
Oxidative Metabolism as a Cause of Lipid Peroxidation in the Execution of Ferroptosis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Overview of the Ferroptotic Pathway
2.1. Presence of Iron Pushes Lipid Peroxidation beyond the Threshold for Cellular Protection
2.2. Elevation in Cell Rupture as a Result of Phospholipid Peroxidation
2.3. Glutathione/GPX4 System as a Primary Protector against Lipid Peroxidation and Ferroptosis
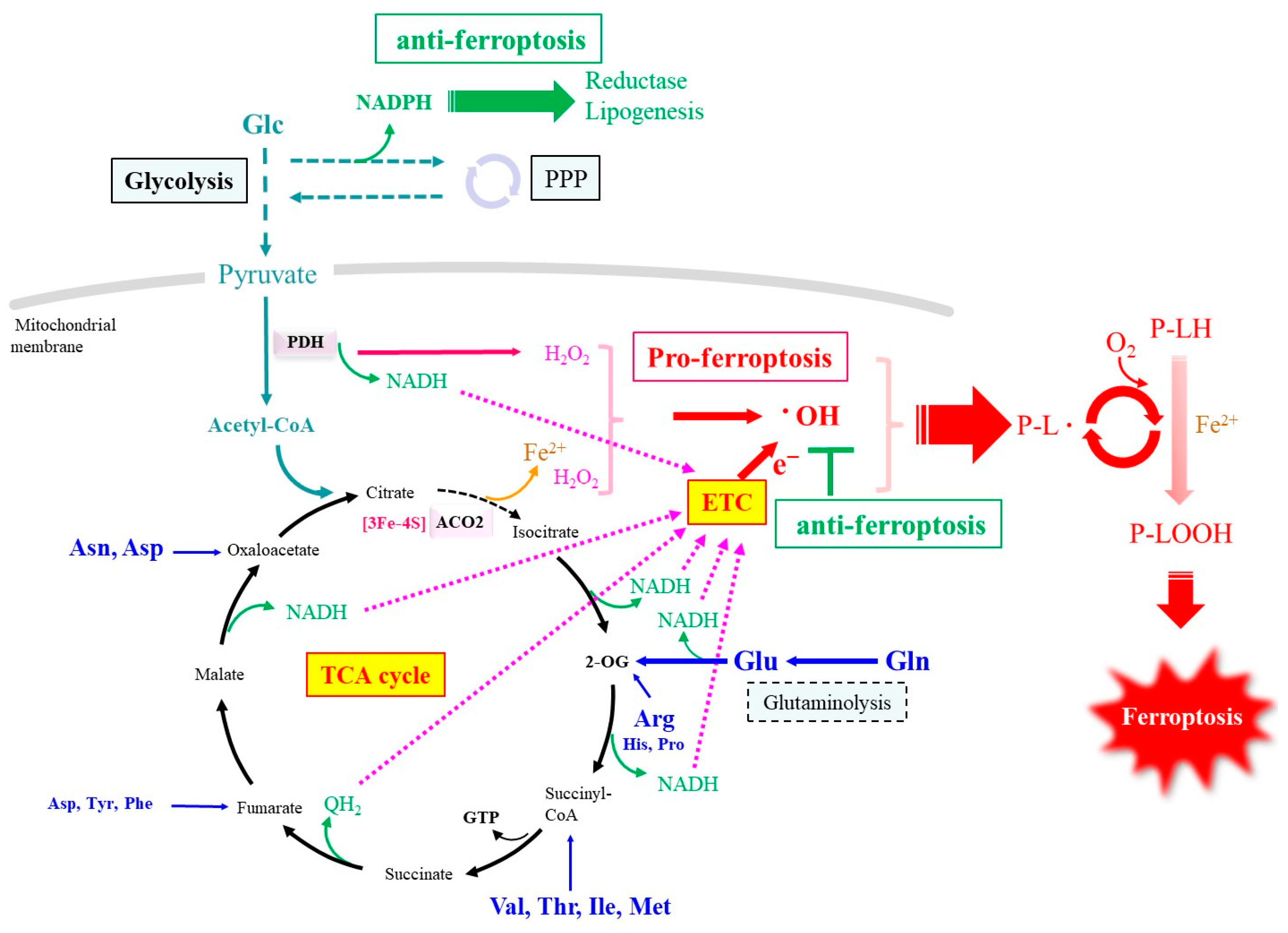
3. Alteration in the Metabolism of Carbon and Nitrogen Compounds upon Ferroptotic Stimulation
3.1. Association of ROS Production with Cell Proliferation
3.2. Iron Release in Association with Cellular Metabolism
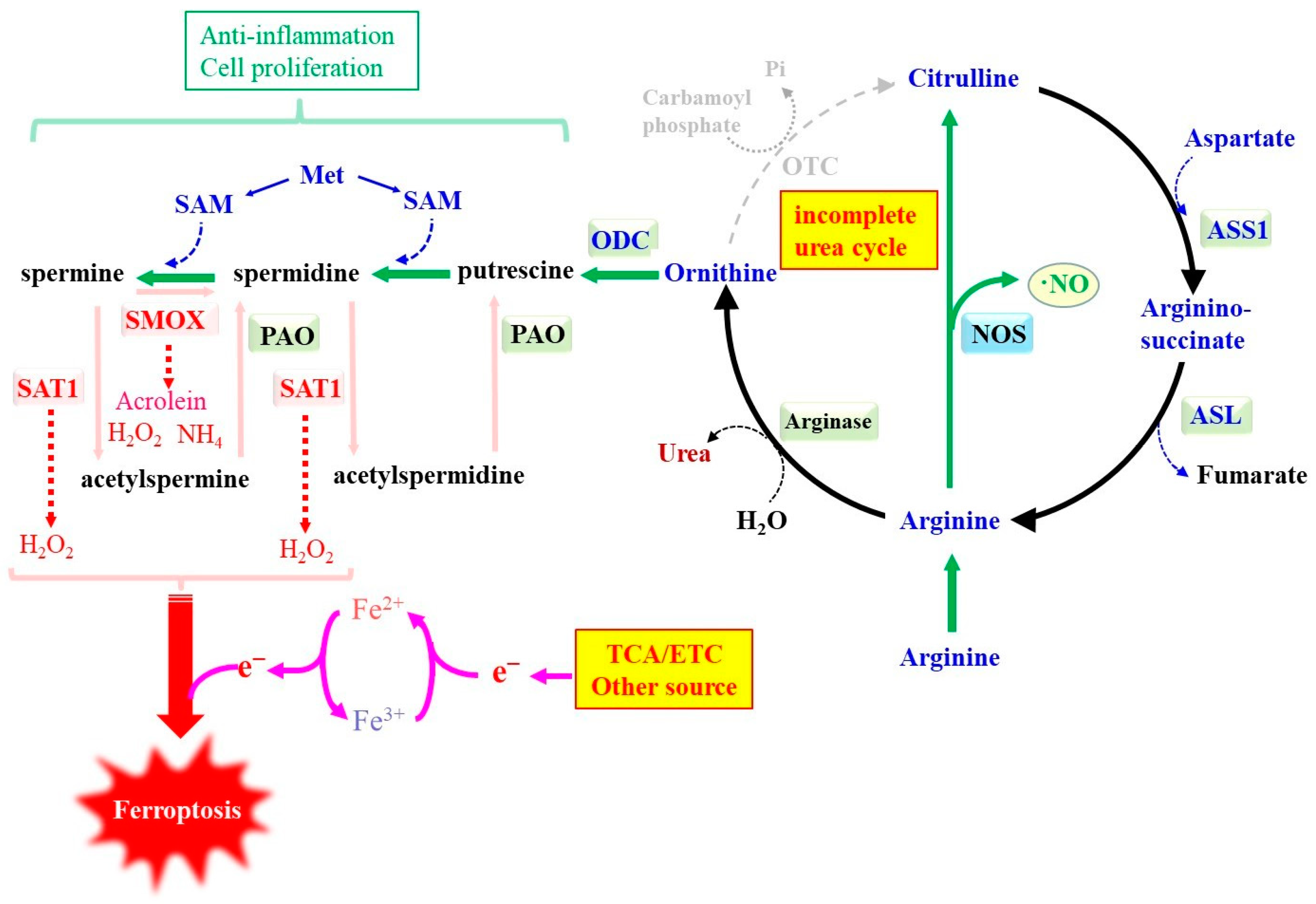
3.3. Nitrogen Metabolism-Associated ROS Formation
3.4. Ferroptosis Suppression by the Redox System
4. ROS from Other Enzymatic Sources
4.1. Arachidonate-Specific Lipoxygenase (ALOX)
4.2. P450 Oxidoreductase (POR)
4.3. Potential Involvement of Other Oxidase/Oxygenase
4.4. Iron-Independent Lipid Peroxidation Induced Cell Death (Lipoxytosis) Regulated by GPX4
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Sauler, M.; Bazan, I.S.; Lee, P.J. Cell Death in the Lung: The Apoptosis-Necroptosis Axis. Annu. Rev. Physiol. 2019, 81, 375–402. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Ofengeim, D. A guide to cell death pathways. Nat. Rev. Mol. Cell Biol. 2024, 25, 379–395. [Google Scholar] [CrossRef] [PubMed]
- Crawford, E.D.; Wells, J.A. Caspase substrates and cellular remodeling. Annu. Rev. Biochem. 2011, 80, 1055–1087. [Google Scholar] [CrossRef] [PubMed]
- Nagata, S. Apoptosis and Clearance of Apoptotic Cells. Annu. Rev. Immunol. 2018, 36, 489–517. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Bayır, H.; Anthonymuthu, T.S.; Tyurina, Y.Y.; Patel, S.J.; Amoscato, A.A.; Lamade, A.M.; Yang, Q.; Vladimirov, G.K.; Philpott, C.C.; Kagan, V.E. Achieving Life through Death: Redox Biology of Lipid Peroxidation in Ferroptosis. Cell Chem. Biol. 2020, 27, 387–408. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 2022, 185, 2401–2421. [Google Scholar] [CrossRef] [PubMed]
- Mishima, E.; Conrad, M. Nutritional and Metabolic Control of Ferroptosis. Annu. Rev. Nutr. 2022, 42, 275–309. [Google Scholar] [CrossRef]
- Dai, E.; Chen, X.; Linkermann, A.; Jiang, X.; Kang, R.; Kagan, V.E.; Bayir, H.; Yang, W.S.; Garcia-Saez, A.J.; Ioannou, M.S.; et al. A guideline on the molecular ecosystem regulating ferroptosis. Nat. Cell Biol. 2024. [Google Scholar] [CrossRef]
- Dixon, S.J.; Olzmann, J.A. The cell biology of ferroptosis. Nat. Rev. Mol. Cell Biol. 2024, 25, 424–442. [Google Scholar] [CrossRef]
- Tarangelo, A.; Magtanong, L.; Bieging-Rolett, K.T.; Li, Y.; Ye, J.; Attardi, L.D.; Dixon, S.J. p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell Rep. 2018, 22, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Gan, B. Mitochondrial regulation of ferroptosis. J. Cell Biol. 2021, 220, e202105043. [Google Scholar] [CrossRef] [PubMed]
- Seibt, T.M.; Proneth, B.; Conrad, M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic. Biol. Med. 2019, 133, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Ursini, F.; Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic. Biol. Med. 2020, 152, 175–185. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Conrad, M.; Sato, H. The oxidative stress-inducible cystine/glutamate antiporter, system x (c) (-): Cystine supplier and beyond. Amino Acids 2012, 42, 231–246. [Google Scholar] [CrossRef]
- Feng, H.; Stockwell, B.R. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol. 2018, 16, e2006203. [Google Scholar] [CrossRef]
- Winterbourn, C.C. Toxicity of iron and hydrogen peroxide: The Fenton reaction. Toxicol. Lett. 1995, 82–83, 969–974. [Google Scholar] [CrossRef]
- Niki, E. Lipid peroxidation: Physiological levels and dual biological effects. Free Radic. Biol. Med. 2009, 47, 469–484. [Google Scholar] [CrossRef]
- Fridovich, I. Superoxide radical and superoxide dismutases. Annu. Rev. Biochem. 1995, 64, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Fujii, J.; Homma, T.; Osaki, T. Superoxide Radicals in the Execution of Cell Death. Antioxidants 2022, 11, 501. [Google Scholar] [CrossRef] [PubMed]
- Valgimigli, L. Lipid Peroxidation and Antioxidant Protection. Biomolecules 2023, 13, 1291. [Google Scholar] [CrossRef] [PubMed]
- Scarpellini, C.; Klejborowska, G.; Lanthier, C.; Hassannia, B.; Vanden Berghe, T.; Augustyns, K. Beyond ferrostatin-1: A comprehensive review of ferroptosis inhibitors. Trends Pharmacol. Sci. 2023, 44, 902–916. [Google Scholar] [CrossRef] [PubMed]
- Mittler, R.; Darash-Yahana, M.; Sohn, Y.S.; Bai, F.; Song, L.; Cabantchik, I.; Jennings, P.A.; Onuchic, J.N.; Nechushtai, R. NEET Proteins: A New Link between Iron Metabolism, Reactive Oxygen Species, and Cancer. Antioxid. Redox Signal. 2019, 30, 1083–1095. [Google Scholar] [CrossRef] [PubMed]
