
The Role of Glia in Wilson’s Disease: Clinical, Neuroimaging, Neuropathological and Molecular Perspectives

 ,
,  ,
,  , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Wilson’s Disease—Neuropsychiatric Symptoms
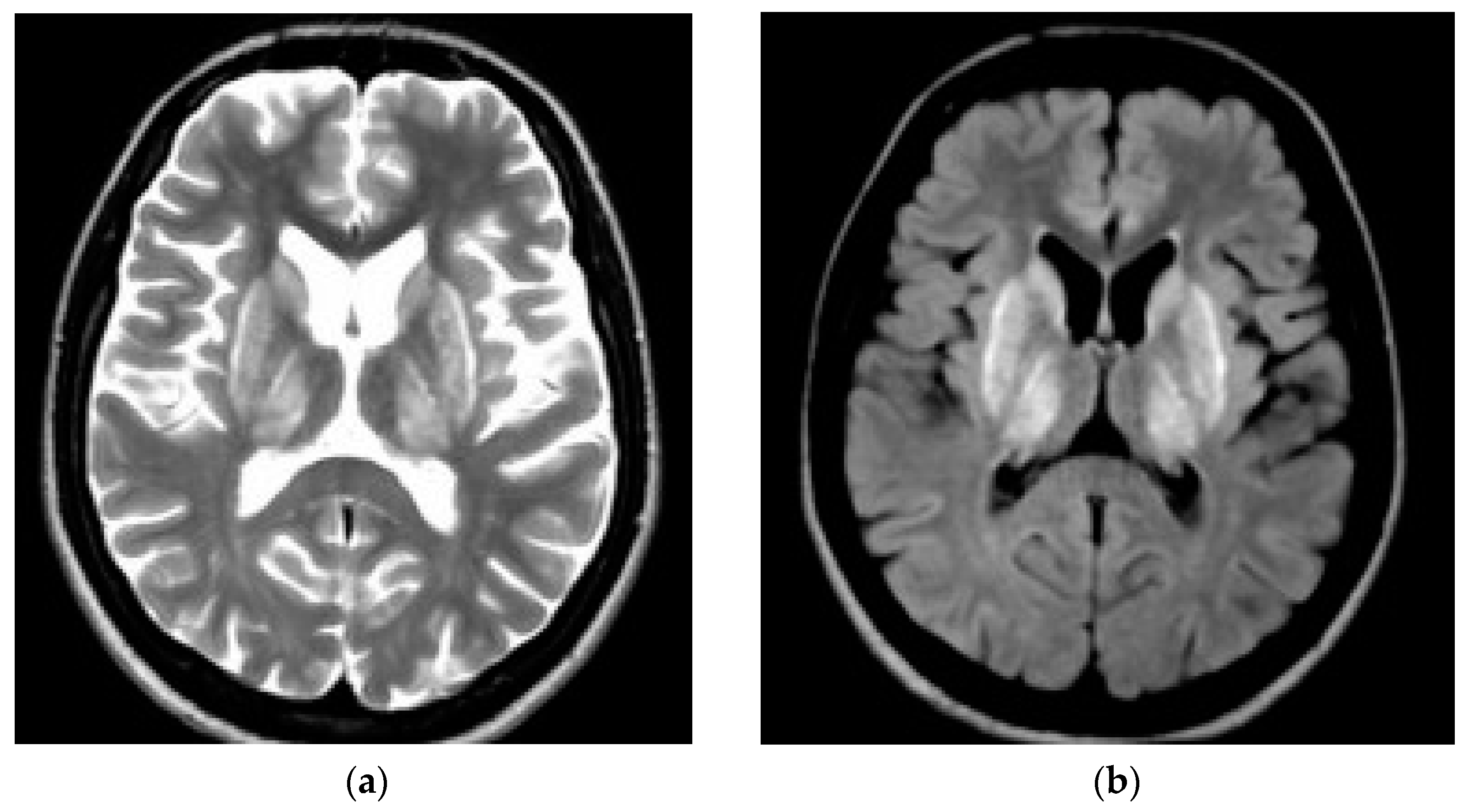
3. CNS Pathology in WD—Neuroimaging Studies
4. Glia in CNS Pathology in WD
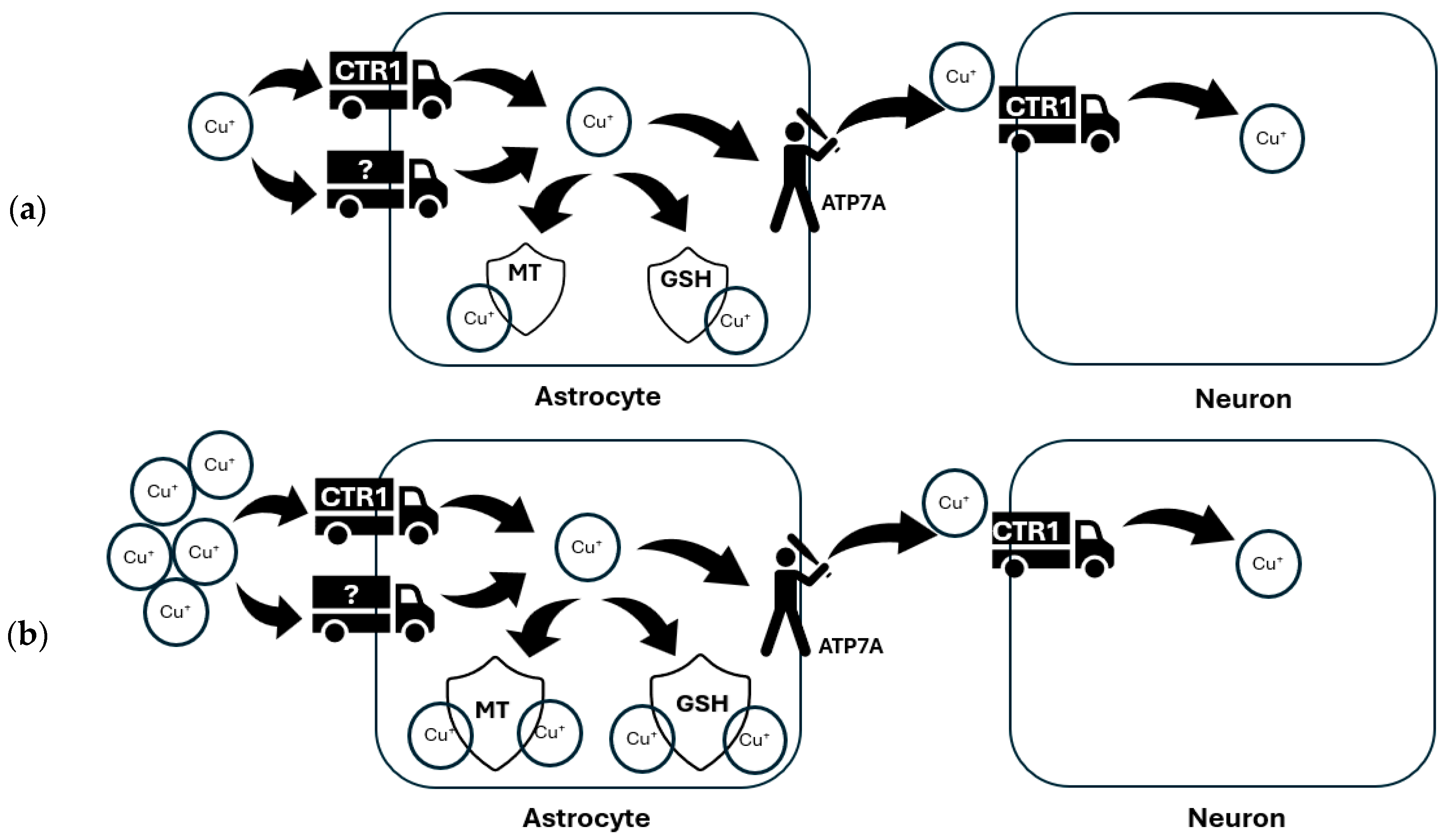
4.1. Glia in Brain Copper Homeostasis
4.2. Glia in Copper-Induced Neuronal Degeneration: Lesson from Alzheimer’s Disease
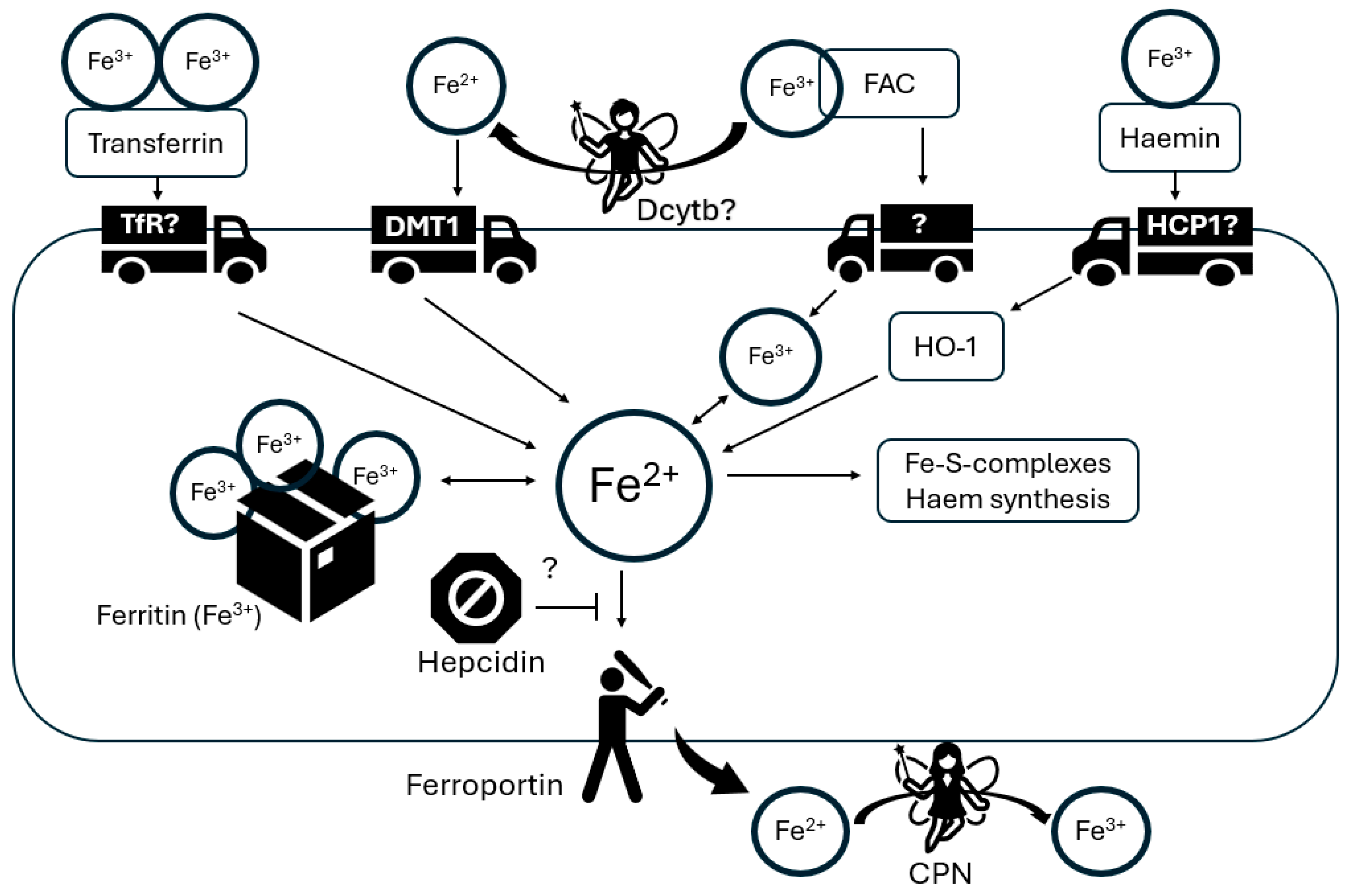
4.3. Glia in Brain Iron Homeostasis
4.4. The Lack of Functional Ceruloplasmin—Role in Neurodegeneration in WD
4.5. Glia in Iron-Induced Neuronal Degeneration: Lesson from Parkinson’s and Alzheimer’s Diseases
4.6. Glial Response and Neuronal Injury in WD—MRS Studies
4.7. Glial Structural Changes in WD—Neuropathological Studies
4.8. Biomarkers of Glia Injury in WD
4.9. Murine Models in Determining Glia Role in WD
5. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Walshe, J.M. History of Wilson’s disease: 1912 to 2000. Mov. Disord. 2006, 21, 142–147. [Google Scholar] [CrossRef]
- Wu, F.; Wang, J.; Pu, C.; Qiao, L.; Jiang, C. Wilson’s disease: A comprehensive review of the molecular mechanisms. Int. J. Mol. Sci. 2015, 16, 6419–6431. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R.; Eickhoff, A. Wilson Disease: Copper-Mediated Cuproptosis, Iron-Related Ferroptosis, and Clinical Highlights, with Comprehensive and Critical Analysis Update. Int. J. Mol. Sci. 2024, 25, 4753. [Google Scholar] [CrossRef] [PubMed]
- Petrukhin, K.; Lutsenko, S.; Chernov, I.; Ross, B.M.; Kaplan, J.H.; Gilliam, T.C. Characterization of the Wilson disease gene encoding a P-type copper transporting ATPase: Genomic organization, alternative splicing, and structure/function predictions. Hum. Mol. Genet. 1994, 3, 1647–1656. [Google Scholar] [CrossRef] [PubMed]
- Tanzi, R.E.; Petrukhin, K.; Chernov, I.; Pellequer, J.L.; Wasco, W.; Ross, B.; Romano, D.M.; Parano, E.; Pavone, L.; Brzustowicz, L.M.; et al. The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nat. Genet. 1993, 5, 344–350. [Google Scholar] [CrossRef]
- Gromadzka, G.; Bendykowska, M.; Przybyłkowski, A. Wilson’s Disease-Genetic Puzzles with Diagnostic Implications. Diagnostics 2023, 13, 1287. [Google Scholar] [CrossRef]
- Stremmel, W.; Weiskirchen, R. Therapeutic strategies in Wilson disease: Pathophysiology and mode of action. Ann. Transl. Med. 2021, 9, 732. [Google Scholar] [CrossRef]
- Roberts, E.A.; Schilsky, M.L. Current and Emerging Issues in Wilson’s Disease. N. Engl. J. Med. 2023, 389, 922–938. [Google Scholar] [CrossRef]
- Ban, X.X.; Wan, H.; Wan, X.X.; Tan, Y.T.; Hu, X.M.; Ban, H.X.; Chen, X.Y.; Huang, K.; Zhang, Q.; Xiong, K. Copper Metabolism and Cuproptosis: Molecular Mechanisms and Therapeutic Perspectives in Neurodegenerative Diseases. Curr. Med. Sci. 2024, 44, 28–50. [Google Scholar] [CrossRef]
- Bertrand, E.; Lewandowska, E.; Szpak, G.M.; Hoogenraad, T. Neuropathological analysis of pathological forms of astroglia in Wilson’s disease. Folia Neuropathol. 2001, 39, 73–79. [Google Scholar]
- Moura, J.; Pinto, C.; Freixo, P.; Alves, H.; Ramos, C.; Santos Silva, E.; Nery, F.; Gandara, J.; Lopes, V.; Ferreira, S.; et al. Correlation between neuroimaging, neurological phenotype, and functional outcomes in Wilson’s disease. Neurol. Sci. 2024, 45, 3201–3208. [Google Scholar] [CrossRef] [PubMed]
- Litwin, T.; Dusek, P.; Antos, A.; Członkowska, A.; Bembenek, J. Tackling the neurological manifestations in Wilson’s disease—Currently available treatment options. Expert Rev. Neurother. 2023, 23, 1249–1259. [Google Scholar] [CrossRef] [PubMed]
- Wungjiranirun, M.; Sharzehi, K. Wilson’s Disease. Semin. Neurol. 2023, 43, 626–633. [Google Scholar] [CrossRef]
- Zimny, S.; Bourhis, H.; Weber, S.; Reiter, F.P.; Hohenester, S.; Kraft, E.; Mohr, I.; Merle, U.; Weiss, K.H.; Denk, G. Medical care of patients with Wilson disease in Germany: A multidisciplinary survey among university centers. Orphanet J. Rare Dis. 2023, 18, 122. [Google Scholar] [CrossRef] [PubMed]
- Członkowska, A.; Rodo, M.; Gromadzka, G. Late onset Wilson’s disease: Therapeutic implications. Mov. Disord. 2008, 23, 896–898. [Google Scholar] [CrossRef] [PubMed]
- Jopowicz, A.; Tarnacka, B. Neurological Wilson’s Disease Signs-Hepatic Encephalopathy or Copper Toxicosis? Diagnostics 2023, 13, 893. [Google Scholar] [CrossRef] [PubMed]
- Prayer, L.; Wimberger, D.; Kramer, J.; Grimm, G.; Oder, W.; Imhof, H. Cranial MRI in Wilson’s disease. Neuroradiology 1990, 32, 211–214. [Google Scholar] [CrossRef]
- Sinha, S.; Taly, A.B.; Ravishankar, S.; Prashanth, L.K.; Venugopal, K.S.; Arunodaya, G.R.; Vasudev, M.K.; Swamy, H.S. Wilson’s disease: Cranial MRI observations and clinical correlation. Neuroradiology 2006, 48, 613–621. [Google Scholar] [CrossRef]
- Kozic, D.; Svetel, M.; Petrovic, B.; Dragasević, N.; Semnic, R.; Kostić, V.S. MR imaging of the brain in patients with hepatic form of Wilson’s disease. Eur. J. Neurol. 2003, 10, 587–592. [Google Scholar] [CrossRef]
- Redzia-Ogrodnik, B.; Czlonkowska, A.; Bembenek, J.; Antos, A.; Kurkowska-Jastrzębska, I.; Skowrońska, M.; Smoliński, Ł.; Litwin, T. Brain magnetic resonance imaging and severity of neurological disease in Wilson’s disease—The neuroradiological correlations. Neurol. Sci. 2022, 43, 4405–4412. [Google Scholar] [CrossRef]
