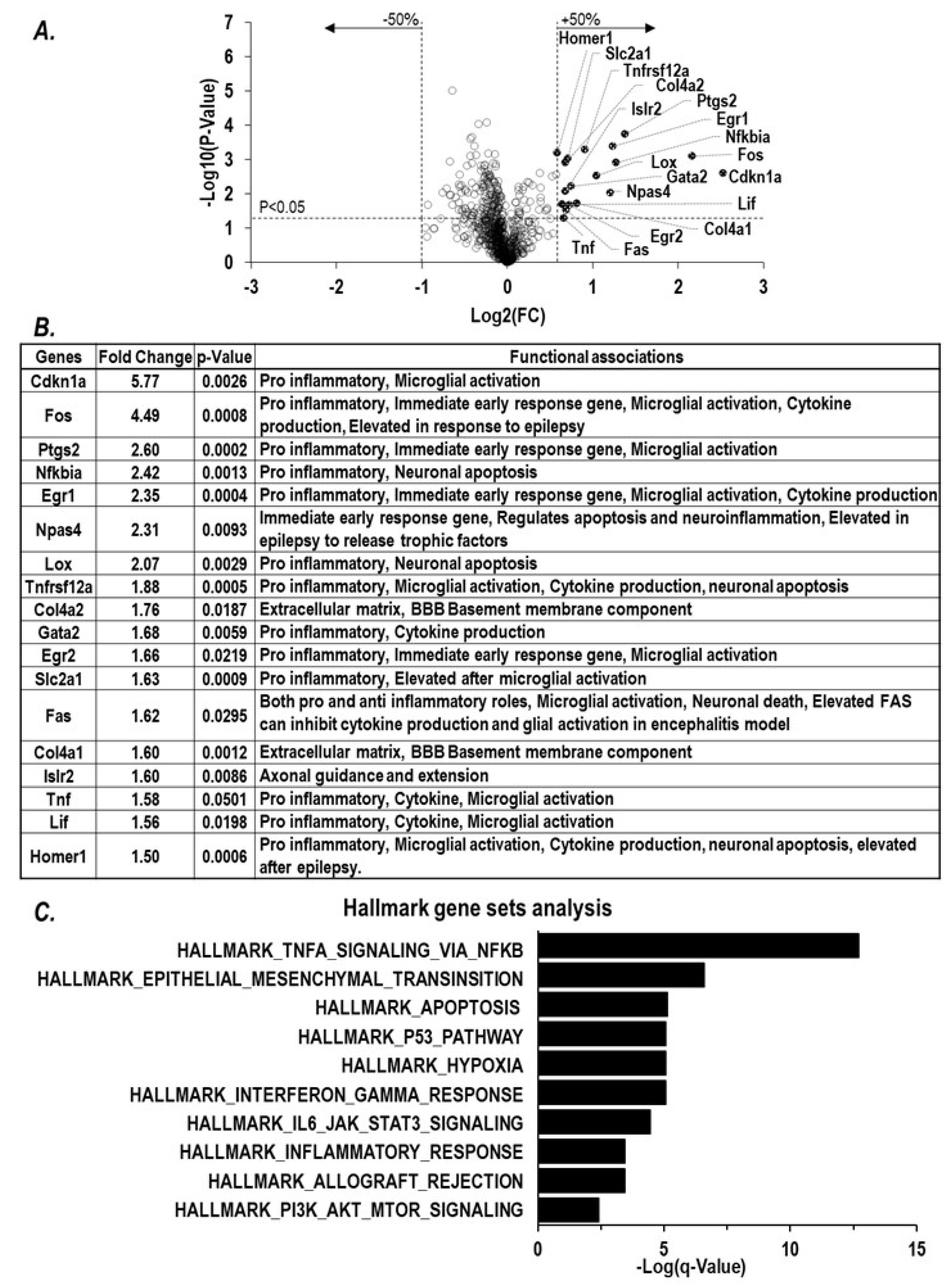
2.1. Paraoxon Exposure Induces Inflammation in the Brain
2.2. Inflammatory Chemokine CCL2 Level Corelates with Extent of Cholinergic Symptoms
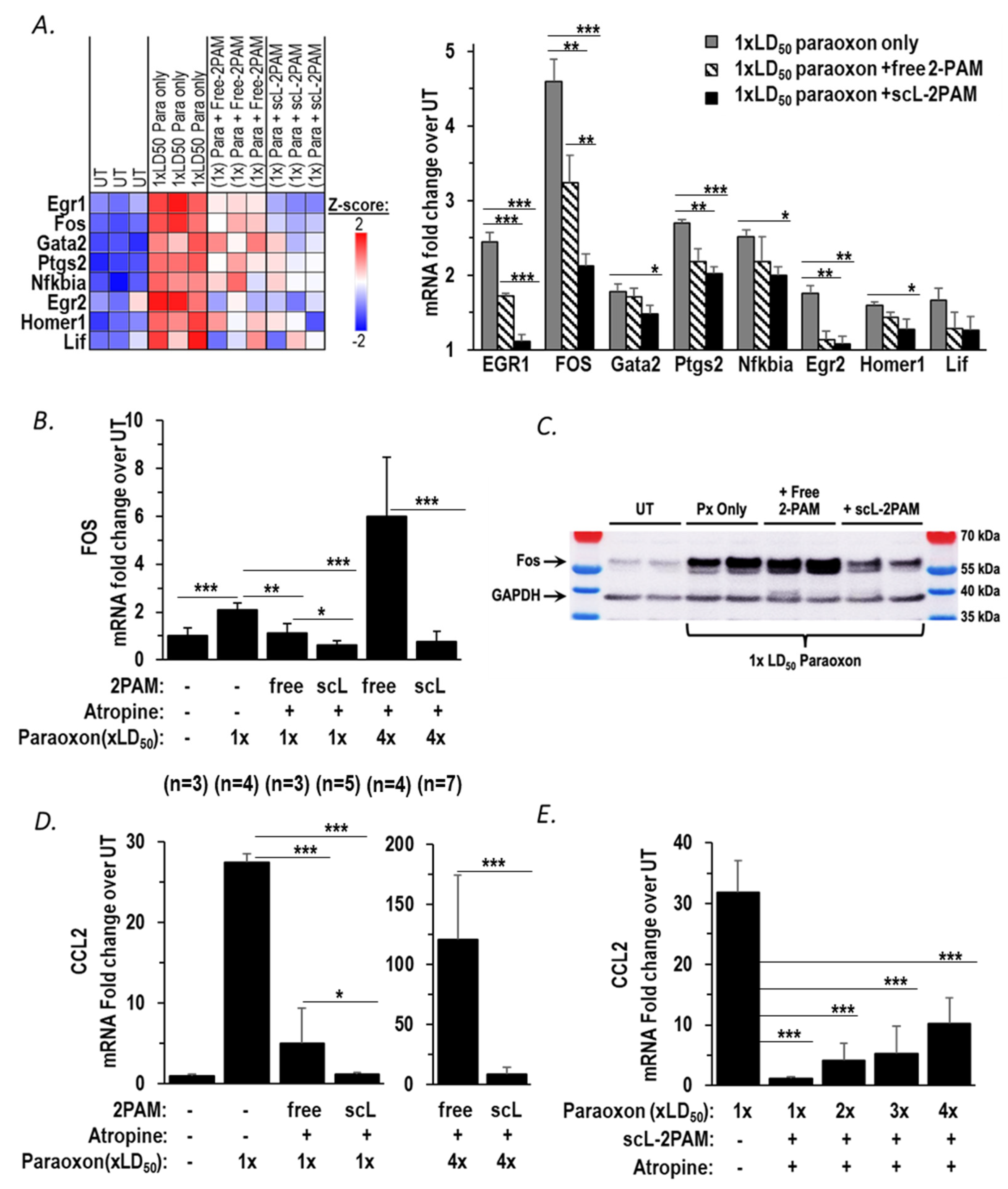
2.3. scL-2PAM Is Very Effective in Reducing Neuroinflammation in the Brain
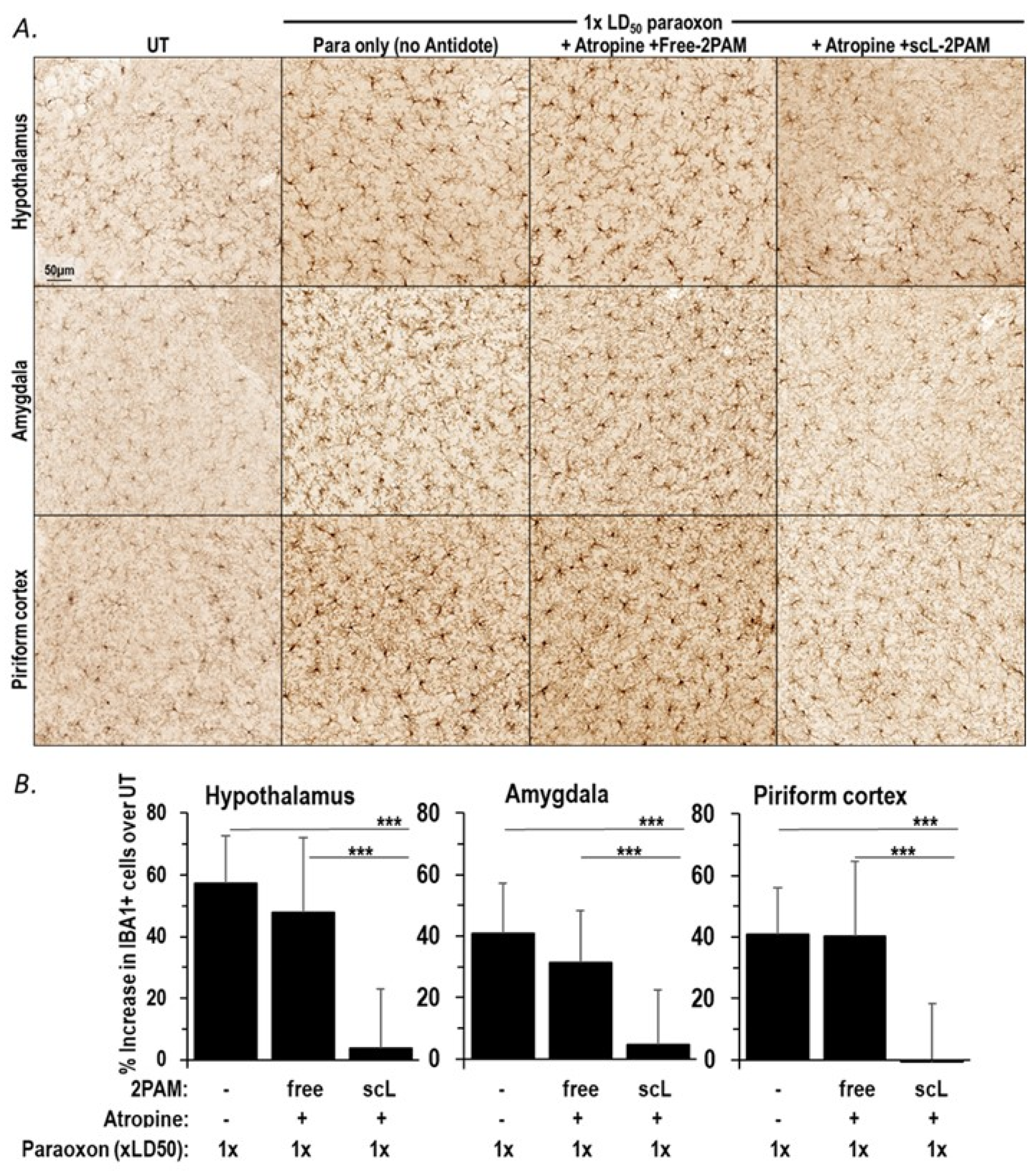
2.4. scL-2PAM Treatment Curbs the Paraoxon-Driven Microglial Activation
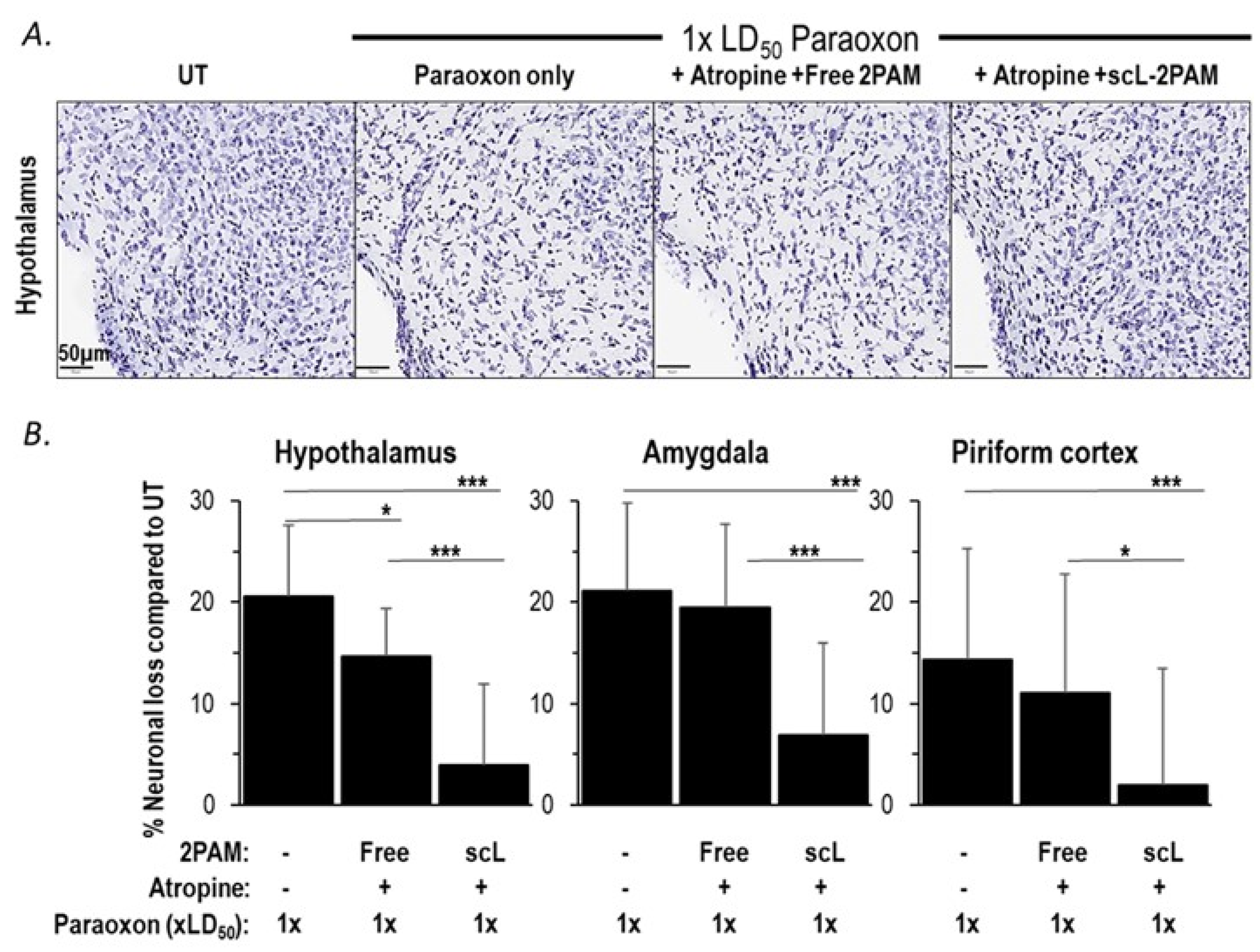
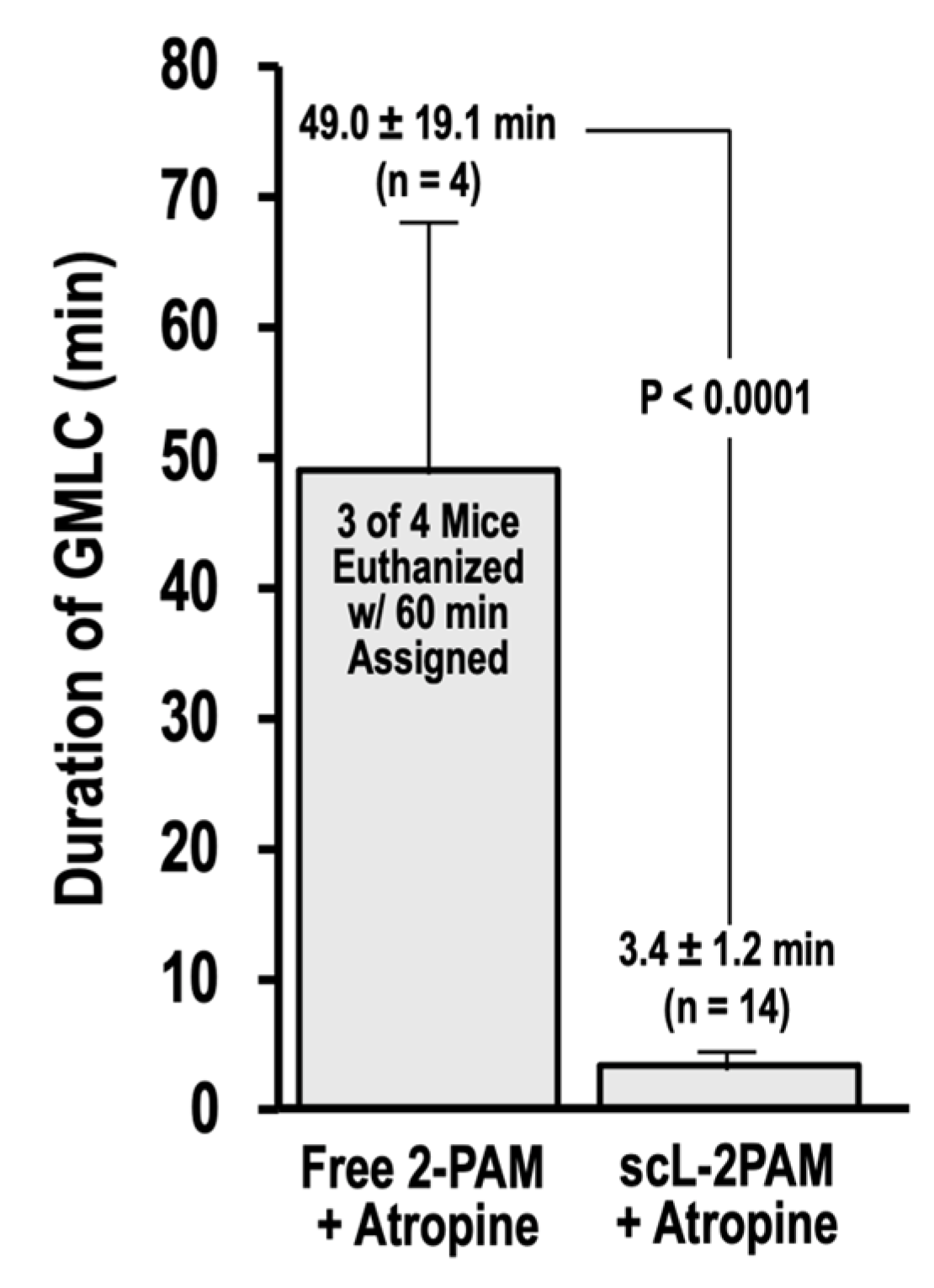
2.5. scL-2PAM Treatment Ameliorates Brain Damage after Paraoxon Exposure
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Paraoxon Preparation
4.3. Preparation of scL Nanocomplex Encapsulating 2-PAM
4.4. Animal Studies
4.5. Assessment of Cholinergic Crisis with Racine Scale
4.6. Tissue Collection for Gene Expression Analyses
4.7. Quantitative RT-PCR
4.8. NanoString nCounter Gene Expression Analysis
4.9. CCL2 Protein ELISA
4.10. Western Blot
4.11. Histology
4.12. Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, Y.; Lein, P.J.; Liu, C.; Bruun, D.A.; Tewolde, T.; Ford, G.; Ford, B.D. Spatiotemporal pattern of neuronal injury induced by DFP in rats: A model for delayed neuronal cell death following acute OP intoxication. Toxicol. Appl. Pharmacol. 2011, 253, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Kuruba, R.; Wu, X.; Reddy, D.S. Benzodiazepine-refractory status epilepticus, neuroinflammation, and interneuron neurodegeneration after acute organophosphate intoxication. Biochim. Biophys. Acta-Mol. Basis Dis. 2018, 1864, 2845–2858. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Bohnert, S.; Bouchard, M.; Vair, C.; Farrell, J.S.; Teskey, G.C.; Mikler, J.; Dunn, J.F. Quantitative T2 MRI is predictive of neurodegeneration following organophosphate exposure in a rat model. Sci. Rep. 2020, 10, 13007. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.D.; Wu, X.; Kuruba, R.; Sridhar, V.; Reddy, D.S. Magnetic resonance imaging analysis of long-term neuropathology after exposure to the nerve agent soman: Correlation with histopathology and neurological dysfunction. Ann. N. Y. Acad. Sci. 2020, 1480, 116–135. [Google Scholar] [CrossRef] [PubMed]
- Jett, D.A.; Spriggs, S.M. Translational research on chemical nerve agents. Neurobiol. Dis. 2020, 133, 104335. [Google Scholar] [CrossRef]
- Calsbeek, J.J.; González, E.A.; Bruun, D.A.; Guignet, M.A.; Copping, N.; Dawson, M.E.; Yu, A.J.; MacMahon, J.A.; Saito, N.H.; Harvey, D.J.; et al. Persistent neuropathology and behavioral deficits in a mouse model of status epilepticus induced by acute intoxication with diisopropylfluorophosphate. NeuroToxicology 2021, 87, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Cavalcante, S.F.d.A.; Simas, A.B.C.; Barcellos, M.C.; de Oliveira, V.G.M.; Sousa, R.B.; Cabral, P.A.d.M.; Kuča, K.; França, T.C.C. Acetylcholinesterase: The “Hub” for Neurodegenerative Diseases and Chemical Weapons Convention. Biomolecules 2020, 10, 414. [Google Scholar] [CrossRef] [PubMed]