- Dutt, S.; Hamza, I.; Bartnikas, T.B. Molecular Mechanisms of Iron and Heme Metabolism. Annu. Rev. Nutr. 2022, 42, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Philpott, C.C.; Patel, S.J.; Protchenko, O. Management versus miscues in the cytosolic labile iron Pool: The varied functions of iron chaperones. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118830. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Wang, T.; Li, Y.; Zhou, Y.; Wang, X.; Yu, X.; Ren, X.; An, Y.; Wu, Y.; Sun, W.; et al. DHA inhibits proliferation and induces ferroptosis of leukemia cells through autophagy dependent degradation of ferritin. Free Radic. Biol. Med. 2019, 131, 356–369. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Tsvetkov, A.S.; Shen, H.M.; Isidoro, C.; Ktistakis, N.T.; Linkermann, A.; Koopman, W.J.H.; Simon, H.U.; Galluzzi, L.; Luo, S.; et al. International consensus guidelines for the definition, detection, and interpretation of autophagy-dependent ferroptosis. Autophagy 2024, 20, 1213–1246. [Google Scholar] [CrossRef]
- Torii, S.; Shintoku, R.; Kubota, C.; Yaegashi, M.; Torii, R.; Sasaki, M.; Suzuki, T.; Mori, M.; Yoshimoto, Y.; Takeuchi, T.; et al. An essential role for functional lysosomes in ferroptosis of cancer cells. Biochem. J. 2016, 473, 769–777. [Google Scholar] [CrossRef]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J., 3rd; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef] [PubMed]
- Latunde-Dada, G.O. Ferroptosis: Role of lipid peroxidation, iron and ferritinophagy. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1893–1900. [Google Scholar] [CrossRef]
- Kaur, J.; Debnath, J. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. 2015, 16, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Galy, B.; Conrad, M.; Muckenthaler, M. Mechanisms controlling cellular and systemic iron homeostasis. Nat. Rev. Mol. Cell Biol. 2024, 25, 133–155. [Google Scholar] [CrossRef]
- Saito, Y. Diverse cytoprotective actions of vitamin E isoforms- role as peroxyl radical scavengers and complementary functions with selenoproteins. Free Radic. Biol. Med. 2021, 175, 121–129. [Google Scholar] [CrossRef]
- Wang, X.; Quinn, P.J. Vitamin E and its function in membranes. Prog. Lipid Res. 1999, 38, 309–336. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Geng, Y.; Lu, X.; Shi, Y.; Wu, G.; Zhang, M.; Shan, B.; Pan, H.; Yuan, J. Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc. Natl. Acad. Sci. USA 2019, 116, 2996–3005. [Google Scholar] [CrossRef]
- Yoshida, M.; Minagawa, S.; Araya, J.; Sakamoto, T.; Hara, H.; Tsubouchi, K.; Hosaka, Y.; Ichikawa, A.; Saito, N.; Kadota, T.; et al. Involvement of cigarette smoke-induced epithelial cell ferroptosis in COPD pathogenesis. Nat. Commun. 2019, 10, 3145. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhang, Y. Role of Mammalian DNA Methyltransferases in Development. Annu. Rev. Biochem. 2020, 89, 135–158. [Google Scholar] [CrossRef]
- Gryzik, M.; Asperti, M.; Denardo, A.; Arosio, P.; Poli, M. NCOA4-mediated ferritinophagy promotes ferroptosis induced by erastin, but not by RSL3 in HeLa cells. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118913. [Google Scholar] [CrossRef]
- Pryor, W.A.; Porter, N.A. Suggested mechanisms for the production of 4-hydroxy-2-nonenal from the autoxidation of polyunsaturated fatty acids. Free Radic. Biol. Med. 1990, 8, 541–543. [Google Scholar] [CrossRef] [PubMed]
- Uchida, K. 4-Hydroxy-2-nonenal: A product and mediator of oxidative stress. Prog. Lipid Res. 2003, 42, 318–343. [Google Scholar] [CrossRef]
- Tudek, B.; Zdżalik-Bielecka, D.; Tudek, A.; Kosicki, K.; Fabisiewicz, A.; Speina, E. Lipid peroxidation in face of DNA damage, DNA repair and other cellular processes. Free Radic. Biol. Med. 2017, 107, 77–89. [Google Scholar] [CrossRef]
- Eckl, P.M.; Bresgen, N. Genotoxicity of lipid oxidation compounds. Free Radic. Biol. Med. 2017, 111, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Jaganjac, M.; Milkovic, L.; Gegotek, A.; Cindric, M.; Zarkovic, K.; Skrzydlewska, E.; Zarkovic, N. The relevance of pathophysiological alterations in redox signaling of 4-hydroxynonenal for pharmacological therapies of major stress-associated diseases. Free Radic. Biol. Med. 2020, 157, 128–153. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; Zhong, H.; Xia, L.; Tao, Y.; Yin, H. Pathophysiology of mitochondrial lipid oxidation: Role of 4-hydroxynonenal (4-HNE) and other bioactive lipids in mitochondria. Free Radic. Biol. Med. 2017, 111, 316–327. [Google Scholar] [CrossRef]
- Zhang, X.; Hou, L.; Guo, Z.; Wang, G.; Xu, J.; Zheng, Z.; Sun, K.; Guo, F. Lipid peroxidation in osteoarthritis: Focusing on 4-hydroxynonenal, malondialdehyde, and ferroptosis. Cell Death Discov. 2023, 9, 320. [Google Scholar] [CrossRef]
- Fujii, J.; Yamada, K.I. Defense systems to avoid ferroptosis caused by lipid peroxidation-mediated membrane damage. Free Radic. Res. 2023, 57, 353–372. [Google Scholar] [CrossRef]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef]
- Magtanong, L.; Ko, P.J.; To, M.; Cao, J.Y.; Forcina, G.C.; Tarangelo, A.; Ward, C.C.; Cho, K.; Patti, G.J.; Nomura, D.K.; et al. Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem. Biol. 2019, 26, 420–432.e9. [Google Scholar] [CrossRef]
- Ma, M.; Kong, P.; Huang, Y.; Wang, J.; Liu, X.; Hu, Y.; Chen, X.; Du, C.; Yang, H. Activation of MAT2A-ACSL3 pathway protects cells from ferroptosis in gastric cancer. Free Radic. Biol. Med. 2022, 181, 288–299. [Google Scholar] [CrossRef]
- Qiu, B.; Zandkarimi, F.; Bezjian, C.T.; Reznik, E.; Soni, R.K.; Gu, W.; Jiang, X.; Stockwell, B.R. Phospholipids with two polyunsaturated fatty acyl tails promote ferroptosis. Cell 2024, 187, 1177–1190.e18. [Google Scholar] [CrossRef]