- Litwin, T.; Gromadzka, G.; Członkowska, A.; Gołębiowski, M.; Poniatowska, R. The effect of gender on brain MRI pathology in Wilson’s disease. Metab. Brain Dis. 2013, 28, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Magalhaes, A.C.; Caramelli, P.; Menezes, J.R.; Lo, L.S.; Bacheschi, L.A.; Barbosa, E.R.; Rosemberg, L.A.; Magalhaes, A. Wilson’s disease: MRI with clinical correlation. Neuroradiology 1994, 36, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Shribman, S.; Bocchetta, M.; Sudre, C.H.; Acosta-Cabronero, J.; Burrows, M.; Cook, P.; Thomas, D.L.; Gillett, G.T.; Tsochatzis, E.A.; Bandmann, O.; et al. Neuroimaging correlates of brain injury in Wilson’s disease: A multimodal, whole brain MRI study. Brain 2022, 145, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Ferenci, P.; Caca, K.; Loudianos, G.; Mieli-Vergani, G.; Tanner, S.; Sternlieb, I.; Schilsky, M.; Cox, D.; Berr, F. Diagnosis and phenotypic classification of Wilson disease. Liver Int. 2003, 23, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Dusek, P.; Smolinski, L.; Redzia-Ogrodnik, B.; Golebiowski, M.; Skowronska, M.; Poujois, A.; Laurencin, C.; Jastrzebska-Kurkowska, I.; Litwin, T.; Członkowska, A. Semiquantitative scale for assessing brain MRI abnormalities in Wilson disease: A validation study. Mov. Disord. 2020, 35, 994–1001. [Google Scholar] [CrossRef] [PubMed]
- Smoliński, Ł.; Ziemssen, T.; Akgun, K.; Antos, A.; Skowrońska, M.; Kurkowska-Jastrzębska, I.; Członkowska, A.; Litwin, T. Brain Atrophy Is Substantially Accelerated in Neurological Wilson’s Disease: A Longitudinal Study. Mov. Disord. 2022, 37, 2446–2451. [Google Scholar] [CrossRef]
- Dusek, P.; Lescinskij, A.; Ruzicka, F.; Acosta-Cabronero, J.; Bruha, R.; Sieger, T.; Hajek, M.; Dezortova, M. Asscociations of brain atrophy and cerebral iron accumulation at MRI with clinical severity in Wilson disease. Radiology 2021, 299, 662–672. [Google Scholar] [CrossRef]
- Dusek, P.; Bahn, E.; Litwin, T.; Jabłonka-Salach, K.; Łuciuk, A.; Huelnhagen, T.; Madai, V.I.; Dieringer, M.A.; Bulska, E.; Knauth, M.; et al. Brain iron accumulation in Wilson disease: A post-mortem 7 Tesla MRI—Histopathological study. Neuropathol. Appl. Neurobiol. 2017, 43, 514–532. [Google Scholar] [CrossRef] [PubMed]
- Rędzia-Ogrodnik, B.; Członkowska, A.; Antos, A.; Bembenek, J.; Kurkowska-Jastrzębska, I.; Przybyłkowski, A.; Skowrońska, M.; Smoliński, Ł.; Litwin, T. Pathognomonic neuroradiological signs in Wilson’s disease—Truth or myth? Park. Rel. Disord. 2023, 107, 105247. [Google Scholar] [CrossRef]
- Brewer, G.J. Risks of copper and iron toxicity during aging in humans. Chem. Res. Toxicol. 2010, 23, 319–326. [Google Scholar] [CrossRef]
- Gromadzka, G.; Wierzbicka, D.; Litwin, T.; Przybyłkowski, A. Difference in iron metabolism may partly explain sex-related variability in the manifestation of Wilson’s disease. J. Trace Elem. Med. Biol. 2020, 62, 126637. [Google Scholar] [CrossRef]
- Gromadzka, G.; Wierzbicka, D.; Litwin, T.; Przybyłkowski, A. Iron metabolism is disturbed and anti-copper treatment improves but does not normalize iron metabolism in Wilson’s disease. Biometals 2021, 34, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Litwin, T.; Gromadzka, G.; Członkowska, A. Wilson’s disease: Does iron metabolism impact phenotypic presentation? Liver Int. 2012, 32, 869–870. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Yano, M.; Fujita, Y.; Wakusawa, S. Compound overload of copper and iron in patients with Wilson’s disease. Med. Mol. Morphol. 2006, 39, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Shiono, Y.; Wakusawa, S.; Hayashi, H.; Takikawa, T.; Yano, M.; Okada, T.; Mabuchi, H.; Kono, S.; Miyajima, H. Iron accumulation in the liver of male patients with Wilson’s disease. Am. J. Gastroenterol. 2001, 96, 3147–3151. [Google Scholar] [CrossRef] [PubMed]
- Medici, V.; di Leo, V.; Lamboglia, F.; Bowlus, C.L.; Tseng, S.C.; D’Incà, R.; Irato, P.; Burra, P.; Martines, D.; Sturniolo, G.C. Effect of penicillamine and zinc on iron metabolism in Wilson’s disease. Scand. J. Gastroenterol. 2007, 42, 1495–1500. [Google Scholar] [CrossRef] [PubMed]
- Hafkemeyer, P.; Schupp, M.; Storch, M.; Gerok, W.; Haaussinger, D. Excessive iron storage in a patient with Wilson’s disease. Clin. Investig. 1994, 72, 134–136. [Google Scholar] [CrossRef]
- McNeill, A.; Birchall, D.; Hayflick, S.J.; Gregory, A.; Schenk, J.F.; Zimmerman, E.A.; Shang, H.; Miyajima, H.; Chinnery, P.F. T2* and FSE MRI distinguishes four subtypes of neurodegeneration with brain iron accumulation. Neurology 2008, 70, 1614–1619. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Bydder, G.M. Brain Atrophy Is a Better Biomarker than Susceptibility for Evaluating Clinical Severity in Wilson Disease. Radiology 2021, 299, 673–674. [Google Scholar] [CrossRef]
- Bruehleimer, M.; Leenders, K.L.; Vontobel, P.; Calonder, C.; Antonini, A.; Weindl, A. Increased cerebral iron uptake in Wilson’s disease: A 52Fe-citrate PET study. J. Nucl. Med. 2000, 41, 781–787. [Google Scholar]
- Litwin, T.; Karliński, M.; Skowrońska, M.; Dzieżyc, K.; Gołębiowski, M.; Członkowska, A. MR image mimicking the “eye of the tiger” sign in Wilson’s disease. J. Neurol. 2014, 261, 1025–1027. [Google Scholar] [CrossRef] [PubMed]
- Litwin, T.; Gromadzka, G.; Szpak, G.M.; Jabłonka-Salach, K.; Bulska, E.; Członkowska, A. Brain metal accumulation in Wilson’s disease. J. Neurol. Sci. 2013, 329, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Tang, D.; Wang, Y.; Li, X.; Bao, H.; Tang, C.; Dong, X.; Li, X.; Yang, Q.; Yan, Y.; et al. The mechanism of ferroptosis and its related diseases. Mol. Biomed. 2023, 16, 33. [Google Scholar]