- Dolgin, E. Syrian gas attack reinforces need for better anti-sarin drugs. Nat. Med. 2013, 19, 1194–1195. [Google Scholar] [CrossRef]
- Eddleston, M. Novel Clinical Toxicology and Pharmacology of Organophosphorus Insecticide Self-Poisoning. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 341–360. [Google Scholar] [CrossRef]
- Murata, K.; Araki, S.; Yokoyama, K.; Okumura, T.; Ishimatsu, S.; Takasu, N.; White, R.F. Asymptomatic sequelae to acute sarin poisoning in the central and autonomic nervous system 6 months after the Tokyo subway attack. J. Neurol. 1997, 244, 601–606. [Google Scholar] [CrossRef]
- Nishiwaki, Y.; Maekawa, K.; Ogawa, Y.; Asukai, N.; Minami, M.; Omae, K.; Yamaguchi, T.; Hirata, M.; Takahashi, M.; Katsumata, M.; et al. Effects of sarin on the nervous system in rescue team staff members and police officers 3 years after the Tokyo subway sarin attack. Environ. Health Perspect. 2001, 109, 1169–1173. [Google Scholar] [CrossRef] [PubMed]
- Loh, Y.; Swanberg, M.M.; Ingram, M.V.; Newmark, J. Case report: Long-term cognitive sequelae of sarin exposure. NeuroToxicology 2010, 31, 244–246. [Google Scholar] [CrossRef]
- Pereira, E.F.R.; Aracava, Y.; DeTolla, L.J.; Beecham, E.J.; Basinger, G.W.; Wakayama, E.J.; Albuquerque, E.X. Animal models that best reproduce the clinical manifestations of human intoxication with organophosphorus compounds. J. Pharmacol. Exp. Ther. 2014, 350, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Yamasue, H.; Abe, O.; Kasai, K.; Suga, M.; Iwanami, A.; Yamada, H.; Tochigi, M.; Ohtani, T.; Rogers, M.A.; Sasaki, T.; et al. Human brain structural change related to acute single exposure to sarin. Ann. Neurol. 2007, 61, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Heaton, K.J.; Palumbo, C.L.; Proctor, S.P.; Killiany, R.J.; Yurgelun-Todd, D.A.; White, R.F. Quantitative magnetic resonance brain imaging in US army veterans of the 1991 Gulf War potentially exposed to sarin and cyclosarin. NeuroToxicology 2007, 28, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Terry, A.V. Functional consequences of repeated organophosphate exposure: Potential non-cholinergic mechanisms. Pharmacol. Ther. 2012, 134, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, A.; Kunis, G.; Berkutzki, T.; Ronen, A.; Krivoy, A.; Yoles, E.; Last, D.; Mardor, Y.; Van Shura, K.; McFarland, E.; et al. Immunomodulation by poly-YE reduces organophosphate-induced brain damage. Brain Behav. Immun. 2012, 26, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Pearson-Smith, J.N.; Liang, L.P.; Rowley, S.D.; Day, B.J.; Patel, M. Oxidative Stress Contributes to Status Epilepticus Associated Mortality. Neurochem. Res. 2017, 42, 2024–2032. [Google Scholar] [CrossRef]
- Liang, L.P.; Pearson-Smith, J.N.; Huang, J.; McElroy, P.; Day, B.J.; Patel, M. Neuroprotective effects of AEOL10150 in a rat organophosphate model. Toxicol. Sci. 2018, 162, 611–621. [Google Scholar] [CrossRef]