- Petan, T.; Manček-Keber, M. Half is enough: Oxidized lysophospholipids as novel bioactive molecules. Free Radic. Biol. Med. 2022, 188, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Beharier, O.; Tyurin, V.A.; Goff, J.P.; Guerrero-Santoro, J.; Kajiwara, K.; Chu, T.; Tyurina, Y.Y.; St Croix, C.M.; Wallace, C.T.; Parry, S.; et al. PLA2G6 guards placental trophoblasts against ferroptotic injury. Proc. Natl. Acad. Sci. USA 2020, 117, 27319–27328. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Xu, J.; Liu, Y.; Lin, Y.; Wang, F.; Han, Y.; Zhang, S.; Gao, X.; Xu, C.; Yuan, H. Exogenous spermidine alleviates diabetic cardiomyopathy via suppressing reactive oxygen species, endoplasmic reticulum stress, and Pannexin-1-mediated ferroptosis. Biomol. Biomed. 2023, 23, 825–837. [Google Scholar] [CrossRef]
- Chen, D.; Chu, B.; Yang, X.; Liu, Z.; Jin, Y.; Kon, N.; Rabadan, R.; Jiang, X.; Stockwell, B.R.; Gu, W. iPLA2beta-mediated lipid detoxification controls p53-driven ferroptosis independent of GPX4. Nat. Commun. 2021, 12, 3644. [Google Scholar] [CrossRef]
- Scuderi, M.R.; Anfuso, C.D.; Lupo, G.; Motta, C.; Romeo, L.; Guerra, L.; Cappellani, A.; Ragusa, N.; Cantarella, G.; Alberghina, M. Expression of Ca2+-independent and Ca2+-dependent phospholipases A2 and cyclooxygenases in human melanocytes and malignant melanoma cell lines. Biochim. Biophys. Acta 2008, 1781, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Duffy, D.L.; Zhu, G.; Li, X.; Sanna, M.; Iles, M.M.; Jacobs, L.C.; Evans, D.M.; Yazar, S.; Beesley, J.; Law, M.H.; et al. Novel pleiotropic risk loci for melanoma and nevus density implicate multiple biological pathways. Nat. Commun. 2018, 9, 4774. [Google Scholar] [CrossRef]
- Fujita, H.; Tanaka, Y.K.; Ogata, S.; Suzuki, N.; Kuno, S.; Barayeu, U.; Akaike, T.; Ogra, Y.; Iwai, K. PRDX6 augments selenium utilization to limit iron toxicity and ferroptosis. Nat. Struct. Mol. Biol. 2024, 1–9. [Google Scholar] [CrossRef]
- Chen, Z.; Inague, A.; Kaushal, K.; Fazeli, G.N.; Xavier da Silva, T.; Ferreira Dos Santos, A.; Cheytan, T.; Porto Freitas, F.; Yildiz, U.; Gasparello Viviani, L.; et al. PRDX6 contributes to selenocysteine metabolism and ferroptosis resistance. bioRxiv. 2024. preprint. [Google Scholar] [CrossRef]
- Hirata, Y.; Cai, R.; Volchuk, A.; Steinberg, B.E.; Saito, Y.; Matsuzawa, A.; Grinstein, S.; Freeman, S.A. Lipid peroxidation increases membrane tension, Piezo1 gating, and cation permeability to execute ferroptosis. Curr. Biol. 2023, 33, 1282–1294.e5. [Google Scholar] [CrossRef]
- Hirata, Y.; Mishima, E. Membrane Dynamics and Cation Handling in Ferroptosis. Physiology 2024, 39, 73–87. [Google Scholar] [CrossRef]
- Phillips, R.; Ursell, T.; Wiggins, P.; Sens, P. Emerging roles for lipids in shaping membrane-protein function. Nature 2009, 459, 379–385. [Google Scholar] [CrossRef]
- Levental, I.; Lyman, E. Regulation of membrane protein structure and function by their lipid nano-environment. Nat. Rev. Mol. Cell Biol. 2023, 24, 107–122. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Imai, H.; Matsuoka, M.; Kumagai, T.; Sakamoto, T.; Koumura, T. Lipid Peroxidation-Dependent Cell Death Regulated by GPx4 and Ferroptosis. Curr. Top. Microbiol. Immunol. 2017, 403, 143–170. [Google Scholar] [CrossRef]
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Porto Freitas, F.; Seibt, T.; et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 2018, 172, 409–422.e21. [Google Scholar] [CrossRef]
- Imai, H.; Hirao, F.; Sakamoto, T.; Sekine, K.; Mizukura, Y.; Saito, M.; Kitamoto, T.; Hayasaka, M.; Hanaoka, K.; Nakagawa, Y. Early embryonic lethality caused by targeted disruption of the mouse PHGPx gene. Biochem. Biophys. Res. Commun. 2003, 305, 278–286. [Google Scholar] [CrossRef]
- Skouta, R.; Dixon, S.J.; Wang, J.; Dunn, D.E.; Orman, M.; Shimada, K.; Rosenberg, P.A.; Lo, D.C.; Weinberg, J.M.; Linkermann, A.; et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am. Chem. Soc. 2014, 136, 4551–4556. [Google Scholar] [CrossRef]
- Zilka, O.; Shah, R.; Li, B.; Friedmann Angeli, J.P.; Griesser, M.; Conrad, M.; Pratt, D.A. On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Cent. Sci. 2017, 3, 232–243. [Google Scholar] [CrossRef]
- McBean, G.J. The transsulfuration pathway: A source of cysteine for glutathione in astrocytes. Amino Acids 2012, 42, 199–205. [Google Scholar] [CrossRef]
- Zhang, H.F.; Klein Geltink, R.I.; Parker, S.J.; Sorensen, P.H. Transsulfuration, minor player or crucial for cysteine homeostasis in cancer. Trends Cell Biol. 2022, 32, 800–814. [Google Scholar] [CrossRef]
- Ikeda, Y.; Fujii, J. The Emerging Roles of γ-Glutamyl Peptides Produced by γ-Glutamyltransferase and the Glutathione Synthesis System. Cells 2023, 12, 2831. [Google Scholar] [CrossRef]
- Ishii, T.; Mann, G.E. Redox status in mammalian cells and stem cells during culture in vitro: Critical roles of Nrf2 and cystine transporter activity in the maintenance of redox balance. Redox Biol. 2014, 2, 786–794. [Google Scholar] [CrossRef]
- Chen, D.; Fan, Z.; Rauh, M.; Buchfelder, M.; Eyupoglu, I.Y.; Savaskan, N. ATF4 promotes angiogenesis and neuronal cell death and confers ferroptosis in a xCT-dependent manner. Oncogene 2017, 36, 5593–5608. [Google Scholar] [CrossRef]
- Sato, H.; Shiiya, A.; Kimata, M.; Maebara, K.; Tamba, M.; Sakakura, Y.; Makino, N.; Sugiyama, F.; Yagami, K.; Moriguchi, T.; et al. Redox imbalance in cystine/glutamate transporter-deficient mice. J. Biol. Chem. 2005, 280, 37423–37429. [Google Scholar] [CrossRef]