- Kardos, J.; Héja, L.; Simon, Á.; Jablonkai, I.; Kovács, R.; Jemnitz, K. Copper signalling: Causes and consequences. Cell Commun. Signal. 2018, 16, 71, Erratum in Cell Commun. Signal. 2018, 16, 80. [Google Scholar] [CrossRef] [PubMed]
- Prohaska, J.R. Functions of trace elements in brain metabolism. Physiol. Rev. 1987, 67, 858–901. [Google Scholar] [CrossRef] [PubMed]
- Dobrowolska, J.; Dehnhardt, M.; Matusch, A.; Zoriy, M.; Palomero-Gallagher, N.; Koscielniak, P.; Becker, J.S. Quantitative imaging of zinc, copper and lead in three distinct regions of the human brain by laser ablation inductively coupled plasma mass spectrometry. Talanta 2008, 74, 717–723. [Google Scholar] [CrossRef] [PubMed]
- Zecca, L.; Stroppolo, A.; Gatti, A.; Tampellini, D.; Toscani, M.; Gallorini, M.; Giaveri, G.; Arosio, P.; Santambrogio, P.; Fariello, R.G. The role of iron and copper molecules in the neuronal vulnerability of locus coeruleus and substantia nigra during aging. Proc. Natl. Acad. Sci. USA 2004, 101, 9843–9848. [Google Scholar] [CrossRef]
- Rahil-Khazen, R.; Bolann, B.J.; Myking, A.; Ulvik, R.J. Multi-element analysis of trace element levels in human autopsy tissues by using inductively coupled atomic emission spectrometry technique (ICP-AES). J. Trace Elem. Med. Biol. 2002, 16, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Lutsenko, S.; Washington-Hughes, C.; Ralle, M.; Schmidt, K. Copper and the brain noradrenergic system. J. Biol. Inorg. Chem. 2019, 24, 1179–1188. [Google Scholar] [CrossRef]
- Collins, J.F. Copper: Molecular, Genetic, and Nutritional Aspects of Major and Trace Minerals; Academic Press: Cambridge, MA, USA, 2017; pp. 69–83. [Google Scholar]
- Buffoni, F.; Ignesti, G. The copper-containing amine oxidases: Biochemical aspects and functional role. Mol. Genet. Metab. 2000, 71, 559–564. [Google Scholar] [CrossRef]
- Bakavayev, S.; Chetrit, N.; Zvagelsky, T.; Mansour, R.; Vyazmensky, M.; Barak, Z.; Israelson, A.; Engel, S. Cu/Zn-superoxide dismutase and wild-type like fALS SOD1 mutants produce cytotoxic quantities of H2O2 via cysteine-dependent redox short-circuit. Sci. Rep. 2019, 9, 10826. [Google Scholar] [CrossRef] [PubMed]
- Öhrvik, H.; Thiele, D.J. The role of Ctr1 and Ctr2 in mammalian copper homeostasis and platinum-based chemotherapy. J. Trace Elem. Med. Biol. 2015, 31, 178–182. [Google Scholar] [CrossRef] [PubMed]
- La Fontaine, S.; Mercer, J.F. Trafficking of the copper-ATPases, ATP7A and ATP7B: Role in copper homeostasis. Arch Biochem Biophys. 2007, 15, 149–167. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.S.; Zheng, W. Copper transport to the brain by the blood-brain barrier and blood-CSF barrier. Brain Res. 2009, 1248, 14–21. [Google Scholar] [CrossRef] [PubMed]
- La Fontaine, S. New insights into CNS requirements for the copper-ATPase, ATP7A. Focus on “Autonomous requirements of the Menkes disease protein in the nervous system”. Am. J. Physiol. Cell Physiol. 2015, 309, C719–C721. [Google Scholar] [CrossRef] [PubMed]
- Tiffany-Castiglioni, E.; Hong, S.; Qian, Y. Copper handling by astrocytes: Insights into neurodegenerative diseases. Int. J. Dev. Neurosci. 2011, 29, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Scheiber, I.F.; Mercer, J.F.; Dringen, R. Copper accumulation by cultured astrocytes. Neurochem. Int. 2010, 56, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Scheiber, I.F.; Mercer, J.F.; Dringen, R. Metabolism and functions of copper in brain. Prog. Neurobiol. 2014, 116, 33–57. [Google Scholar] [CrossRef]
- Reddy, P.V.; Rao, K.V.; Norenberg, M.D. The mitochondrial permeability transition, and oxidative and nitrosative stress in the mechanism of copper toxicity in cultured neurons and astrocytes. Lab. Investig. 2008, 88, 816–830. [Google Scholar] [CrossRef]
- Witt, B.; Stiboller, M.; Raschke, S.; Friese, S.; Ebert, F.; Schwerdtle, T. Characterizing effects of excess copper levels in a human astrocytic cell line with focus on oxidative stress markers. J. Trace Elem. Med. Biol. 2021, 65, 126711. [Google Scholar] [CrossRef]
- Dringen, R.; Scheiber, I.F.; Mercer, J.F. Copper metabolism of astrocytes. Front. Aging Neurosci. 2013, 5, 9. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Peña, M.M.; Nose, Y.; Thiele, D.J. Biochemical characterization of the human copper transporter Ctr1. J. Biol. Chem. 2002, 277, 4380–4387. [Google Scholar] [CrossRef] [PubMed]
- Arredondo, M.; Muñoz, P.; Mura, C.V.; Nùñez, M.T. DMT1, a physiologically relevant apical Cu1+ transporter of intestinal cells. Am. J. Physiol.-Cell Physiol. 2003, 284, C1525–C1530. [Google Scholar] [CrossRef] [PubMed]
- Del Rio, R.; Quintanilla, R.A.; Orellana, J.A.; Retamal, M.A. Neuron-Glia Crosstalk in the Autonomic Nervous System and Its Possible Role in the Progression of Metabolic Syndrome: A New Hypothesis. Front. Physiol. 2015, 6, 350. [Google Scholar] [CrossRef] [PubMed]
- Scheiber, I.F.; Dringen, R. Copper-treatment increases the cellular GSH content and accelerates GSH export from cultured rat astrocytes. Neurosci. Lett. 2011, 498, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Scheiber, I.F.; Schmidt, M.M.; Dringen, R. Copper export from cultured astrocytes. Neurochem. Int. 2012, 60, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Huster, D. Wilson disease. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 531–539. [Google Scholar] [CrossRef]
- Bakkar, N.; Starr, A.; Rabichow, B.E.; Lorenzini, I.; McEachin, Z.T.; Kraft, R.; Chaung, M.; Macklin-Isquierdo, S.; Wingfield, T.; Carhart, B.; et al. The M1311V variant of ATP7A is associated with impaired trafficking and copper homeostasis in models of motor neuron disease. Neurobiol. Dis. 2021, 149, 105228. [Google Scholar] [CrossRef] [PubMed]