- Putra, M.; Sharma, S.; Gage, M.; Gasser, G.; Hinojo-Perez, A.; Olson, A.; Gregory-Flores, A.; Puttachary, S.; Wang, C.; Anantharam, V.; et al. Inducible nitric oxide synthase inhibitor, 1400W, mitigates DFP-induced long-term neurotoxicity in the rat model. Neurobiol. Dis. 2020, 133, 104443. [Google Scholar] [CrossRef] [PubMed]
- Rojas, A.; Ganesh, T.; Wang, W.; Wang, J.; Dingledine, R. A rat model of organophosphate-induced status epilepticus and the beneficial effects of EP2 receptor inhibition. Neurobiol. Dis. 2020, 133, 104399. [Google Scholar] [CrossRef]
- Prager, E.M.; Aroniadou-Anderjaska, V.; Almeida-Suhett, C.P.; Figueiredo, T.H.; Apland, J.P.; Braga, M.F.M. Acetylcholinesterase inhibition in the basolateral amygdala plays a key role in the induction of status epilepticus after soman exposure. NeuroToxicology 2013, 38, 84–90. [Google Scholar] [CrossRef]
- Aroniadou-Anderjaska, V.; Figueiredo, T.H.; de Araujo Furtado, M.; Pidoplichko, V.I.; Braga, M.F.M. Mechanisms of Organophosphate Toxicity and the Role of Acetylcholinesterase Inhibition. Toxics 2023, 11, 866. [Google Scholar] [CrossRef] [PubMed]
- Andrew, P.M.; Lein, P.J. Neuroinflammation as a Therapeutic Target for Mitigating the Long-Term Consequences of Acute Organophosphate Intoxication. Front. Pharmacol. 2021, 12, 674325. [Google Scholar] [CrossRef] [PubMed]
- Konsman, J. Cytokines in the Brain and Neuroinflammation: We Didn’t Starve the Fire! Pharmaceuticals 2022, 15, 140. [Google Scholar] [CrossRef] [PubMed]
- Rojas, A.; Ganesh, T.; Lelutiu, N.; Gueorguieva, P.; Dingledine, R. Inhibition of the prostaglandin EP2 receptor is neuroprotective and accelerates functional recovery in a rat model of organophosphorus induced status epilepticus. Neuropharmacology 2015, 93, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Sisó, S.; Hobson, B.A.; Harvey, D.J.; Bruun, D.A.; Rowland, D.J.; Garbow, J.R.; Lein, P.J. Spatiotemporal progression and remission of lesions in the rat brain following acute intoxication with diisopropylfluorophosphate. Toxicol. Sci. 2017, 157, 330–341. [Google Scholar] [CrossRef]
- Hobson, B.A.; Rowland, D.J.; Sisó, S.; Guignet, M.A.; Harmany, Z.T.; Bandara, S.B.; Saito, N.; Harvey, D.J.; Bruun, D.A.; Garbow, J.R.; et al. TSPO PET Using [18F]PBR111 Reveals Persistent Neuroinflammation Following Acute Diisopropylfluorophosphate Intoxication in the Rat. Toxicol. Sci. 2019, 170, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Naughton, S.X.; Terry, A.V. Neurotoxicity in acute and repeated organophosphate exposure. Toxicology 2018, 408, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Guignet, M.; Lein, P.J. Neuroinflammation in organophosphate-induced neurotoxicity. In Advances in Neurotoxicology; Academic Press: Cambridge, MA, USA, 2019; Volume 3, pp. 35–79. [Google Scholar]
- Rojas, A.; Ganesh, T.; Manji, Z.; O’Neill, T.; Dingledine, R. Inhibition of the prostaglandin E2 receptor EP2 prevents status epilepticus-induced deficits in the novel object recognition task in rats. Neuropharmacology 2016, 110, 419–430. [Google Scholar] [CrossRef]