- Lee, J.; Kang, E.S.; Kobayashi, S.; Homma, T.; Sato, H.; Seo, H.G.; Fujii, J. The viability of primary hepatocytes is maintained under a low cysteine-glutathione redox state with a marked elevation in ophthalmic acid production. Exp. Cell Res. 2017, 361, 178–191. [Google Scholar] [CrossRef]
- Upadhyayula, P.S.; Higgins, D.M.; Mela, A.; Banu, M.; Dovas, A.; Zandkarimi, F.; Patel, P.; Mahajan, A.; Humala, N.; Nguyen, T.T.T.; et al. Dietary restriction of cysteine and methionine sensitizes gliomas to ferroptosis and induces alterations in energetic metabolism. Nat. Commun. 2023, 14, 1187. [Google Scholar] [CrossRef]
- Hedley, D.W.; McCulloch, E.A.; Minden, M.D.; Chow, S.; Curtis, J. Antileukemic action of buthionine sulfoximine: Evidence for an intrinsic death mechanism based on oxidative stress. Leukemia 1998, 12, 1545–1552. [Google Scholar] [CrossRef]
- Anderson, C.P.; Tsai, J.M.; Meek, W.E.; Liu, R.M.; Tang, Y.; Forman, H.J.; Reynolds, C.P. Depletion of glutathione by buthionine sulfoxine is cytotoxic for human neuroblastoma cell lines via apoptosis. Exp. Cell Res. 1999, 246, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Harris, I.S.; Endress, J.E.; Coloff, J.L.; Selfors, L.M.; McBrayer, S.K.; Rosenbluth, J.M.; Takahashi, N.; Dhakal, S.; Koduri, V.; Oser, M.G.; et al. Deubiquitinases Maintain Protein Homeostasis and Survival of Cancer Cells upon Glutathione Depletion. Cell Metab. 2019, 29, 1166–1181.e6. [Google Scholar] [CrossRef]
- Homma, T.; Kobayashi, S.; Fujii, J. Cysteine preservation confers resistance to glutathione-depleted cells against ferroptosis via CDGSH iron sulphur domain-containing proteins (CISDs). Free Radic. Res. 2020, 54, 397–407. [Google Scholar] [CrossRef]
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 864–868. [Google Scholar] [CrossRef]
- Bachhawat, A.K.; Kaur, A. Glutathione Degradation. Antioxid. Redox Signal. 2017, 27, 1200–1216. [Google Scholar] [CrossRef]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 2014, 3, e02523. [Google Scholar] [CrossRef]
- Xue, Y.; Lu, F.; Chang, Z.; Li, J.; Gao, Y.; Zhou, J.; Luo, Y.; Lai, Y.; Cao, S.; Li, X.; et al. Intermittent dietary methionine deprivation facilitates tumoral ferroptosis and synergizes with checkpoint blockade. Nat. Commun. 2023, 14, 4758. [Google Scholar] [CrossRef]
- Li, J.; Lu, M.; Ahn, Y.; Cao, K.; Pinkus, C.A.; Stansfield, J.C.; Wu, Z.; Zhang, B.B. CHAC1 inactivation is effective to preserve muscle glutathione but is insufficient to protect against muscle wasting in cachexia. PLoS ONE 2023, 18, e0283806. [Google Scholar] [CrossRef]
- Fang, X.; Ardehali, H.; Min, J.; Wang, F. The molecular and metabolic landscape of iron and ferroptosis in cardiovascular disease. Nat. Rev. Cardiol. 2023, 20, 7–23. [Google Scholar] [CrossRef]
- Lei, G.; Zhuang, L.; Gan, B. The roles of ferroptosis in cancer: Tumor suppression, tumor microenvironment, and therapeutic interventions. Cancer Cell. 2024, 42, 513–534. [Google Scholar] [CrossRef]
- Escuder-Rodríguez, J.J.; Liang, D.; Jiang, X.; Sinicrope, F.A. Ferroptosis: Biology and Role in Gastrointestinal Disease. Gastroenterology 2024, 167, 231–249. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.W.; Amante, J.J.; Goel, H.L.; Mercurio, A.M. The α6β4 integrin promotes resistance to ferroptosis. J. Cell Biol. 2017, 216, 4287–4297. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.W.; Amante, J.J.; Mercurio, A.M. Cell clustering mediated by the adhesion protein PVRL4 is necessary for alpha6beta4 integrin-promoted ferroptosis resistance in matrix-detached cells. J. Biol. Chem. 2018, 293, 12741–12748. [Google Scholar] [CrossRef] [PubMed]
- Havens, C.G.; Ho, A.; Yoshioka, N.; Dowdy, S.F. Regulation of late G1/S phase transition and APC Cdh1 by reactive oxygen species. Mol. Cell. Biol. 2006, 26, 4701–4711. [Google Scholar] [CrossRef] [PubMed]
- Mitra, K.; Wunder, C.; Roysam, B.; Lin, G.; Lippincott-Schwartz, J. A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc. Natl. Acad. Sci. USA 2009, 106, 11960–11965. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.H.; Finkel, T. Metabolic regulation of the cell cycle. Curr. Opin. Cell Biol. 2013, 25, 724–729. [Google Scholar] [CrossRef] [PubMed]
- Patterson, J.C.; Joughin, B.A.; van de Kooij, B.; Lim, D.C.; Lauffenburger, D.A.; Yaffe, M.B. ROS and Oxidative Stress Are Elevated in Mitosis during Asynchronous Cell Cycle Progression and Are Exacerbated by Mitotic Arrest. Cell Syst. 2019, 8, 163–167.e2. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.F.; Dong, Q.; Bai, Y.; Yuan, J.; Xu, Q.; Cao, C.; Liu, X. Oxidative stress induces mitotic arrest by inhibiting Aurora A-involved mitotic spindle formation. Free Radic. Biol. Med. 2017, 103, 177–187. [Google Scholar] [CrossRef]
- Odle, R.I.; Walker, S.A.; Oxley, D.; Kidger, A.M.; Balmanno, K.; Gilley, R.; Okkenhaug, H.; Florey, O.; Ktistakis, N.T.; Cook, S.J. An mTORC1-to-CDK1 Switch Maintains Autophagy Suppression during Mitosis. Mol. Cell 2020, 77, 228–240.e7. [Google Scholar] [CrossRef]
