
Pim Kinase Inhibition Disrupts CXCR4 Signalling in Megakaryocytes and Platelets by Reducing Receptor Availability at the Surface

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
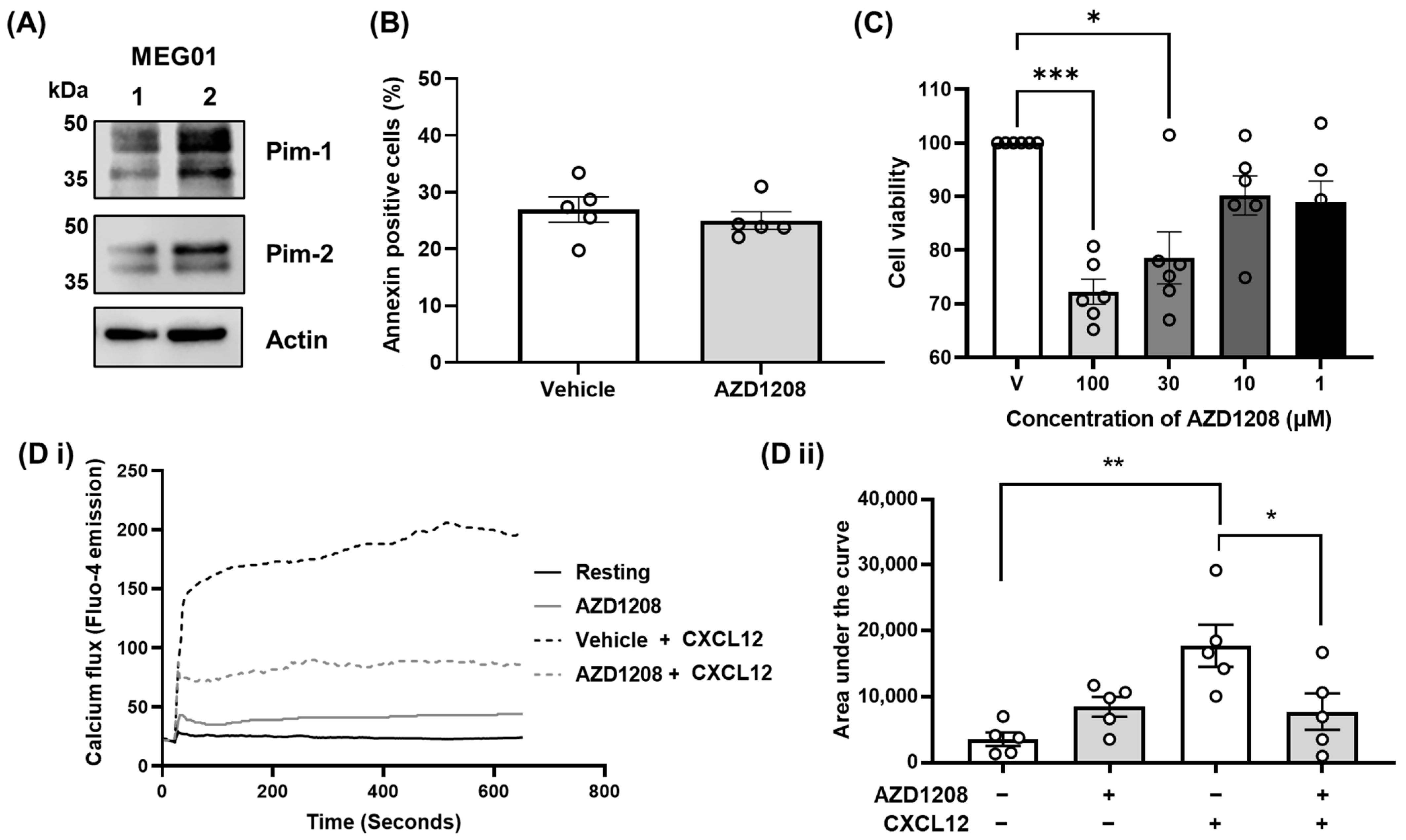
2.1. Pim Kinase Regulates CXCR4 Signalling Events in Megakaryocytes
2.2. Pim Kinase Positively Regulates Megakaryocyte Homing and Migration to CXCL12
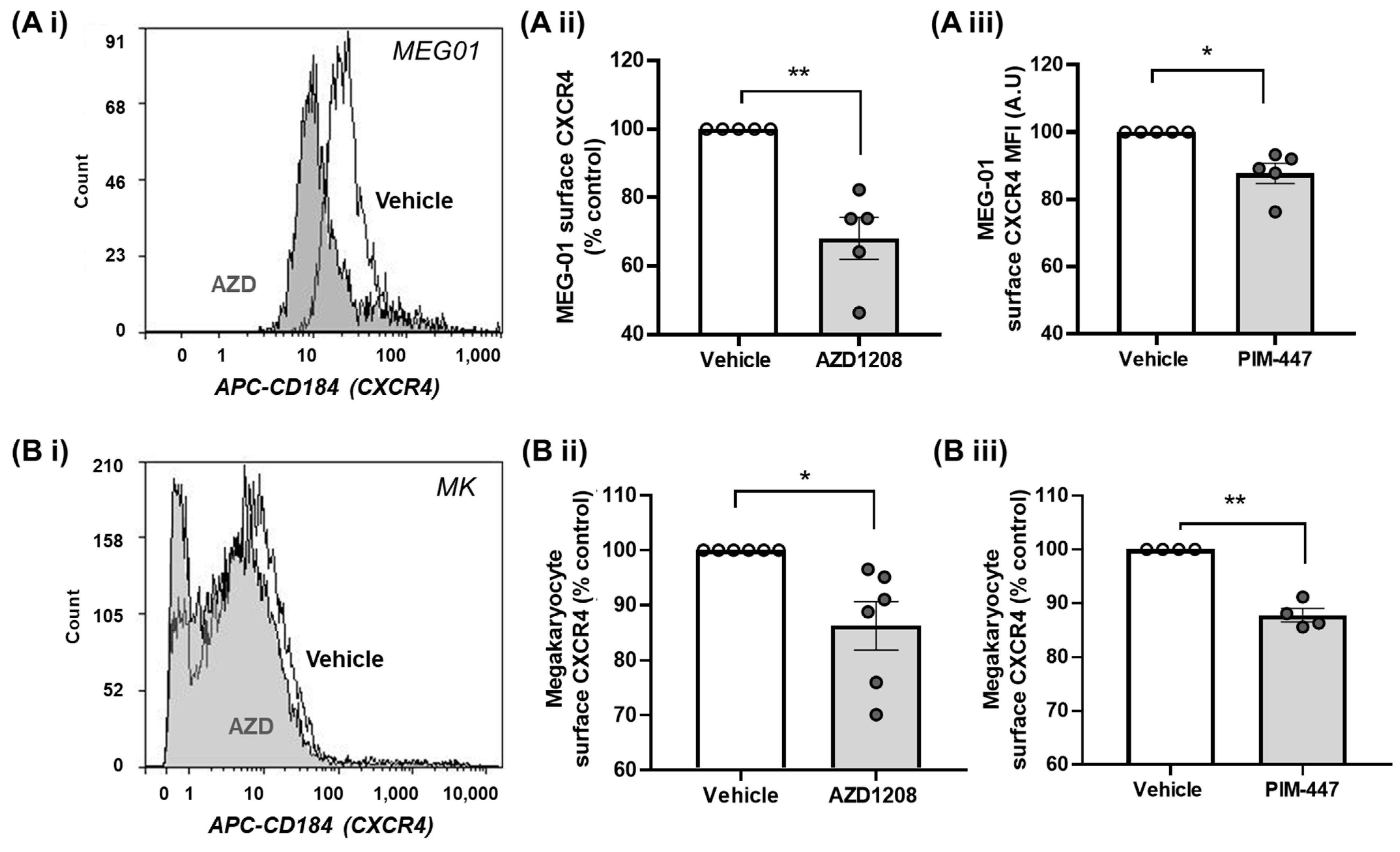
2.3. Pim Kinase Inhibition Reduces CXCR4 Surface Expression Levels in Megakaryocytes
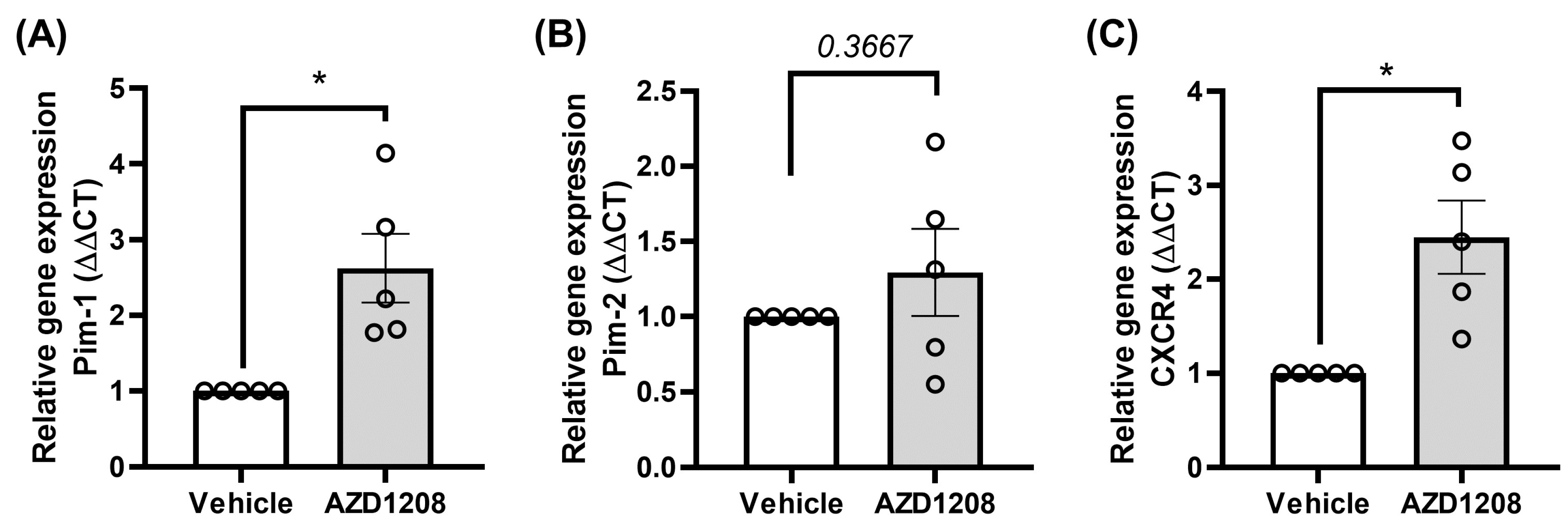
2.4. Megakaryocyte Cells Attempt to Overcome Pim Kinase Inhibition by Increasing Pim-1 and CXCR4 Expression
2.5. Pim Kinase Inhibition Reduces Platelet CXCR4 Responses via Receptor Internalisation
2.6. Pim Kinase Inhibitors Induce Rapid Receptor Internalisation and Prevent Recycling to the Plasma Membrane
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Platelet Preparation
4.3. Isolation and Culture of Bone Marrow-Derived Megakaryocytes
4.4. Cell Culture
4.5. Flow Cytometry
4.5.1. CXCR4 Surface Expression
4.5.2. CD62P Exposure and Fibrinogen Binding
4.6. Calcium Assays
4.7. Transwell Migration Assay
4.8. Live Cell migration/Motility Assay
4.9. Gene Expression Studies
4.10. Western Blotting
4.11. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stone, A.P.; Nascimento, T.F.; Barrachina, M.N. The bone marrow niche from the inside out: How megakaryocytes are shaped by and shape hematopoiesis. Blood 2022, 139, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Kowalska, M.A.; Ratajczak, J.; Hoxie, J.; Brass, L.F.; Gewirtz, A.; Poncz, M.; Ratajczak, M.Z. Megakaryocyte precursors, megakaryocytes and platelets express the HIV co-receptor CXCR4 on their surface: Determination of response to stromal-derived factor-1 by megakaryocytes and platelets. Br. J. Haematol. 