- Eddleston, M.; Buckley, N.A.; Eyer, P.; Dawson, A.H. Management of acute organophosphorus pesticide poisoning. Lancet 2008, 371, 597–607. [Google Scholar] [CrossRef]
- Deshpande, L.S.; DeLorenzo, R.J. Novel therapeutics for treating organophosphate-induced status epilepticus co-morbidities, based on changes in calcium homeostasis. Neurobiol. Dis. 2020, 133, 104418. [Google Scholar] [CrossRef] [PubMed]
- Jokanović, M. Pyridinium Oximes in the Treatment of Poisoning with Organophosphorus Compounds; Elsevier: Amsterdam, The Netherlands, 2015; pp. 1057–1070. [Google Scholar]
- Sakurada, K.; Ohta, H. No promising antidote 25 years after the Tokyo subway sarin attack: A review. Leg. Med. 2020, 47, 101761. [Google Scholar] [CrossRef] [PubMed]
- Pirollo, K.; Moghe, M.; Guan, M.; Rait, A.; Wang, A.; Kim, S.-S.; Chang, E.; Harford, J. A Pralidoxime Nanocomplex Formulation Targeting Transferrin Receptors for Reactivation of Brain Acetylcholinesterase After Exposure of Mice to an Anticholinesterase Organophosphate. Int. J. Nanomed. 2024, 19, 307–326. [Google Scholar] [CrossRef]
- Shrot, S.; Ramaty, E.; Biala, Y.; Bar-Klein, G.; Daninos, M.; Kamintsky, L.; Makarovsky, I.; Statlender, L.; Rosman, Y.; Krivoy, A.; et al. Prevention of organophosphate-induced chronic epilepsy by early benzodiazepine treatment. Toxicology 2014, 323, 19–25. [Google Scholar] [CrossRef]
- Iha, H.A.; Kunisawa, N.; Shimizu, S.; Onishi, M.; Nomura, Y.; Matsubara, N.; Iwai, C.; Ogawa, M.; Hashimura, M.; Sato, K.; et al. Mechanism Underlying Organophosphate Paraoxon-Induced Kinetic Tremor. Neurotox. Res. 2019, 35, 575–583. [Google Scholar] [CrossRef]
- Ito, D.; Imai, Y.; Ohsawa, K.; Nakajima, K.; Fukuuchi, Y.; Kohsaka, S. Microglia-specific localisation of a novel calcium binding protein, Iba1. Mol. Brain Res. 1998, 57, 1–9. [Google Scholar] [CrossRef]
- Flannery, B.M.; Bruun, D.A.; Rowland, D.J.; Banks, C.N.; Austin, A.T.; Kukis, D.L.; Li, Y.; Ford, B.D.; Tancredi, D.J.; Silverman, J.L.; et al. Persistent neuroinflammation and cognitive impairment in a rat model of acute diisopropylfluorophosphate intoxication. J. Neuroinflamm. 2016, 13, 267. [Google Scholar] [CrossRef] [PubMed]
- Parakalan, R.; Jiang, B.; Nimmi, B.; Janani, M.; Jayapal, M.; Lu, J.; Tay, S.S.W.; Ling, E.-A.; Dheen, S.T. Transcriptome analysis of amoeboid and ramified microglia isolated from the corpus callosum of rat brain. BMC Neurosci. 2012, 13, 64. [Google Scholar] [CrossRef]
- Vidal-Itriago, A.; Radford, R.A.W.; Aramideh, J.A.; Maurel, C.; Scherer, N.M.; Don, E.K.; Lee, A.; Chung, R.S.; Graeber, M.B.; Morsch, M. Microglia morphophysiological diversity and its implications for the CNS. Front. Immunol. 2022, 13, 997786. [Google Scholar] [CrossRef] [PubMed]
- Ring, R.H.; Valo, Z.; Gao, C.; Barish, M.E.; Singer-Sam, J. The Cdkn1a gene (p21Waf1/Cip1) is an inflammatory response gene in the mouse central nervous system. Neurosci. Lett. 2003, 350, 73–76. [Google Scholar] [CrossRef]