- Nowosad, A.; Besson, A. Lysosomes at the Crossroads of Cell Metabolism, Cell Cycle, and Stemness. Int. J. Mol. Sci. 2022, 23, 2290. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez Sawicki, L.; Garcia, K.A.; Corsico, B.; Scaglia, N. De novo lipogenesis at the mitotic exit is used for nuclear envelope reassembly/expansion. Implications for combined chemotherapy. Cell Cycle 2019, 18, 1646–1659. [Google Scholar] [CrossRef] [PubMed]
- Scaglia, N.; Tyekucheva, S.; Zadra, G.; Photopoulos, C.; Loda, M. De novo fatty acid synthesis at the mitotic exit is required to complete cellular division. Cell Cycle 2014, 13, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Storck, E.M.; Özbalci, C.; Eggert, U.S. Lipid Cell Biology: A Focus on Lipids in Cell Division. Annu. Rev. Biochem. 2018, 87, 839–869. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, P. Methionine Dependence of Cancer. Biomolecules 2020, 10, 568. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. The magic of a methyl group: Biochemistry at the service of medicine. Biochem. Pharmacol. 2023, 216, 115786. [Google Scholar] [CrossRef]
- Homma, T.; Kobayashi, S.; Fujii, J. Methionine Deprivation Reveals the Pivotal Roles of Cell Cycle Progression in Ferroptosis That Is Induced by Cysteine Starvation. Cells 2022, 11, 1603. [Google Scholar] [CrossRef] [PubMed]
- Rodencal, J.; Kim, N.; He, A.; Li, V.L.; Lange, M.; He, J.; Tarangelo, A.; Schafer, Z.T.; Olzmann, J.A.; Long, J.Z.; et al. Sensitization of cancer cells to ferroptosis coincident with cell cycle arrest. Cell Chem. Biol. 2024, 31, 234–248.e13. [Google Scholar] [CrossRef]
- Imai, H.; Nakagawa, Y. Biological significance of phospholipid hydroperoxide glutathione peroxidase (PHGPx, GPx4) in mammalian cells. Free Radic. Biol. Med. 2003, 34, 145–169. [Google Scholar] [CrossRef]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of Mitochondria in Ferroptosis. Mol. Cell 2019, 73, 354–363.e3. [Google Scholar] [CrossRef] [PubMed]
- Griffith, O.W. Biologic and pharmacologic regulation of mammalian glutathione synthesis. Free Radic. Biol. Med. 1999, 27, 922–935. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Ikeda, Y.; Shigeno, Y.; Konno, H.; Fujii, J. γ-Glutamylcysteine synthetase and γ-glutamyl transferase as differential enzymatic sources of γ-glutamylpeptides in mice. Amino Acids 2020, 52, 555–566. [Google Scholar] [CrossRef]
- Soga, T.; Baran, R.; Suematsu, M.; Ueno, Y.; Ikeda, S.; Sakurakawa, T.; Kakazu, Y.; Ishikawa, T.; Robert, M.; Nishioka, T.; et al. Differential metabolomics reveals ophthalmic acid as an oxidative stress biomarker indicating hepatic glutathione consumption. J. Biol. Chem. 2006, 281, 16768–16776. [Google Scholar] [CrossRef] [PubMed]
- Yamada, N.; Karasawa, T.; Kimura, H.; Watanabe, S.; Komada, T.; Kamata, R.; Sampilvanjil, A.; Ito, J.; Nakagawa, K.; Kuwata, H.; et al. Ferroptosis driven by radical oxidation of n-6 polyunsaturated fatty acids mediates acetaminophen-induced acute liver failure. Cell Death Dis. 2020, 11, 144. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, Y.; Takahashi, M.; Sugiura, Y.; Izumi, Y.; Nishiyama, K.; Nishida, M.; Suematsu, M.; Bamba, T.; Yamada, K.I. Structural library and visualization of endogenously oxidized phosphatidylcholines using mass spectrometry-based techniques. Nat. Commun. 2021, 12, 6339. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.P.; Mockabee-Macias, A.; Jiang, C.; Falzone, A.; Prieto-Farigua, N.; Stone, E.; Harris, I.S.; DeNicola, G.M. Non-canonical Glutamate-Cysteine Ligase Activity Protects against Ferroptosis. Cell Metab. 2021, 33, 174–189.e7. [Google Scholar] [CrossRef] [PubMed]
- Soga, T.; Sugimoto, M.; Honma, M.; Mori, M.; Igarashi, K.; Kashikura, K.; Ikeda, S.; Hirayama, A.; Yamamoto, T.; Yoshida, H.; et al. Serum metabolomics reveals γ-glutamyl dipeptides as biomarkers for discrimination among different forms of liver disease. J. Hepatol. 2011, 55, 896–905. [Google Scholar] [CrossRef]
- Chen, M.S.; Wang, S.F.; Hsu, C.Y.; Yin, P.H.; Yeh, T.S.; Lee, H.C.; Tseng, L.M. CHAC1 degradation of glutathione enhances cystine-starvation-induced necroptosis and ferroptosis in human triple negative breast cancer cells via the GCN2-eIF2α-ATF4 pathway. Oncotarget 2017, 8, 114588–114602. [Google Scholar] [CrossRef]
- Gaschler, M.M.; Hu, F.; Feng, H.; Linkermann, A.; Min, W.; Stockwell, B.R. Determination of the Subcellular Localization and Mechanism of Action of Ferrostatins in Suppressing Ferroptosis. ACS Chem. Biol. 2018, 13, 1013–1020. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.J.; Ikeda, M.; Ide, T.; Hur, K.Y.; Lee, M.S. Mitochondrial event as an ultimate step in ferroptosis. Cell Death Discov. 2022, 8, 414. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Du, Y.; Zhou, Y.; Chen, Q.; Luo, Z.; Ren, Y.; Chen, X.; Chen, G. Iron and copper: Critical executioners of ferroptosis, cuproptosis and other forms of cell death. Cell Commun. Signal. 2023, 21, 327. [Google Scholar] [CrossRef] [PubMed]
- Read, A.D.; Bentley, R.E.; Archer, S.L.; Dunham-Snary, K.J. Mitochondrial iron-sulfur clusters: Structure, function, and an emerging role in vascular biology. Redox Biol. 2021, 47, 102164. [Google Scholar] [CrossRef] [PubMed]
- Selvanathan, A.; Parayil Sankaran, B. Mitochondrial iron-sulfur cluster biogenesis and neurological disorders. Mitochondrion 2022, 62, 41–49. [Google Scholar] [CrossRef]