1999, 104, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Hamada, T.; Möhle, R.; Hesselgesser, J.; Hoxie, J.; Nachman, R.L.; Moore, M.A.; Rafii, S. Transendothelial migration of megakaryocytes in response to stromal cell-derived factor 1 (SDF-1) enhances platelet formation. J. Exp. Med. 1998, 188, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Avecilla, S.T.; Hattori, K.; Heissig, B.; Tejada, R.; Liao, F.; Shido, K.; Jin, D.K.; Dias, S.; Zhang, F.; Hartman, T.E.; et al. Chemokine-mediated interaction of hematopoietic progenitors with the bone marrow vascular niche is required for thrombopoiesis. Nat. Med. 2004, 10, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Niswander, L.M.; Fegan, K.H.; Kingsley, P.D.; McGrath, K.E.; Palis, J. SDF-1 dynamically mediates megakaryocyte niche occupancy and thrombopoiesis at steady state and following radiation injury. Blood 2014, 124, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Pitchford, S.C.; Lodie, T.; Rankin, S.M. VEGFR1 stimulates a CXCR4-dependent translocation of megakaryocytes to the vascular niche, enhancing platelet production in mice. Blood 2012, 120, 2787–2795. [Google Scholar] [CrossRef] [PubMed]
- Suraneni, P.K.; Corey, S.J.; Hession, M.J.; Ishaq, R.; Awomolo, A.; Hasan, S.; Shah, C.; Liu, H.; Wickrema, A.; Debili, N.; et al. Dynamins 2 and 3 control the migration of human megakaryocytes by regulating CXCR4 surface expression and ITGB1 activity. Blood Adv. 2018, 2, 3540–3552. [Google Scholar] [CrossRef] [PubMed]
- Salim, J.P.; Goette, N.P.; Lev, P.R.; Chazarreta, C.D.; Heller, P.G.; Alvarez, C.; Molinas, F.C.; Marta, R.F. Dysregulation of stromal derived factor 1/CXCR4 axis in the megakaryocytic lineage in essential thrombocythemia. Br. J. Haematol. 2009, 144, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Clemetson, K.J.; Clemetson, J.M.; Proudfoot, A.E.; Power, C.A.; Baggiolini, M.; Wells, T.N. Functional expression of CCR1, CCR3, CCR4, and CXCR4 chemokine receptors on human platelets. Blood 2000, 96, 4046–4054. [Google Scholar] [CrossRef]
- Walsh, T.G.; Harper, M.T.; Poole, A.W. SDF-1alpha is a novel autocrine activator of platelets operating through its receptor CXCR4. Cell Signal 2015, 27, 37–46. [Google Scholar] [CrossRef]
- Chatterjee, M.; Gawaz, M. Platelet-derived CXCL12 (SDF-1α): Basic mechanisms and clinical implications. J. Thromb. Haemost. 2013, 11, 1954–1967. [Google Scholar] [CrossRef]