- Kaler, S.G. ATP7A-related copper transport diseases-emerging concepts and future trends. Nat. Rev. Neurol. 2011, 7, 15–29. [Google Scholar] [CrossRef]
- Palm, R.; Wahlström, G.; Hallmans, G. Age related changes in weight and the concentrations of zinc and copper in the brain of the adult rat. Lab. Anim. 1990, 24, 240–245. [Google Scholar] [CrossRef]
- Montes, S.; Rivera-Mancia, S.; Diaz-Ruiz, A.; Tristan-Lopez, L.; Rios, C. Copper and copper proteins in Parkinson’s disease. Oxidative Med. Cell. Longev. 2014, 2014, 147251. [Google Scholar] [CrossRef] [PubMed]
- Scheiber, I.F.; Dringen, R. Copper accelerates glycolytic flux in cultured astrocytes. Neurochem. Res. 2011, 36, 894–903. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, J.; García, A.; Oliva, A.M.; Giralt, M.; Gasull, T.; González, B.; Milnerowicz, H.; Wood, A.; Bremner, I. Effect of zinc, copper and glucocorticoids on metallothionein levels of cultured neurons and astrocytes from rat brain. Chem.-Biol. Interact. 1994, 93, 197–219. [Google Scholar] [CrossRef]
- Pope, S.A.; Milton, R.; Heales, S.J. Astrocytes protect against copper-catalysed loss of extracellular glutathione. Neurochem. Res. 2008, 33, 1410–1418. [Google Scholar] [CrossRef]
- Wang, X.F.; Cynader, M.S. Pyruvate released by astrocytes protects neurons from copper-catalyzed cysteine neurotoxicity. J. Neurosci. 2001, 21, 3322–3331. [Google Scholar] [CrossRef]
- Cobine, P.A.; Brady, D.C. Cuproptosis: Cellular and molecular mechanisms underlying copper-induced cell death. Mol. Cell 2022, 82, 1786–1787. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, P.; Coy, S.; Petrova, B.; Dreishpoon, M.; Verma, A.; Abdusamad, M.; Rossen, J.; Joesch-Cohen, L.; Humeidi, R.; Spangler, R.D.; et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 2022, 375, 1254–1261. [Google Scholar] [CrossRef]
- Zheng, Z.; White, C.; Lee, J.; Peterson, T.S.; Bush, A.I.; Sun, G.Y.; Weisman, G.A.; Petris, M.J. Altered microglial copper homeostasis in a mouse model of Alzheimer’s disease. J. Neurochem. 2010, 114, 1630–1638. [Google Scholar] [CrossRef]
- Hu, Z.; Yu, F.; Gong, P.; Qiu, Y.; Zhou, W.; Cui, Y.; Li, J. Subneurotoxic copper(II)-induced NF-κB-dependent microglial activation is associated with mitochondrial ROS. Toxicol. Appl. Pharmacol. 2014, 276, 95–103. [Google Scholar] [CrossRef]
- Rossi-George, A.; Guo, C.J. Copper modulates the phenotypic response of activated BV2 microglia through the release of nitric oxide. Nitric Oxide 2012, 27, 201–209. [Google Scholar] [CrossRef]
- Rossi-George, A.; Guo, C.J. Copper disrupts S-nitrosothiol signaling in activated BV2 microglia. Neurochem. Int. 2016, 99, 1–8. [Google Scholar] [CrossRef]
- Du, X.; Wang, Z.; Zheng, Y.; Li, H.; Ni, J.; Liu, Q. Inhibitory effect of selenoprotein P on Cu+/Cu2+-induced Aβ42 aggregation and toxicity. Inorg. Chem. 2014, 53, 1672–1678. [Google Scholar] [CrossRef] [PubMed]
- Goch, W.; Bal, W. Numerical Simulations Reveal Randomness of Cu(II) Induced Aβ Peptide Dimerization under Conditions Present in Glutamatergic Synapses. PLoS ONE 2017, 12, e0170749. [Google Scholar] [CrossRef] [PubMed]
- Kitazawa, M.; Hsu, H.W.; Medeiros, R. Copper Exposure Perturbs Brain Inflammatory Responses and Impairs Clearance of Amyloid-Beta. Toxicol. Sci. 2016, 152, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Venneti, S.; Lopresti, B.J.; Wang, G.; Hamilton, R.L.; Mathis, C.A.; Klunk, W.E.; Apte, U.M.; Wiley, C.A. PK11195 labels activated microglia in Alzheimer’s disease and in vivo in a mouse model using PET. Neurobiol. Aging 2009, 30, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Guan, H.; Yang, Y.; Luo, S.; Hou, L.; Chen, H.; Li, J. Cu(II) disrupts autophagy-mediated lysosomal degradation of oligomeric Aβ in microglia via mTOR-TFEB pathway. Toxicol. Appl. Pharmacol. 2020, 401, 115090. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V.; Deane, R.; Sagare, A.P.; Bell, R.D.; Winkler, E.A. Low-density lipoprotein receptor-related protein-1: A serial clearance homeostatic mechanism controlling Alzheimer’s amyloid β-peptide elimination from the brain. J. Neurochem. 2010, 115, 1077–1089. [Google Scholar] [CrossRef]
- Lee, K.H.; Yun, S.J.; Nam, K.N.; Gho, Y.S.; Lee, E.H. Activation of microglial cells by ceruloplasmin. Brain Res. 2007, 1171, 1–8. [Google Scholar] [CrossRef]
- Lim, S.L.; Rodriguez-Ortiz, C.J.; Hsu, H.W.; Wu, J.; Zumkehr, J.; Kilian, J.; Vidal, J.; Ayata, P.; Kitazawa, M. Chronic copper exposure directs microglia towards degenerative expression signatures in wild-type and J20 mouse model of Alzheimer’s disease. J. Trace Elem. Med. Biol. 2020, 62, 126578. [Google Scholar] [CrossRef]
- Zucconi, G.G.; Cipriani, S.; Scattoni, R.; Balgkouranidou, I.; Hawkins, D.P.; Ragnarsdottir, K.V. Copper deficiency elicits glial and neuronal response typical of neurodegenerative disorders. Neuropathol. Appl. Neurobiol. 2007, 33, 212–225. [Google Scholar] [CrossRef]
- Bastian, T.W.; Rao, R.; Tran, P.V.; Georgieff, M.K. The Effects of Early-Life Iron Deficiency on Brain Energy Metabolism. Neurosci. Insights 2020, 15, 2633105520935104. [Google Scholar] [CrossRef] [PubMed]
- Fretham, S.J.; Carlson, E.S.; Georgieff, M.K. The role of iron in learning and memory. Adv. Nutr. 2011, 2, 112–121. [Google Scholar] [CrossRef] [PubMed]
- DeBenedictis, C.A.; Raab, A.; Ducie, E.; Howley, S.; Feldmann, J.; Grabrucker, A.M. Concentrations of Essential Trace Metals in the Brain of Animal Species—A Comparative Study. Brain Sci. 2020, 10, 460. [Google Scholar] [CrossRef]