- Kuban, R.J.; Wiesner, R.; Rathman, J.; Veldink, G.; Nolting, H.; Solé, V.A.; Kühn, H. The iron ligand sphere geometry of mammalian 15-lipoxygenases. Biochem. J. 1998, 332, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.; Kuhn, H.; Heydeck, D. Structural and functional biology of arachidonic acid 15-lipoxygenase-1 (ALOX15). Gene 2015, 573, 1–32. [Google Scholar] [CrossRef]
- Rouault, T.A. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat. Chem. Biol. 2006, 2, 406–414. [Google Scholar] [CrossRef]
- Levi, S.; Ripamonti, M.; Dardi, M.; Cozzi, A.; Santambrogio, P. Mitochondrial Ferritin: Its Role in Physiological and Pathological Conditions. Cells 2021, 10, 1969. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Q.; Chang, S.Y.; Wu, Q.; Gou, Y.J.; Jia, L.; Cui, Y.M.; Yu, P.; Shi, Z.H.; Wu, W.S.; Gao, G.; et al. The Protective Role of Mitochondrial Ferritin on Erastin-Induced Ferroptosis. Front. Aging Neurosci. 2016, 8, 308. [Google Scholar] [CrossRef]
- Wang, P.; Cui, Y.; Ren, Q.; Yan, B.; Zhao, Y.; Yu, P.; Gao, G.; Shi, H.; Chang, S.; Chang, Y.Z. Mitochondrial ferritin attenuates cerebral ischaemia/reperfusion injury by inhibiting ferroptosis. Cell Death Dis. 2021, 12, 447. [Google Scholar] [CrossRef] [PubMed]
- Na, Y.R.; Je, S.; Seok, S.H. Metabolic features of macrophages in inflammatory diseases and cancer. Cancer Lett. 2018, 413, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.D.; Diotallevi, M.; Nicol, T.; McNeill, E.; Shaw, A.; Chuaiphichai, S.; Hale, A.; Starr, A.; Nandi, M.; Stylianou, E.; et al. Nitric Oxide Modulates Metabolic Remodeling in Inflammatory Macrophages through TCA Cycle Regulation and Itaconate Accumulation. Cell Rep. 2019, 28, 218–230.e7. [Google Scholar] [CrossRef]
- Castro, L.; Tórtora, V.; Mansilla, S.; Radi, R. Aconitases: Non-redox Iron-Sulfur Proteins Sensitive to Reactive Species. Accounts Chem. Res. 2019, 52, 2609–2619. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, E.M.; Gonzalez-Cotto, M.; Baseler, W.A.; Davies, L.C.; Ghesquière, B.; Maio, N.; Rice, C.M.; Rouault, T.A.; Cassel, T.; Higashi, R.M.; et al. Nitric oxide orchestrates metabolic rewiring in M1 macrophages by targeting aconitase 2 and pyruvate dehydrogenase. Nat. Commun. 2020, 11, 698. [Google Scholar] [CrossRef] [PubMed]
- Jastroch, M.; Divakaruni, A.S.; Mookerjee, S.; Treberg, J.R.; Brand, M.D. Mitochondrial proton and electron leaks. Essays Biochem. 2010, 47, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, R.J. Teaching the fundamentals of electron transfer reactions in mitochondria and the production and detection of reactive oxygen species. Redox Biol. 2015, 4, 381–398. [Google Scholar] [CrossRef] [PubMed]
- Gardner, P.R.; Nguyen, D.D.; White, C.W. Aconitase is a sensitive and critical target of oxygen poisoning in cultured mammalian cells and in rat lungs. Proc. Natl. Acad. Sci. USA 1994, 91, 12248–12252. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.J.; Artyomov, M.N. Itaconate: The poster child of metabolic reprogramming in macrophage function. Nat. Rev. Immunol. 2019, 19, 273–281. [Google Scholar] [CrossRef]
- Marelja, Z.; Leimkühler, S.; Missirlis, F. Iron Sulfur and Molybdenum Cofactor Enzymes Regulate the Drosophila Life Cycle by Controlling Cell Metabolism. Front. Physiol. 2018, 9, 50. [Google Scholar] [CrossRef]
- Cao, K.; Xu, J.; Cao, W.; Wang, X.; Lv, W.; Zeng, M.; Zou, X.; Liu, J.; Feng, Z. Assembly of mitochondrial succinate dehydrogenase in human health and disease. Free Radic. Biol. Med. 2023, 207, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Haschka, D.; Hoffmann, A.; Weiss, G. Iron in immune cell function and host defense. Semin. Cell Dev. Biol. 2021, 115, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Baik, A.H.; Haribowo, A.G.; Chen, X.; Queliconi, B.B.; Barrios, A.M.; Garg, A.; Maishan, M.; Campos, A.R.; Matthay, M.A.; Jain, I.H. Oxygen toxicity causes cyclic damage by destabilizing specific Fe-S cluster-containing protein complexes. Mol. Cell 2023, 83, 942–960.e9. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, S.W.; Sviderskiy, V.O.; Terzi, E.M.; Papagiannakopoulos, T.; Moreira, A.L.; Adams, S.; Sabatini, D.M.; Birsoy, K.; Possemato, R. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature 2017, 551, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Zhou, Y.; Li, Y.; Xia, J.; Chen, Y.; Chen, S.; Wang, X.; Sun, W.; Wang, T.; Ren, X.; et al. Identification of Frataxin as a regulator of ferroptosis. Redox Biol. 2020, 32, 101483. [Google Scholar] [CrossRef] [PubMed]
- Nechushtai, R.; Rowland, L.; Karmi, O.; Marjault, H.B.; Nguyen, T.T.; Mittal, S.; Ahmed, R.S.; Grant, D.; Manrique-Acevedo, C.; Morcos, F.; et al. CISD3/MiNT is required for complex I function, mitochondrial integrity, and skeletal muscle maintenance. Proc. Natl. Acad. Sci. USA 2024, 121, e2405123121. [Google Scholar] [CrossRef] [PubMed]
- Ryall, J.; Nguyen, M.; Bendayan, M.; Shore, G.C. Expression of nuclear genes encoding the urea cycle enzymes, carbamoyl-phosphate synthetase I and ornithine carbamoyl transferase, in rat liver and intestinal mucosa. Eur. J. Biochem. 1985, 152, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, R.W.; Rodriguez, P.C.; Toque, H.A.; Narayanan, S.P.; Caldwell, R.B. Arginase: A Multifaceted Enzyme Important in Health and Disease. Physiol. Rev. 2018, 98, 641–665. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Dai, J.; Zhang, Z.; Yu, H.; Zhang, J.; Zhu, X.; Qin, Y.; Zhang, L.; Zhang, P. ASS1-Mediated Reductive Carboxylation of Cytosolic Glutamine Confers Ferroptosis Resistance in Cancer Cells. Cancer Res. 2023, 83, 1646–1665. [Google Scholar] [CrossRef]