- Chatterjee, M.; Seizer, P.; Borst, O.; Schönberger, T.; Mack, A.; Geisler, T.; Langer, H.F.; May, A.E.; Vogel, S.; Lang, F.; et al. SDF-1α induces differential trafficking of CXCR4-CXCR7 involving cyclophilin A, CXCR7 ubiquitination and promotes platelet survival. FASEB J. 2014, 28, 2864–2878. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, M.; Rath, D.; Gawaz, M. Role of chemokine receptors CXCR4 and CXCR7 for platelet function. Biochem. Soc. Trans. 2015, 43, 720–726. [Google Scholar] [CrossRef]
- Leberzammer, J.; von Hundelshausen, P. Chemokines, molecular drivers of thromboinflammation and immunothrombosis. Front. Immunol. 2023, 14, 1276353. [Google Scholar] [CrossRef] [PubMed]
- Nanki, T.; Hayashida, K.; El-Gabalawy, H.S.; Suson, S.; Shi, K.; Girschick, H.J.; Yavuz, S.; Lipsky, P.E. Stromal cell-derived factor-1-CXC chemokine receptor 4 interactions play a central role in CD4+ T cell accumulation in rheumatoid arthritis synovium. J. Immunol. 2000, 165, 6590–6598. [Google Scholar] [CrossRef] [PubMed]
- Chong, B.F.; Mohan, C. Targeting the CXCR4/CXCL12 axis in systemic lupus erythematosus. Expert Opin. Ther. Targets 2009, 13, 1147–1153. [Google Scholar] [CrossRef]
- Martirosyan, A.; Poghosyan, D.; Ghonyan, S.; Mkrtchyan, N.; Amaryan, G.; Manukyan, G. Transmigration of Neutrophils from Patients with Familial Mediterranean Fever Causes Increased Cell Activation. Front. Immunol. 2021, 12, 672728. [Google Scholar] [CrossRef]
- Aksu, K.; Donmez, A.; Keser, G. Inflammation-induced thrombosis: Mechanisms, disease associations and management. Curr. Pharm. Des. 2012, 18, 1478–1493. [Google Scholar] [CrossRef]
- Wegner, S.A.; Ehrenberg, P.K.; Chang, G.; Dayhoff, D.E.; Sleeker, A.L.; Michael, N.L. Genomic organization and functional characterization of the chemokine receptor CXCR4, a major entry co-receptor for human immunodeficiency virus type 1. J. Biol. Chem. 1998, 273, 4754–4760. [Google Scholar] [CrossRef]
- Loetscher, M.; Geiser, T.; O’Reilly, T.; Zwahlen, R.; Baggiolini, M.; Moser, B. Cloning of a human seven-transmembrane domain receptor, LESTR, that is highly expressed in leukocytes. J. Biol. Chem. 1994, 269, 232–237. [Google Scholar] [CrossRef]