- Zonis, S.; Ljubimov, V.A.; Mahgerefteh, M.; Pechnick, R.N.; Wawrowsky, K.; Chesnokova, V. p21 Cip restrains hippocampal neurogenesis and protects neuronal progenitors from apoptosis during acute systemic inflammation. Hippocampus 2013, 23, 1383–1394. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, D.; McQuade, J.S.; Behbehani, M.; Tsien, J.Z.; Xu, M. C-Fos Regulates Neuronal Excitability and Survival. Nat. Genet. 2002, 30, 416–420. [Google Scholar] [CrossRef] [PubMed]
- Lara Aparicio, S.Y.; Laureani Fierro, Á.d.J.; Aranda Abreu, G.E.; Toledo Cárdenas, R.; García Hernández, L.I.; Coria Ávila, G.A.; Rojas Durán, F.; Aguilar, M.E.H.; Manzo Denes, J.; Chi-Castañeda, L.D.; et al. Current Opinion on the Use of c-Fos in Neuroscience. NeuroSci 2022, 3, 687–702. [Google Scholar] [CrossRef]
- Wei, C.J.; Li, Y.L.; Zhu, Z.L.; Jia, D.M.; Fan, M.L.; Li, T.; Wang, X.J.; Li, Z.G.; Ma, H.S. Inhibition of activator protein 1 attenuates neuroinflammation and brain injury after experimental intracerebral hemorrhage. CNS Neurosci. Ther. 2019, 25, 1182–1188. [Google Scholar] [CrossRef]
- Lu, Y.; Li, B.; Xu, A.; Liang, X.; Xu, T.; Jin, H.; Xie, Y.; Wang, R.; Liu, X.; Gao, X.; et al. NF-κB and AP-1 are required for the lipopolysaccharide-induced expression of MCP-1, CXCL1, and Cx43 in cultured rat dorsal spinal cord astrocytes. Front. Mol. Neurosci. 2022, 15, 859558. [Google Scholar] [CrossRef] [PubMed]
- Tureyen, K.; Brooks, N.; Bowen, K.; Svaren, J.; Vemuganti, R. Transcription factor early growth response-1 induction mediates inflammatory gene expression and brain damage following transient focal ischemia. J. Neurochem. 2008, 105, 1313–1324. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Huang, Q.; Du, X.; Xu, S.; Li, M.; Ma, S. Early activation of Egr-1 promotes neuroinflammation and dopaminergic neurodegeneration in an experimental model of Parkinson’s disease. Exp. Neurol. 2018, 302, 145–154. [Google Scholar] [CrossRef]
- Yan, Y.; Tan, X.; Wu, X.; Shao, B.; Wu, X.; Cao, J.; Xu, J.; Jin, W.; Li, L.; Xu, W.; et al. Involvement of early growth response-2 (Egr-2) in lipopolysaccharide-induced neuroinflammation. J. Mol. Histol. 2013, 44, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Choy, F.C.; Klarić, T.S.; Koblar, S.A.; Lewis, M.D. The role of the neuroprotective factor Npas4 in cerebral ischemia. Int. J. Mol. Sci. 2015, 16, 29011–29028. [Google Scholar] [CrossRef] [PubMed]
- O’Callaghan, J.P.; Kelly, K.A.; Locker, A.R.; Miller, D.B.; Lasley, S.M. Corticosterone primes the neuroinflammatory response to DFP in mice: Potential animal model of Gulf War Illness. J. Neurochem. 2015, 133, 708–721. [Google Scholar] [CrossRef]
- Michalovicz, L.T.; Locker, A.R.; Kelly, K.A.; Miller, J.V.; Barnes, Z.; Fletcher, M.A.; Miller, D.B.; Klimas, N.G.; Morris, M.; Lasley, S.M.; et al. Corticosterone and pyridostigmine/DEET exposure attenuate peripheral cytokine expression: Supporting a dominant role for neuroinflammation in a mouse model of Gulf War Illness. NeuroToxicology 2019, 70, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Verna, L.; Hardy, S.; Forsayeth, J.; Zhu, Y.; Stemerman, M.B. Adenovirus-mediated overexpression of c-Jun and c-Fos induces intercellular adhesion molecule-1 and monocyte chemoattractant protein-1 in human endothelial cells. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2078–2084. [Google Scholar] [CrossRef]