- Madsen, E.; Gitlin, J.D. Copper and iron disorders of the brain. Annu. Rev. Neurosci. 2007, 30, 317–337. [Google Scholar] [CrossRef] [PubMed]
- Kulaszyńska, M.; Kwiatkowski, S.; Skonieczna-Żydecka, K. The Iron Metabolism with a Specific Focus on the Functioning of the Nervous System. Biomedicines 2024, 12, 595. [Google Scholar] [CrossRef] [PubMed]
- Salvador, G.A.; Uranga, R.M.; Giusto, N.M. Iron and mechanisms of neurotoxicity. Int. J. Alzheimer’s Dis. 2011, 2011, 720658. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Dringen, R.; Bishop, G.M.; Koeppe, M.; Dang, T.N.; Robinson, S.R. The pivotal role of astrocytes in the metabolism of iron in the brain. Neurochem. Res. 2007, 32, 1884–1890. [Google Scholar] [CrossRef]
- Crichton, R.R.; Dexter, D.T.; Ward, R.J. Brain iron metabolism and its perturbation in neurological diseases. Monatshefte Chem.—Chem. Mon. 2011, 142, 341–355. [Google Scholar] [CrossRef]
- Moos, T.; Nielsen, T.R.; Skjørringe, T.; Morgan, E.H. Iron trafficking inside the brain. J. Neurochem. 2007, 103, 1730–1740. [Google Scholar] [CrossRef]
- Moos, T. Immunohistochemical localization of intraneuronal transferrin receptor immunoreactivity in the adult mouse central nervous system. J. Comp. Neurol. 1996, 375, 675–692. [Google Scholar] [CrossRef]
- Tulpule, K.; Robinson, S.R.; Bishop, G.M.; Dringen, R. Uptake of ferrous iron by cultured rat astrocytes. J. Neurosci. Res. 2010, 88, 563–571. [Google Scholar] [CrossRef]
- Bishop, G.M.; Scheiber, I.F.; Dringen, R.; Robinson, S.R. Synergistic accumulation of iron and zinc by cultured astrocytes. J Neural. Transm. 2010, 117, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Pelizzoni, I.; Zacchetti, D.; Campanella, A.; Grohovaz, F.; Codazzi, F. Iron uptake in quiescent and inflammation-activated astrocytes: A potentially neuroprotective control of iron burden. Biochim. Biophys. Acta 2013, 1832, 1326–1333. [Google Scholar] [CrossRef] [PubMed]
- Burdo, J.R.; Menzies, S.L.; Simpson, I.A.; Garrick, L.M.; Garrick, M.D.; Dolan, K.G.; Haile, D.J.; Beard, J.L.; Connor, J.R. Distribution of divalent metal transporter 1 and metal transport protein 1 in the normal and Belgrade rat. J. Neurosci. Res. 2001, 66, 1198–1207. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.F.; Ong, W.Y.; Connor, J.R. A light and electron microscopic study of the iron transporter protein DMT-1 in the monkey cerebral neocortex and hippocampus. J. Neurocytol. 2001, 30, 353–360. [Google Scholar] [CrossRef]
- Musci, G.; Polticelli, F.; Bonaccorsi di Patti, M.C. Ceruloplasmin-ferroportin system of iron traffic in vertebrates. World J. Biol. Chem. 2014, 5, 204–215. [Google Scholar]
- Jeong, S.Y.; David, S. Glycosylphosphatidylinositol-anchored ceruloplasmin is required for iron efflux from cells in the central nervous system. J. Biol. Chem. 2003, 278, 27144–27148. [Google Scholar] [CrossRef]
- Chen, Z.; Jiang, R.; Chen, M.; Zheng, J.; Chen, M.; Braidy, N.; Liu, S.; Liu, G.; Maimaitiming, Z.; Shen, T.; et al. Multi-copper ferroxidase deficiency leads to iron accumulation and oxidative damage in astrocytes and oligodendrocytes. Sci. Rep. 2019, 9, 9437. [Google Scholar] [CrossRef]
- Riemer, J.; Hoepken, H.H.; Czerwinska, H.; Robinson, S.R.; Dringen, R. Colorimetric ferrozine-based assay for the quantitation of iron in cultured cells. Anal. Biochem. 2004, 33, 370–375. [Google Scholar] [CrossRef]
- Connor, J.R. Iron acquisition and expression of iron regulatory proteins in the developing brain: Manipulation by ethanol exposure, iron deprivation and cellular dysfunction. Dev. Neurosci. 1994, 16, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Connor, J.R.; Menzies, S.L.; St Martin, S.M.; Mufson, E.J. Cellular distribution of transferrin, ferritin, and iron in normal and aged human brains. J. Neurosci. Res. 1990, 27, 595–611. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M.; Vininsky, R.; Brull, R.; Small, L.; Brawer, J.R. Astrocyte mitochondria: A substrate for iron deposition in the aging rat substantia nigra. Exp. Neurol. 1998, 152, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Kaur, C.; Ling, E.A. Increased expression of transferrin receptors and iron in amoeboid microglial cells in postnatal rats following an exposure to hypoxia. Neurosci. Lett. 1999, 262, 183–186. [Google Scholar] [CrossRef]
- LeVine, S.M. Exploring Potential Mechanisms Accounting for Iron Accumulation in the Central Nervous System of Patients with Alzheimer’s Disease. Cells 2024, 13, 689. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, M.S.; Andersen, M.V.; Christoffersen, P.R.; Jensen, M.D.; Lichota, J.; Moos, T. Neurodegeneration with inflammation is accompanied by accumulation of iron and ferritin in microglia and neurons. Neurobiol. Dis. 2015, 81, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Gaasch, J.A.; Lockman, P.R.; Geldenhuys, W.J.; Allen, D.D.; Van der Schyf, C.J. Brain iron toxicity: Differential responses of astrocytes, neurons, and endothelial cells. Neurochem. Res. 2007, 32, 1196–1208. [Google Scholar] [CrossRef]
- Nnah, I.C.; Wessling-Resnick, M. Brain iron homeostasis: A focus on microglial iron. Pharmaceuticals 2018, 11, 129. [Google Scholar] [CrossRef]
- Connor, J.R.; Boeshore, K.L.; Benkovic, S.A.; Menzies, S.L. Isoforms of ferritin have a specific cellular distribution in the brain. J. Neurosci. Res. 1994, 37, 461–465. [Google Scholar] [CrossRef]
- Han, J.; Day, J.R.; Connor, J.R.; Beard, J.L. H and L ferritin subunit mRNA expression differs in brains of control and iron-deficient rats. J. Nutr. 2002, 132, 2769–2774. [Google Scholar] [CrossRef]