- Ganju, R.K.; Brubaker, S.A.; Meyer, J.; Dutt, P.; Yang, Y.; Qin, S.; Newman, W.; Groopman, J.E. The alpha-chemokine, stromal cell-derived factor-1alpha, binds to the transmembrane G-protein-coupled CXCR-4 receptor and activates multiple signal transduction pathways. J. Biol. Chem. 1998, 273, 23169–23175. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, J.; Lei, W.; Wang, H.; Ni, Y.; Liu, Y.; Yan, H.; Tian, Y.; Wang, Z.; Yang, Z.; et al. CXCL12-CXCR4/CXCR7 Axis in Cancer: From Mechanisms to Clinical Applications. Int. J. Biol. Sci. 2023, 19, 3341–3359. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, Y.; Crecelius, J.M.; Marchese, A. G protein-coupled receptor kinase phosphorylation of distal C-tail sites specifies βarrestin1-mediated signaling by chemokine receptor CXCR4. J. Biol. Chem. 2022, 298, 102351. [Google Scholar] [CrossRef]
- DeNies, M.S.; Smrcka, A.V.; Schnell, S.; Liu, A.P. β-arrestin mediates communication between plasma membrane and intracellular GPCRs to regulate signaling. Commun. Biol. 2020, 3, 789. [Google Scholar] [CrossRef]
- Wernig, G.; Gonneville, J.R.; Crowley, B.J.; Rodrigues, M.S.; Reddy, M.M.; Hudon, H.E.; Walz, C.; Reiter, A.; Podar, K.; Royer, Y.; et al. The Jak2V617F oncogene associated with myeloproliferative diseases requires a functional FERM domain for transformation and for expression of the Myc and Pim proto-oncogenes. Blood 2008, 111, 3751–3759. [Google Scholar] [CrossRef] [PubMed]
- Wingett, D.; Reeves, R.; Magnuson, N.S. Stability changes in pim-1 proto-oncogene mRNA after mitogen stimulation of normal lymphocytes. J. Immunol. 1991, 147, 3653–3659. [Google Scholar] [CrossRef]
- An, N.; Lin, Y.W.; Mahajan, S.; Kellner, J.N.; Wang, Y.; Li, Z.; Kraft, A.S.; Kang, Y. Pim1 serine/threonine kinase regulates the number and functions of murine hematopoietic stem cells. Stem Cells 2013, 31, 1202–1212. [Google Scholar] [CrossRef]