- Wang, H.N.; Ji, K.; Zhang, L.N.; Xie, C.C.; Li, W.Y.; Zhao, Z.F.; Chen, J.J. Inhibition of c-Fos expression attenuates IgE-mediated mast cell activation and allergic inflammation by counteracting an inhibitory AP1/Egr1/IL-4 axis. J. Transl. Med. 2021, 19, 261. [Google Scholar] [CrossRef] [PubMed]
- Gorbacheva, A.M.; Uvarova, A.N.; Ustiugova, A.S.; Bhattacharyya, A.; Korneev, K.V.; Kuprash, D.V.; Mitkin, N.A. EGR1 and RXRA transcription factors link TGF-β pathway and CCL2 expression in triple negative breast cancer cells. Sci. Rep. 2021, 11, 14120. [Google Scholar] [CrossRef]
- Trizzino, M.; Zucco, A.; Deliard, S.; Wang, F.; Barbieri, E.; Veglia, F.; Gabrilovich, D.; Gardini, A. EGR1 is a gatekeeper of inflammatory enhancers in human macrophages. Sci. Adv. 2021, 7, eaaz8836. [Google Scholar] [CrossRef] [PubMed]
- Piovesana, R.; Salazar Intriago, M.S.; Dini, L.; Tata, A.M. Cholinergic Modulation of Neuroinflammation: Focus on α7 Nicotinic Receptor. Int. J. Mol. Sci. 2021, 22, 4912. [Google Scholar] [CrossRef]
- RamaRao, G.; Afley, P.; Acharya, J.; Bhattacharya, B.K. Efficacy of antidotes (midazolam, atropine and HI-6) on nerve agent induced molecular and neuropathological changes. BMC Neurosci. 2014, 15, 47. [Google Scholar] [CrossRef]
- Kruijer, W.; Cooper, J.A.; Hunter, T.; Verma, I.M. Platelet-derived growth factor induces rapid but transient expression of the c-fos gene and protein. Nature 1984, 312, 711–716. [Google Scholar] [CrossRef]
- Zare, Z.; Zarbakhsh, S.; Mashhadban, S.; Moradgholi, A.; Mohammadi, M. Apoptosis is involved in paraoxon-induced histological changes in rat cerebellum. Drug Chem. Toxicol. 2022, 45, 2554–2560. [Google Scholar] [CrossRef]
- Figueiredo, T.H.; Apland, J.P.; Braga, M.F.M.; Marini, A.M. Acute and long-term consequences of exposure to organophosphate nerve agents in humans. Epilepsia 2018, 59, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Trinka, E.; Cock, H.; Hesdorffer, D.; Rossetti, A.O.; Scheffer, I.E.; Shinnar, S.; Shorvon, S.; Lowenstein, D.H. A definition and classification of status epilepticus—Report of the ILAE Task Force on Classification of Status Epilepticus. Epilepsia 2015, 56, 1515–1523. [Google Scholar] [CrossRef] [PubMed]
- McDonough, J.H.; Shih, T.-M. Neuropharmacological Mechanisms of Nerve Agent-induced Seizure and Neuropathology. Neurosci. Biobehav. Rev. 1997, 21, 559–579. [Google Scholar] [CrossRef] [PubMed]
- McDonough, J.H., Jr.; McMonagle, J.; Copeland, T.; Zoeffel, D.; Shih, T.-M. Comparative evaluation of benzodiazepines for control of soman-induced seizures. Arch. Toxicol. 1999, 73, 473–478. [Google Scholar] [CrossRef]