- Bishop, G.M.; Dang, T.N.; Dringen, R.; Robinson, S.R. Accumulation of non-transferrin-bound iron by neurons, astrocytes, and microglia. Neurotox. Res. 2011, 19, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.J.; Dexter, D.T.; Crichton, R.R. Iron, neuroinflammation and neurodegeneration. Int. J. Mol. Sci. 2022, 23, 7267. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, R.C.; Sosa, J.C.; Gardeck, A.M.; Baez, A.S.; Lee, C.H.; Wessling-Resnick, M. Inflammation-induced iron transport and metabolism by brain microglia. J. Biol. Chem. 2018, 293, 7853–7863. [Google Scholar] [CrossRef]
- Roy, S.; Lutsenko, S. Mechanism of Cu entry into the brain: Many unanswered questions. Neural Regen. Res. 2024, 19, 2421–2429. [Google Scholar] [CrossRef] [PubMed]
- Roemhild, K.; von Maltzahn, F.; Weiskirchen, R.; Knüchel, R.; von Stillfried, S.; Lammers, T. Iron metabolism: Pathophysiology and pharmacology. Trends Pharmacol. Sci. 2021, 42, 640–656. [Google Scholar] [CrossRef]
- Bresgen, N.; Eckl, P.M. Oxidative stress and the homeodynamics of iron metabolism. Biomolecules 2015, 5, 808–847. [Google Scholar] [CrossRef] [PubMed]
- Baader, S.L.; Bruchelt, G.; Carmine, T.C.; Lode, H.N.; Rieth, A.G.; Niethammer, D. Ascorbic-acid-mediated iron release from cellular ferritin and its relation to the formation of DNA strand breaks in neuroblastoma cells. J. Cancer Res. Clin. Oncol. 1994, 120, 415–421. [Google Scholar] [CrossRef]
- Galaris, D.; Barbouti, A.; Pantopoulos, K. Iron homeostasis and oxidative stress: An intimate relationship. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 118535. [Google Scholar] [CrossRef]
- Urati, A.; Dey, M.; Gautam, A.S.; Singh, R.K. Iron-induced cellular in vitro neurotoxic responses in rat C6 cell line. Environ. Toxicol. 2022, 37, 1968–1978. [Google Scholar] [CrossRef]
- Zhang, Z.; Wei, T.; Hou, J.; Li, G.; Yu, S.; Xin, W. Iron-induced oxidative damage and apoptosis in cerebellar granule cells: Attenuation by tetramethylpyrazine and ferulic acid. Eur. J. Pharmacol. 2003, 467, 41–47. [Google Scholar] [CrossRef]
- Li, L.B.; Chai, R.; Zhang, S.; Xu, S.F.; Zhang, Y.H.; Li, H.L.; Fan, Y.G.; Guo, C. Iron Exposure and the Cellular Mechanisms Linked to Neuron Degeneration in Adult Mice. Cells 2019, 8, 198. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Min, L.; Chen, H.; Deng, W.; Tan, M.; Liu, H.; Hou, J. Iron overload in the motor cortex induces neuronal ferroptosis following spinal cord injury. Redox Biol. 2021, 43, 101984. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019, 23, 101107. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.Y.; Huang, B.Y.; Nie, H.F.; Chen, X.Y.; Zhou, Y.; Yang, T.; Cheng, S.W.; Mei, Z.G.; Ge, J.W. The Interplay between Mitochondrial Dysfunction and Ferroptosis during Ischemia-Associated Central Nervous System Diseases. Brain Sci. 2023, 13, 1367. [Google Scholar] [CrossRef] [PubMed]
- Baoyinna, B.; Miao, J.; Oliver, P.J.; Ye, Q.; Shaheen, N.; Kalin, T.; He, J.; Parinandi, N.L.; Zhao, Y.; Zhao, J. Non-Lethal Doses of RSL3 Impair Microvascular Endothelial Barrier through Degradation of Sphingosie-1-Phosphate Receptor 1 and Cytoskeletal Arrangement in A Ferroptosis-Independent Manner. Biomedicines 2023, 11, 2451. [Google Scholar] [CrossRef]
- Chen, X.; Yu, C.; Kang, R.; Kroemer, G.; Tang, D. Cellular degradation systems in ferroptosis. Cell Death Differ. 2021, 28, 1135–1148. [Google Scholar] [CrossRef]
- Darius, J.R.; Metselaar, L.B.; Greenough, M.; Bush, A.; Ayton, S.J. Ferroptosis and NRF2: An emerging battlefield in the neurodegeneration of Alzheimer’s disease. Essays Biochem. 2021, 65, 925–940. [Google Scholar]
- Cardoso, B.; Hare, D.; Bush, A.; Roberts, B.R. Glutathione peroxidase 4: A new player in neurodegeneration? Mol. Psychiatry 2017, 22, 328–335. [Google Scholar] [CrossRef]
- Wei, R.; Wei, P.; Yuan, H.; Yi, X.; Aschner, M.; Jiang, Y.M.; Li, S.J. Inflammation in Metal-Induced Neurological Disorders and Neurodegenerative Diseases. Biol. Trace Elem. Res. 2024; Epub ahead of print. [Google Scholar] [CrossRef]
- Friedrich, I.; Reimann, K.; Jankuhn, S.; Kirilina, E.; Stieler, J.; Sonntag, M.; Meijer, J.; Weiskopf, N.; Reinert, T.; Arendt, T.; et al. Cell specific quantitative iron mapping on brain slices by immuno-µPIXE in healthy elderly and Parkinson’s disease. Acta Neuropathol. Commun. 2021, 9, 47. [Google Scholar] [CrossRef]
- Martin-Bastida, A.; Tilley, B.S.; Bansal, S.; Gentleman, S.M. Iron and inflammation: In vivo and post-mortem studies in Parkinson’s disease. J. Neural Transm. 2021, 128, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Wood, H. α-Synuclein-activated microglia are implicated in PD pathogenesis. Nat. Rev. Neurol. 2022, 18, 188. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Phillips, K.; Wielgus, A.R.; Liu, J.; Albertini, A.; Zucca, F.A.; Faust, R.; Qian, S.Y.; Miller, D.S.; Chignell, C.F.; et al. Neuromelanin activates microglia and induces degeneration of dopaminergic neurons: Implications for progression of Parkinson’s disease. Neurotox. Res. 2011, 19, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; You, L.; Zhang, J.; Chang, Y.Z.; Yu, P. Brain Iron Metabolism, Redox Balance and Neurological Diseases. Antioxidants 2023, 12, 1289. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Song, N.; Jiang, H.; Wang, J.; Xie, J. Pro-inflammatory cytokines modulate iron regulatory protein 1 expression and iron transportation through reactive oxygen/nitrogen species production in ventral mesencephalic neurons. Biochim. Biophys. Acta 2013, 1832, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Faucheux, B.A.; Martin, M.E.; Beaumont, C.; Hunot, S.; Hauw, J.J.; Agid, Y.; Hirsch, E.C. Lack of up-regulation of ferritin is associated with sustained iron regulatory protein-1 binding activity in the substantia nigra of patients with Parkinson’s disease. J. Neurochem. 