- Grundler, R.; Brault, L.; Gasser, C.; Bullock, A.N.; Dechow, T.; Woetzel, S.; Pogacic, V.; Villa, A.; Ehret, S.; Berridge, G.; et al. Dissection of PIM serine/threonine kinases in FLT3-ITD-induced leukemogenesis reveals PIM1 as regulator of CXCL12-CXCR4-mediated homing and migration. J. Exp. Med. 2009, 206, 1957–1970. [Google Scholar] [CrossRef]
- Decker, S.; Finter, J.; Forde, A.J.; Kissel, S.; Schwaller, J.; Mack, T.S.; Kuhn, A.; Gray, N.; Follo, M.; Jumaa, H.; et al. PIM Kinases Are Essential for Chronic Lymphocytic Leukemia Cell Survival (PIM2/3) and CXCR4-Mediated Microenvironmental Interactions (PIM1). Mol. Cancer Ther. 2014, 13, 1231–1245. [Google Scholar] [CrossRef]
- Mikkers, H.; Nawijn, M.; Allen, J.; Brouwers, C.; Verhoeven, E.; Jonkers, J.; Berns, A. Mice deficient for all PIM kinases display reduced body size and impaired responses to hematopoietic growth factors. Mol. Cell. Biol. 2004, 24, 6104–6115. [Google Scholar] [CrossRef]
- Nock, S.; Karim, E.; Unsworth, A.J. Pim Kinases: Important Regulators of Cardiovascular Disease. Int. J. Mol. Sci. 2023, 24, 11582. [Google Scholar] [CrossRef] [PubMed]
- Raab, M.S.; Thomas, S.K.; Ocio, E.M.; Guenther, A.; Goh, Y.-T.; Talpaz, M.; Hohmann, N.; Zhao, S.; Xiang, F.; Simon, C.; et al. The first-in-human study of the pan-PIM kinase inhibitor PIM447 in patients with relapsed and/or refractory multiple myeloma. Leukemia 2019, 33, 2924–2933. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Laguna, I.; Dillon, P.M.; Anthony, S.P.; Janat-Amsbury, M.; Ashenbramer, N.; Warner, S.L.; Mouritsen, L.; Wade, M.L.; Whatcott, C.; Bearss, D. A phase I, first-in-human, open-label, dose-escalation, safety, pharmacokinetic, and pharmacodynamic study of oral TP-3654 administered daily for 28 days to patients with advanced solid tumors. J. Clin. Oncol. 2020, 38, 3586. [Google Scholar] [CrossRef]
- Unsworth, A.J.; Bye, A.P.; Sage, T.; Gaspar, R.S.; Eaton, N.; Drew, C.; Stainer, A.; Kriek, N.; Volberding, P.J.; Hutchinson, J.L.; et al. Antiplatelet properties of Pim kinase inhibition are mediated through disruption of thromboxane A2 receptor signaling. Haematologica 2021, 106, 1968–1978. [Google Scholar] [CrossRef] [PubMed]