- Shih, T.-M.; Duniho, S.M.; McDonough, J.H. Control of nerve agent-induced seizures is critical for neuroprotection and survival. Toxicol. Appl. Pharmacol. 2003, 188, 69–80. [Google Scholar] [CrossRef] [PubMed]
- De Araujo Furtado, M.; Rossetti, F.; Chanda, S.; Yourick, D. Exposure to nerve agents: From status epilepticus to neuroinflammation, brain damage, neurogenesis and epilepsy. NeuroToxicology 2012, 33, 1476–1490. [Google Scholar] [CrossRef] [PubMed]
- Raveh, L.; Eisenkraft, A.; Weissman, B.A. Caramiphen edisylate: An optimal antidote against organophosphate poisoning. Toxicology 2014, 325, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Colovic, M.B.; Krstic, D.Z.; Lazarevic-Pasti, T.D.; Bondzic, A.M.; Vasic, V.M. Acetylcholinesterase Inhibitors: Pharmacology and Toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef] [PubMed]
- Carey, J.L.; Dunn, C.; Gaspari, R.J. Central respiratory failure during acute organophosphate poisoning. Respir. Physiol. Neurobiol. 2013, 189, 403–410. [Google Scholar] [CrossRef]
- Sil, S.; Ghosh, T. Role of cox-2 mediated neuroinflammation on the neurodegeneration and cognitive impairments in colchicine induced rat model of Alzheimer’s Disease. J. Neuroimmunol. 2016, 291, 115–124. [Google Scholar] [CrossRef]
- Rogers, A.; Schmuck, G.; Scholz, G.; Williams, D.C. c-fos mRNA Expression in Rat Cortical Neurons During Glutamate-Mediated Excitotoxicity. Toxicol. Sci. 2004, 82, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Rojas, A.; Abreu-Melon, J.M.; Wang, S.; Dingledine, R. Time-dependent neuropathology in rats following organophosphate-induced status epilepticus. NeuroToxicology 2022, 91, 45–59. [Google Scholar] [CrossRef]
- Yang, J.; Bruun, D.A.; Wang, C.; Wan, D.; McReynolds, C.B.; Phu, K.; Inceoglu, B.; Lein, P.J.; Hammock, B.D. Lipidomes of brain from rats acutely intoxicated with diisopropylfluorophosphate identifies potential therapeutic targets. Toxicol. Appl. Pharmacol. 2019, 382, 114749. [Google Scholar] [CrossRef]
- Rosman, Y.; Eisenkraft, A.; Krivoy, A.; Schein, O.; Makarovski, I.; Shrot, S.; Ramaty, E.; Shilderman, E.B.; Kapon, J.; Gilat, E.; et al. Using MRI for the assessment of paraoxon-induced brain damage and efficacy of antidotal treatment. J. Appl. Toxicol. 2012, 32, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Zare, Z.; Zarbakhsh, S.; Tehrani, M.; Mohammadi, M. Paraoxon-induced damage in rat hippocampus is associated with alterations in the expression of apoptosis-related proteins. Pestic. Biochem. Physiol. 2020, 166, 104580. [Google Scholar] [CrossRef]
- Deshpande, L.S.; Carter, D.S.; Phillips, K.F.; Blair, R.E.; DeLorenzo, R.J. Development of status epilepticus, sustained calcium elevations and neuronal injury in a rat survival model of lethal paraoxon intoxication. NeuroToxicology 2014, 44, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Shrot, S.; Tauber, M.; Shiyovich, A.; Milk, N.; Rosman, Y.; Eisenkraft, A.; Kadar, T.; Kassirer, M.; Cohen, Y. Early brain magnetic resonance imaging can predict short and long-term outcomes after organophosphate poisoning in a rat model. NeuroToxicology 2015, 48, 206–216. [Google Scholar] [CrossRef]