2002, 83, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Ayton, S.; Portbury, S.; Kalinowski, P.; Agarwal, P.; Diouf, I.; Schneider, J.A.; Morris, M.C.; Bush, A.I. Regional brain iron associated with deterioration in Alzheimer’s disease: A large cohort study and theoretical significance. Alzheimers Dement. 2021, 17, 1244–1256. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, L.; Camaschella, C. A potential pathogenetic role of iron in Alzheimer’s disease. J. Cell Mol. Med. 2008, 12, 1548–1550. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.T.; Randall, J.D.; Cahill, C.M.; Eder, P.S.; Huang, X.; Gunshin, H.; Leiter, L.; McPhee, J.; Sarang, S.S.; Utsuki, T.; et al. An iron-responsive element type II in the 5′-untranslated region of the Alzheimer’s amyloid precursor protein transcript. J. Biol. Chem. 2002, 277, 45518–45528. [Google Scholar] [CrossRef]
- Connor, J.R.; Snyder, B.S.; Beard, J.L.; Fine, R.E.; Mufson, E.J. Regional distribution of iron and iron-regulatory proteins in the brain in aging and Alzheimer’s disease. J. Neurosci. Res. 1992, 31, 327–335. [Google Scholar] [CrossRef]
- Cui, Z.; Zhong, Z.; Yang, Y.; Wang, B.; Sun, Y.; Sun, Q.; Yang, G.Y.; Bian, L. Ferrous Iron Induces Nrf2 Expression in Mouse Brain Astrocytes to Prevent Neurotoxicity. J. Biochem. Mol. Toxicol. 2016, 30, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Liddell, J.R.; Robinson, S.R.; Dringen, R. Endogenous glutathione and catalase protect cultured rat astrocytes from the iron-mediated toxicity of hydrogen peroxide. Neurosci. Lett. 2004, 364, 164–167. [Google Scholar] [CrossRef] [PubMed]
- Codazzi, F.; Pelizzoni, I.; Zacchetti, D.; Grohovaz, F. Iron entry in neurons and astrocytes: A link with synaptic activity. Front. Mol. Neurosci. 2015, 8, 18. [Google Scholar] [CrossRef] [PubMed]
- Rathore, K.I.; Redensek, A.; David, S. Iron homeostasis in astrocytes and microglia is differentially regulated by TNF-α and TGF-β1. Glia 2012, 60, 738–750. [Google Scholar] [CrossRef] [PubMed]
- Urrutia, P.; Aguirre, P.; Esparza, A.; Tapia, V.; Mena, N.P.; Arredondo, M.; González-Billault, C.; Núñez, M.T. Inflammation alters the expression of DMT1, FPN1 and hepcidin, and it causes iron accumulation in central nervous system cells. J. Neurochem. 2013, 126, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Regan, R.F.; Kumar, N.; Gao, F.; Guo, Y. Ferritin induction protects cortical astrocytes from heme-mediated oxidative injury. Neuroscience 2002, 113, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Tarnacka, B.; Szeszkowski, W.; Gołębiowski, M.; Członkowska, A. Metabolic changes in 37 newly diagnosed Wilson’s disease patients assessed by Magnetic Resonance Spectroscopy. Park. Relat. Disord. 2009, 15, 582–586. [Google Scholar] [CrossRef]
- Tarnacka, B.; Szeszkowski, W.; Gołębiowski, M.; Członkowska, A. MR spectroscopy in monitoring the treatment of Wilson’s disease patients. Mov. Disord. 2008, 23, 1560–1566. [Google Scholar] [CrossRef] [PubMed]
- Poujois, A.; Mikol, J.; Woimant, F. Wilson disease: Brain pathology. Handb. Clin. Neurol. 2017, 142, 77–89. [Google Scholar] [PubMed]
- Jaeger, V.; DeMorrow, S.; McMillin, S. The direct contribution of astrocytes and microglia to the pathogenesis of hepatic encephalopathy. J. Clin. Transl. Hepatol. 2019, 7, 352–361. [Google Scholar] [CrossRef]
- Kida, H.; Anraku, S. Histopathological study on astrocyte in various liver diseases part I. Kurume Med. J. 1979, 26, 69–73. [Google Scholar] [CrossRef]
- Litwin, T.; Dzieżyc, K.; Członkowska, A. Wilson disease—Treatment perspectives. Ann. Transl. Med. 2019, 7, S68. [Google Scholar] [CrossRef] [PubMed]
- Antos, A.; Członkowska, A.; Bembenek, J.; Skowrońska, M.; Kurkowska-Jastrzębska, I.; Litwin, T. Blood based biomarkers of central nervous system involvement in Wilson’s disease. Diagnostics 2023, 13, 1554. [Google Scholar] [CrossRef]
- Shribman, S.; Heller, C.; Burrows, M.; Heslegrave, A.; Swift, I.; Foiani, M.S.; Gillett, G.T.; Tsochatzis, E.A.; Rowe, J.B.; Gerhard, A.; et al. Plasma neurofilament light as a biomarker of neurological involvement in Wilson’s disease. Mov. Disord. 2021, 36, 50–58. [Google Scholar] [CrossRef]
- Wang, R.M.; Xu, W.Q.; Zheng, Z.W.; Yang, G.M.; Zhang, M.Y.; Ke, H.Z.; Wu, Z.Y. Serum Neurofilament Light Chain in Wilson’s Disease: A Promising Indicator but Unparallel to Real-Time Treatment Response. Mov. Disord. 2022, 37, 1531–1535. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Huang, Z.; Yang, H.; Luo, Y.; You, H.; Chen, D.; Pei, Z.; Li, X. Plasma neurofilament light chain as a biomarker in Wilson’s disease. Park. Relat. Disord. 2022, 95, 5–10. [Google Scholar] [CrossRef]
- Ziemssen, T.; Akgun, K.; Członkowska, A.; Antos, A.; Bembenek, J.; Kurkowska-Jastrzębska, I.; Przybyłkowski, A.; Skowrońska, M.; Smolinski, L.; Litwin, T. Serum neurofilament light chain as a biomarker of brain injury in Wilson’s disease: Clinical and neuroradiological correlations. Mov. Disord. 2022, 37, 1074–1079. [Google Scholar] [CrossRef]
- Ziemssen, T.; Smolinski, L.; Członkowska, A.; Akgun, K.; Antos, A.; Bembenek, J.; Kurkowska-Jastrzębska, I.; Przybyłkowski, A.; Skowrońska, M.; Redzia-Ogrodnik, B.; et al. Serum neurofilament light chain and initial severity of neurological disease predict the early neurological deterioration in Wilson’s disease. Acta Neurol. Belg. 2023, 123, 917–925. [Google Scholar] [CrossRef] [PubMed]