- Ogura, M.; Morishima, Y.; Ohno, R.; Kato, Y.; Hirabayashi, N.; Nagura, H.; Saito, H. Establishment of a Novel Human Megakaryoblastic Leukemia Cell Line, MEG-01, With Positive Philadelphia Chromosome. Blood 1985, 66, 1384–1392. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.; Tamura, K.; DeAngelo, D.J.; de Bono, J.; Lorente, D.; Minden, M.; Uy, G.L.; Kantarjian, H.; Chen, L.S.; Gandhi, V.; et al. Phase I studies of AZD1208, a proviral integration Moloney virus kinase inhibitor in solid and haematological cancers. Br. J. Cancer 2018, 118, 1425–1433. [Google Scholar] [CrossRef] [PubMed]
- Iida, S.; Sunami, K.; Minami, H.; Hatake, K.; Sekiguchi, R.; Natsume, K.; Ishikawa, N.; Rinne, M.; Taniwaki, M. A phase I, dose-escalation study of oral PIM447 in Japanese patients with relapsed and/or refractory multiple myeloma. Int. J. Hematol. 2021, 113, 797–806. [Google Scholar] [CrossRef]
- Byrne, M.; Donnellan, W.; Patel, M.R.; Zeidan, A.M.; Cherry, M.; Baer, M.R.; Fathi, A.T.; Kaplan, J.; Zhou, F.; Zheng, F.; et al. Preliminary Results from an Ongoing Phase 1/2 Study of INCB053914, a Pan-Proviral Integration Sites for Moloney Virus (PIM) Kinase Inhibitor, in Patients with Advanced Hematologic Malignancies. Blood 2017, 130 (Suppl. 1), 2585. [Google Scholar] [CrossRef]
- Novartis Pharmaceuticals, I. A Safety and Efficacy Study of LGH447 in Patients with Acute Myeloid Leukemia (AML) or High Risk Myelodysplastic Syndrome (MDS). 2014. Available online: https://classic.clinicaltrials.gov/show/NCT02078609 (accessed on 2 May 2023).
- Białopiotrowicz, E.; Górniak, P.; Noyszewska-Kania, M.; Puła, B.; Makuch-Łasica, H.; Nowak, G.; Bluszcz, A.; Szydłowski, M.; Jabłonska, E.; Piechna, K.; et al. Microenvironment-induced PIM kinases promote CXCR4-triggered mTOR pathway required for chronic lymphocytic leukaemia cell migration. J. Cell. Mol. Med. 2018, 22, 3548–3559. [Google Scholar] [CrossRef]
- D’Agostino, G.; Artinger, M.; Locati, M.; Perez, L.; Legler, D.F.; Bianchi, M.E.; Rüegg, C.; Thelen, M.; Marchese, A.; Rocchi, M.B.L.; et al. β-Arrestin1 and β-Arrestin2 Are Required to Support the Activity of the CXCL12/HMGB1 Heterocomplex on CXCR4. Front. Immunol. 2020, 11, 550824. [Google Scholar] [CrossRef]