- Bae, D.H.; Lane, D.J.R.; Jansson, P.J.; Richardson, D.R. The old and new biochemistry of polyamines. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 2053–2068. [Google Scholar] [CrossRef]
- Latour, Y.L.; Gobert, A.P.; Wilson, K.T. The role of polyamines in the regulation of macrophage polarization and function. Amino Acids 2020, 52, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Chia, T.Y.; Zolp, A.; Miska, J. Polyamine Immunometabolism: Central Regulators of Inflammation, Cancer and Autoimmunity. Cells 2022, 11, 896. [Google Scholar] [CrossRef]
- Holbert, C.E.; Cullen, M.T.; Casero, R.A., Jr.; Stewart, T.M. Polyamines in cancer: Integrating organismal metabolism and antitumour immunity. Nat. Rev. Cancer 2022, 22, 467–480. [Google Scholar] [CrossRef]
- Cheng, Q.; Ni, L.; Liu, A.; Huang, X.; Xiang, P.; Zhang, Q.; Yang, H. Spermidine protects cartilage from IL-1beta-mediated ferroptosis. Mol. Cell. Biochem. 2023, 1–10. [Google Scholar] [CrossRef]
- Youssef, M.A.M.; Mohamed, T.M.; Bakry, A.A.; El-Keiy, M.M. Synergistic effect of spermidine and ciprofloxacin against Alzheimer’s disease in male rat via ferroptosis modulation. Int. J. Biol. Macromol. 2024, 263, 130387. [Google Scholar] [CrossRef]
- Casero, R.A.; Pegg, A.E. Polyamine catabolism and disease. Biochem. J. 2009, 421, 323–338. [Google Scholar] [CrossRef] [PubMed]
- Murray Stewart, T.; Dunston, T.T.; Woster, P.M.; Casero, R.A., Jr. Polyamine catabolism and oxidative damage. J. Biol. Chem. 2018, 293, 18736–18745. [Google Scholar] [CrossRef]
- Niu, C.; Jiang, D.; Guo, Y.; Wang, Z.; Sun, Q.; Wang, X.; Ling, W.; An, X.; Ji, C.; Li, S.; et al. Spermidine suppresses oxidative stress and ferroptosis by Nrf2/HO-1/GPX4 and Akt/FHC/ACSL4 pathway to alleviate ovarian damage. Life Sci. 2023, 332, 122109. [Google Scholar] [CrossRef]
- Ou, Y.; Wang, S.J.; Li, D.; Chu, B.; Gu, W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc. Natl. Acad. Sci. USA 2016, 113, E6806–E6812. [Google Scholar] [CrossRef] [PubMed]
- Wan, K.; Jia, M.; Zhang, H.; Lan, Y.; Wang, S.; Zhang, K.; Wang, Z.; Zhu, H.; Zheng, X.; Luo, Y.; et al. Electroacupuncture Alleviates Neuropathic Pain by Suppressing Ferroptosis in Dorsal Root Ganglion via SAT1/ALOX15 Signaling. Mol. Neurobiol. 2023, 60, 6121–6132. [Google Scholar] [CrossRef]
- Wei, W.; Lu, Y.; Hu, Q.; Yin, J.; Wang, Y.; Zhang, H.; Zhao, Q.; Liu, L. Synergistic antitumor efficacy of gemcitabine and cisplatin to induce ferroptosis in pancreatic ductal adenocarcinoma via Sp1-SAT1-polyamine metabolism pathway. Cell. Oncol. 2024, 47, 321–341. [Google Scholar] [CrossRef] [PubMed]
- Bi, G.; Liang, J.; Bian, Y.; Shan, G.; Huang, Y.; Lu, T.; Zhang, H.; Jin, X.; Chen, Z.; Zhao, M.; et al. Polyamine-mediated ferroptosis amplification acts as a targetable vulnerability in cancer. Nat. Commun. 2024, 15, 2461. [Google Scholar] [CrossRef] [PubMed]
- Ba, T.; Zhao, D.; Chen, Y.; Zeng, C.; Zhang, C.; Niu, S.; Dai, H. L-Citrulline Supplementation Restrains Ferritinophagy-Mediated Ferroptosis to Alleviate Iron Overload-Induced Thymus Oxidative Damage and Immune Dysfunction. Nutrients 2022, 14, 4549. [Google Scholar] [CrossRef] [PubMed]
- Buettner, G.R. The pecking order of free radicals and antioxidants: Lipid peroxidation, alpha-tocopherol, and ascorbate. Arch. Biochem. Biophys. 1993, 300, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Che, Q.; Espey, M.G.; Sun, A.Y.; Pooput, C.; Kirk, K.L.; Krishna, M.C.; Khosh, D.B.; Drisko, J.; Levine, M. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc. Natl. Acad. Sci. USA 2008, 105, 11105–11109. [Google Scholar] [CrossRef]
- Deshwal, S.; Onishi, M.; Tatsuta, T.; Bartsch, T.; Cors, E.; Ried, K.; Lemke, K.; Nolte, H.; Giavalisco, P.; Langer, T. Mitochondria regulate intracellular coenzyme Q transport and ferroptotic resistance via STARD7. Nat. Cell Biol. 2023, 25, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Mishima, E.; Ito, J.; Wu, Z.; Nakamura, T.; Wahida, A.; Doll, S.; Tonnus, W.; Nepachalovich, P.; Eggenhofer, E.; Aldrovandi, M.; et al. A non-canonical vitamin K cycle is a potent ferroptosis suppressor. Nature 2022, 608, 778–783. [Google Scholar] [CrossRef]
- Kraft, V.A.N.; Bezjian, C.T.; Pfeiffer, S.; Ringelstetter, L.; Müller, C.; Zandkarimi, F.; Merl-Pham, J.; Bao, X.; Anastasov, N.; Kössl, J.; et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent. Sci. 2020, 6, 41–53. [Google Scholar] [CrossRef]
- Freitas, F.P.; Alborzinia, H.; Dos Santos, A.F.; Nepachalovich, P.; Pedrera, L.; Zilka, O.; Inague, A.; Klein, C.; Aroua, N.; Kaushal, K.; et al. 7-Dehydrocholesterol is an endogenous suppressor of ferroptosis. Nature 2024, 626, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ran, Q.; Duan, Q.; Jin, J.; Wang, Y.; Yu, L.; Wang, C.; Zhu, Z.; Chen, X.; Weng, L.; et al. 7-Dehydrocholesterol dictates ferroptosis sensitivity. Nature 2024, 626, 411–418. [Google Scholar] [CrossRef]
- Yamada, N.; Karasawa, T.; Ito, J.; Yamamuro, D.; Morimoto, K.; Nakamura, T.; Komada, T.; Baatarjav, C.; Saimoto, Y.; Jinnouchi, Y.; et al. Inhibition of 7-dehydrocholesterol reductase prevents hepatic ferroptosis under an active state of sterol synthesis. Nat. Commun. 2024, 15, 2195. [Google Scholar] [CrossRef] [PubMed]