- Lekomtseva, Y.; Voloshyn-Gaponov, I.; Gorbach, T. Targeting higher levels of tau protein in Ukrainian patients with Wilson’s disease. Neurol. Ther. 2019, 8, 59–68. [Google Scholar] [CrossRef]
- Lin, J.; Zheng, Y.; Liu, Y.; Lin, Y.; Wang, Q.; Lin, X.H.; Zhu, W.; Lin, W.H.; Wang, N.; Chen, W.J.; et al. Higher concentration of plasma glial fibrillary acidic protein in Wilson disease patients with neurological manifestations. Mov. Disord. 2021, 36, 1446–1450. [Google Scholar] [CrossRef]
- Smoliński, L.; Litwin, T.; Redzia-Ogrodnik, B.; Dzieżyc, K.; Kurkowska-Jastrzębska, I.; Członkowska, A. Brain volume is related to neurological impairment and to copper overload in Wilson’s disease. Neurol. Sci. 2019, 40, 2089–2095. [Google Scholar] [CrossRef] [PubMed]
- Falcone, C.; Penna, E.; Hong, T.; Tarantal, A.F.; Hof, P.R.; Hopkins, W.D.; Sherwood, C.C.; Noctor, S.C.; Martínez-Cerdeño, V. Cortical Interlaminar Astrocytes Are Generated Prenatally, Mature Postnatally, and Express Unique Markers in Human and Nonhuman Primates. Cereb. Cortex 2021, 31, 379–395. [Google Scholar] [CrossRef] [PubMed]
- Tabata, H. Diverse subtypes of astrocytes and their development during corticogenesis. Front. Neurosci. 2015, 9, 114. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, G.; Yang, L.; Li, Z.; Zhang, Z.; Xu, Z.; Cai, Y.; Du, H.; Su, Z.; Wang, Z.; et al. Decoding Cortical Glial Cell Development. Neurosci. Bull. 2021, 37, 440–460. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef] [PubMed]
- Bélanger, M.; Allaman, I.; Magistretti, P.J. Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Noctor, S.C.; Martínez-Cerdeño, V.; Ivic, L.; Kriegstein, A. Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat. Neurosci. 2004, 7, 136–144. [Google Scholar] [CrossRef]
- Malik, S.; Vinukonda, G.; Vose, L.R.; Diamond, D.; Bhimavarapu, B.B.; Hu, F.; Zia, M.T.; Hevner, R.; Zecevic, N.; Ballabh, P. Neurogenesis continues in the third trimester of pregnancy and is suppressed by premature birth. J. Neurosci. 2013, 33, 411–423. [Google Scholar] [CrossRef]
- Li, J.; Pan, L.; Pembroke, W.G.; Rexach, J.E.; Godoy, M.I.; Condro, M.C.; Alvarado, A.G.; Harteni, M.; Chen, Y.W.; Stiles, L.; et al. Conservation and divergence of vulnerability and responses to stressors between human and mouse astrocytes. Nat. Commun. 2021, 12, 3958. [Google Scholar] [CrossRef]
- Kodama, H.; Meguro, Y.; Abe, T.; Rayner, M.H.; Suzuki, K.T.; Kobayashi, S.; Nishimura, M. Genetic expression of Menkes disease in cultured astrocytes of the macular mouse. J. Inherit. Metab. Dis. 1991, 14, 896–901. [Google Scholar] [CrossRef] [PubMed]
- Scheiber, I.F.; Dringen, R. Astrocyte functions in the copper homeostasis of the brain. Neurochem. Int. 2013, 62, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Telianidis, J.; Hung, Y.H.; Materia, S.; Fontaine, S.L. Role of the P-Type ATPases, ATP7A and ATP7B in brain copper homeostasis. Front. Aging Neurosci. 2013, 5, 44. [Google Scholar] [CrossRef] [PubMed]
- Barnes, N.; Tsivkovskii, R.; Tsivkovskaia, N.; Lutsenko, S. The copper-transporting ATPases, menkes and wilson disease proteins, have distinct roles in adult and developing cerebellum. J. Biol. Chem. 2005, 280, 9640–9645. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.M.; Gitschier, J.; Packman, S. Developmental expression of the mouse mottled and toxic milk genes suggests distinct functions for the Menkes and Wilson disease copper transporters. Hum. Mol. Genet. 1997, 6, 1043–1049. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.; Rani, I.; Pawar, A.; Picozza, M.; Rongioletti, M.; Squitti, R. Microglia and Astrocytes in Alzheimer’s Disease in the Context of the Aberrant Copper Homeostasis Hypothesis. Biomolecules 2021, 11, 1598. [Google Scholar] [CrossRef] [PubMed]
- Orre, M.; Kamphuis, W.; Osborn, L.M.; Jansen, A.H.P.; Kooijman, L.; Bossers, K.; Hol, E.M. Isolation of glia from Alzheimer’s mice reveals inflammation and dysfunction. Neurobiol. Aging 2014, 35, 2746–2760. [Google Scholar] [CrossRef]
- Terwel, D.; Löschmann, Y.N.; Schmidt, H.H.; Schöler, H.R.; Cantz, T.; Heneka, M.T. Neuroinflammatory and behavioural changes in the Atp7B mutant mouse model of Wilson’s disease. J. Neurochem. 2011, 118, 105–112. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gromadzka, G.; Wilkaniec, A.; Tarnacka, B.; Hadrian, K.; Bendykowska, M.; Przybyłkowski, A.; Litwin, T. The Role of Glia in Wilson’s Disease: Clinical, Neuroimaging, Neuropathological and Molecular Perspectives. Int. J. Mol. Sci. 2024, 25, 7545. https://doi.org/10.3390/ijms25147545
Gromadzka G, Wilkaniec A, Tarnacka B, Hadrian K, Bendykowska M, Przybyłkowski A, Litwin T. The Role of Glia in Wilson’s Disease: Clinical, Neuroimaging, Neuropathological and Molecular Perspectives. International Journal of Molecular Sciences. 2024; 25(14):7545. https://doi.org/10.3390/ijms25147545
Chicago/Turabian StyleGromadzka, Grażyna, Anna Wilkaniec, Beata Tarnacka, Krzysztof Hadrian, Maria Bendykowska, Adam Przybyłkowski, and Tomasz Litwin. 2024. "The Role of Glia in Wilson’s Disease: Clinical, Neuroimaging, Neuropathological and Molecular Perspectives" International Journal of Molecular Sciences 25, no. 14: 7545. https://doi.org/10.3390/ijms25147545
APA StyleGromadzka, G., Wilkaniec, A., Tarnacka, B., Hadrian, K., Bendykowska, M., Przybyłkowski, A., & Litwin, T. (2024). The Role of Glia in Wilson’s Disease: Clinical, Neuroimaging, Neuropathological and Molecular Perspectives. International Journal of Molecular Sciences, 25(14), 7545. https://doi.org/10.3390/ijms25147545