- Orsini, M.J.; Parent, J.-L.; Mundell, S.J.; Benovic, J.L. Trafficking of the HIV Coreceptor CXCR4: Role of arrestins and identification of residues in the c-terminal tail that mediate receptor internalization. J. Biol. Chem. 1999, 274, 31076–31086. [Google Scholar] [CrossRef] [PubMed]
- Mundell, S.J.; Barton, J.F.; Mayo-Martin, M.B.; Hardy, A.R.; Poole, A.W. Rapid resensitization of purinergic receptor function in human platelets. J. Thromb. Haemost. 2008, 6, 1393–1404. [Google Scholar] [CrossRef] [PubMed]
- Eaton, N.; Drew, C.; Wieser, J.; Munday, A.D.; Falet, H. Dynamin 2 is required for GPVI signaling and platelet hemostatic function in mice. Haematologica 2020, 105, 1414–1423. [Google Scholar] [CrossRef] [PubMed]
- Beautrait, A.; Paradis, J.S.; Zimmerman, B.; Giubilaro, J.; Nikolajev, L.; Armando, S.; Kobayashi, H.; Yamani, L.; Namkung, Y.; Heydenreich, F.M.; et al. A new inhibitor of the β-arrestin/AP2 endocytic complex reveals interplay between GPCR internalization and signalling. Nat. Commun. 2017, 8, 15054. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A.; Bürkle, A. The CXCR4 chemokine receptor in acute and chronic leukaemia: A marrow homing receptor and potential therapeutic target. Br. J. Haematol. 2007, 137, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.F.; Liu, Z.Y.; Groopman, J.E. The alpha-chemokine receptor CXCR4 is expressed on the megakaryocytic lineage from progenitor to platelets and modulates migration and adhesion. Blood 1998, 92, 756–764. [Google Scholar] [CrossRef]
- Leberzammer, J.; Agten, S.M.; Blanchet, X.; Duan, R.; Ippel, H.; Megens, R.T.A.; Schulz, C.; Aslani, M.; Duchene, J.; Döring, Y.; et al. Targeting platelet-derived CXCL12 impedes arterial thrombosis. Blood 2022, 139, 2691–2705. [Google Scholar] [CrossRef] [PubMed]