- TeSlaa, T.; Ralser, M.; Fan, J.; Rabinowitz, J.D. The pentose phosphate pathway in health and disease. Nat. Metab. 2023, 5, 1275–1289. [Google Scholar] [CrossRef]
- Brewer, A.C.; Mustafi, S.B.; Murray, T.V.; Rajasekaran, N.S.; Benjamin, I.J. Reductive stress linked to small HSPs, G6PD, and Nrf2 pathways in heart disease. Antioxid. Redox Signal. 2013, 18, 1114–1127. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef]
- Stoyanovsky, D.A.; Tyurina, Y.Y.; Shrivastava, I.; Bahar, I.; Tyurin, V.A.; Protchenko, O.; Jadhav, S.; Bolevich, S.B.; Kozlov, A.V.; Vladimirov, Y.A.; et al. Iron catalysis of lipid peroxidation in ferroptosis: Regulated enzymatic or random free radical reaction? Free Radic. Biol. Med. 2019, 133, 153–161. [Google Scholar] [CrossRef]
- Song, S.; Su, Z.; Kon, N.; Chu, B.; Li, H.; Jiang, X.; Luo, J.; Stockwell, B.R.; Gu, W. ALOX5-mediated ferroptosis acts as a distinct cell death pathway upon oxidative stress in Huntington’s disease. Genes Dev. 2023, 37, 204–217. [Google Scholar] [CrossRef]
- Wiesner, R.; Suzuki, H.; Walther, M.; Yamamoto, S.; Kuhnm, H. Suicidal inactivation of the rabbit 15-lipoxygenase by 15S-HpETE is paralleled by covalent modification of active site peptides. Free Radic. Biol. Med. 2003, 34, 304–315. [Google Scholar] [CrossRef]
- Kishimoto, K.; Nakamura, M.; Suzuki, H.; Yoshimoto, T.; Yamamoto, S.; Takao, T.; Shimonishi, Y.; Tanabe, T. Suicide inactivation of porcine leukocyte 12-lipoxygenase associated with its incorporation of 15-hydroperoxy-5,8,11,13-eicosatetraenoic acid derivative. Biochim. Biophys. Acta 1996, 1300, 56–62. [Google Scholar] [CrossRef]
- Kapralov, A.A.; Yang, Q.; Dar, H.H.; Tyurina, Y.Y.; Anthonymuthu, T.S.; Kim, R.; St Croix, C.M.; Mikulska-Ruminska, K.; Liu, B.; Shrivastava, I.H. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat. Chem. Biol. 2020, 16, 278–290. [Google Scholar] [CrossRef]
- Percival, M.D.; Denis, D.; Riendeau, D.; Gresser, M.J. Investigation of the mechanism of non-turnover-dependent inactivation of purified human 5-lipoxygenase. Inactivation by H2O2 and inhibition by metal ions. Eur. J. Biochem. 1992, 210, 109–117. [Google Scholar] [CrossRef]
- Nagatani, K.; Abe, Y.; Homma, T.; Fujii, J.; Szuki, T. Copper chelation by d-penicillamine alleviates melanocyte death induced by rhododendrol without inhibiting tyrosinase. Biochem. Biophys. Res. Commun. 2023, 663, 71–77. [Google Scholar] [CrossRef]
- Rouzer, C.A.; Marnett, L.J. Structural and Chemical Biology of the Interaction of Cyclooxygenase with Substrates and Non-Steroidal Anti-Inflammatory Drugs. Chem. Rev. 2020, 120, 7592–7641. [Google Scholar] [CrossRef]
- Iyanagi, T. Molecular mechanism of metabolic NAD(P)H-dependent electron-transfer systems: The role of redox cofactors. Biochim. Biophys. Acta Bioenerg. 2019, 1860, 233–258. [Google Scholar] [CrossRef]
- Zou, Y.; Li, H.; Graham, E.T.; Deik, A.A.; Eaton, J.K.; Wang, W.; Sandoval-Gomez, G.; Clish, C.B.; Doench, J.G.; Schreiber, S.L. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat. Chem. Biol. 2020, 16, 302–309. [Google Scholar] [CrossRef]
- Yan, B.; Ai, Y.; Sun, Q.; Ma, Y.; Cao, Y.; Wang, J.; Zhang, Z.; Wang, X. Membrane Damage during Ferroptosis Is Caused by Oxidation of Phospholipids Catalyzed by the Oxidoreductases POR and CYB5R1. Mol. Cell 2021, 81, 355–369.e10. [Google Scholar] [CrossRef]
- Ryter, S.W. Heme Oxgenase-1, a Cardinal Modulator of Regulated Cell Death and Inflammation. Cells 2021, 10, 515. [Google Scholar] [CrossRef]
- Park, M.W.; Cha, H.W.; Kim, J.; Kim, J.H.; Yang, H.; Yoon, S.; Boonpraman, N.; Yi, S.S.; Yoo, I.D.; Moon, J.S. NOX4 promotes ferroptosis of astrocytes by oxidative stress-induced lipid peroxidation via the impairment of mitochondrial metabolism in Alzheimer’s diseases. Redox Biol. 2021, 41, 101947. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Li, X.; Zhu, S.; Long, Q.; Hu, Y.; Zhang, L.; Liu, Z.; Li, B.; Li, X. Ferroptosis-related NFE2L2 and NOX4 Genes are Potential Risk Prognostic Biomarkers and Correlated with Immunogenic Features in Glioma. Cell Biochem. Biophys. 2023, 81, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Tsuruta, K.; Matsuoka, M.; Harada, S.; Enomoto, A.; Kumagai, T.; Yasuda, S.; Koumura, T.; Yamada, K.I.; Imai, H. Slowly progressive cell death induced by GPx4-deficiency occurs via MEK1/ERK2 activation as a downstream signal after iron-independent lipid peroxidation. J. Clin. Biochem. Nutr. 2024, 74, 97–107. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fujii, J.; Imai, H. Oxidative Metabolism as a Cause of Lipid Peroxidation in the Execution of Ferroptosis. Int. J. Mol. Sci. 2024, 25, 7544. https://doi.org/10.3390/ijms25147544
Fujii J, Imai H. Oxidative Metabolism as a Cause of Lipid Peroxidation in the Execution of Ferroptosis. International Journal of Molecular Sciences. 2024; 25(14):7544. https://doi.org/10.3390/ijms25147544
Chicago/Turabian StyleFujii, Junichi, and Hirotaka Imai. 2024. "Oxidative Metabolism as a Cause of Lipid Peroxidation in the Execution of Ferroptosis" International Journal of Molecular Sciences 25, no. 14: 7544. https://doi.org/10.3390/ijms25147544
APA StyleFujii, J., & Imai, H. (2024). Oxidative Metabolism as a Cause of Lipid Peroxidation in the Execution of Ferroptosis. International Journal of Molecular Sciences, 25(14), 7544. https://doi.org/10.3390/ijms25147544