- Farizatto, K.L.G.; Almeida, M.F.; Long, R.T.; Bahr, B.A. Early Synaptic Alterations and Selective Adhesion Signaling in Hippocampal Dendritic Zones Following Organophosphate Exposure. Sci. Rep. 2019, 9, 6532. [Google Scholar] [CrossRef]
- Dail, M.B.; Leach, C.A.; Meek, E.C.; Olivier, A.K.; Pringle, R.B.; Green, C.E.; Chambers, J.E. Novel brain-penetrating oxime acetylcholinesterase reactivators attenuate organophosphate-induced neuropathology in the rat hippocampus. Toxicol. Sci. 2019, 169, 465–474. [Google Scholar] [CrossRef]
- Rojas, A.; Wang, W.; Glover, A.; Manji, Z.; Fu, Y.; Dingledine, R. Beneficial outcome of urethane treatment following status epilepticus in a rat organophosphorus toxicity model. eNeuro 2018, 5. [Google Scholar] [CrossRef]
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef] [PubMed]
- López-Granero, C.; Cañadas, F.; Cardona, D.; Yu, Y.; Giménez, E.; Lozano, R.; Avila, D.S.; Aschner, M.; Sánchez-Santed, F. Chlorpyrifos-, diisopropylphosphorofluoridate-, and parathion-induced behavioral and oxidative stress effects: Are they mediated by analogous mechanisms of action? Toxicol. Sci. 2013, 131, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Ireland, D.; Zhang, S.; Bochenek, V.; Hsieh, J.-H.; Rabeler, C.; Meyer, Z.; Collins, E.-M.S. Differences in neurotoxic outcomes of organophosphorus pesticides revealed via multi-dimensional screening in adult and regenerating planarians. Front. Toxicol. 2022, 4, 948455. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moghe, M.; Kim, S.-S.; Guan, M.; Rait, A.; Pirollo, K.F.; Harford, J.B.; Chang, E.H. scL-2PAM: A Novel Countermeasure That Ameliorates Neuroinflammation and Neuronal Losses in Mice Exposed to an Anticholinesterase Organophosphate. Int. J. Mol. Sci. 2024, 25, 7539. https://doi.org/10.3390/ijms25147539
Moghe M, Kim S-S, Guan M, Rait A, Pirollo KF, Harford JB, Chang EH. scL-2PAM: A Novel Countermeasure That Ameliorates Neuroinflammation and Neuronal Losses in Mice Exposed to an Anticholinesterase Organophosphate. International Journal of Molecular Sciences. 2024; 25(14):7539. https://doi.org/10.3390/ijms25147539
Chicago/Turabian StyleMoghe, Manish, Sang-Soo Kim, Miaoyin Guan, Antonina Rait, Kathleen F. Pirollo, Joe B. Harford, and Esther H. Chang. 2024. "scL-2PAM: A Novel Countermeasure That Ameliorates Neuroinflammation and Neuronal Losses in Mice Exposed to an Anticholinesterase Organophosphate" International Journal of Molecular Sciences 25, no. 14: 7539. https://doi.org/10.3390/ijms25147539
APA StyleMoghe, M., Kim, S. -S., Guan, M., Rait, A., Pirollo, K. F., Harford, J. B., & Chang, E. H. (2024). scL-2PAM: A Novel Countermeasure That Ameliorates Neuroinflammation and Neuronal Losses in Mice Exposed to an Anticholinesterase Organophosphate. International Journal of Molecular Sciences, 25(14), 7539. https://doi.org/10.3390/ijms25147539