- Rath, D.; Chatterjee, M.; Bongartz, A.; Müller, K.; Droppa, M.; Stimpfle, F.; Borst, O.; Zuern, C.; Vogel, S.; Gawaz, M.; et al. Platelet surface expression of SDF-1 is associated with clinical outcomes in the patients with cardiovascular disease. Platelets 2017, 28, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Rath, D.; Chatterjee, M.; Borst, O.; Müller, K.; Langer, H.; Mack, A.F.; Schwab, M.; Winter, S.; Gawaz, M.; Geisler, T. Platelet surface expression of stromal cell-derived factor-1 receptors CXCR4 and CXCR7 is associated with clinical outcomes in patients with coronary artery disease. J. Thromb. Haemost. 2015, 13, 719–728. [Google Scholar] [CrossRef]
- Sun, S.; Chai, S.; Zhang, F.; Lu, L. Overexpressed microRNA-103a-3p inhibits acute lower-extremity deep venous thrombosis via inhibition of CXCL12. IUBMB Life 2020, 72, 492–504. [Google Scholar] [CrossRef]
- Chen, J.; Yang, Y.; Wang, S.; Zhang, K. Crocin improves lower extremity deep venous thrombosis by regulating the PIM1/FOXO3a axis. Cell. Mol. Biol. 2023, 69, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Xu, X.; Geng, J.; Wan, X.; Dai, H. The autocrine CXCR4/CXCL12 axis contributes to lung fibrosis through modulation of lung fibroblast activity. Exp. Ther. Med. 2020, 19, 1844–1854. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, R.M.; Davis, P.; Jayson, M.I. Thrombocytosis in rheumatoid arthritis. Ann. Rheum. Dis. 1976, 35, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.; Mankin, P.; Gnanamony, M.; de Alarcon, P.A. Evaluation of angiogenic signaling molecules associated with reactive thrombocytosis in an iron-deficient rat model. Pediatr. Res. 2021, 90, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Ngamsri, K.C.; Putri, R.A.; Jans, C.; Schindler, K.; Fuhr, A.; Zhang, Y.; Gamper-Tsigaras, J.; Ehnert, S.; Konrad, F.M. CXCR4 and CXCR7 Inhibition Ameliorates the Formation of Platelet-Neutrophil Complexes and Neutrophil Extracellular Traps through Adora2b Signaling. Int. J. Mol. Sci. 2021, 22, 13576. [Google Scholar] [CrossRef] [PubMed]
- Uy, G.L.; Rettig, M.P.; Motabi, I.H.; McFarland, K.; Trinkaus, K.M.; Hladnik, L.M.; Kulkarni, S.; Abboud, C.N.; Cashen, A.F.; Stockerl-Goldstein, K.E.; et al. A phase 1/2 study of chemosensitization with the CXCR4 antagonist plerixafor in relapsed or refractory acute myeloid leukemia. Blood 2012, 119, 3917–3924. [Google Scholar] [CrossRef] [PubMed]
- Busillo, J.M.; Benovic, J.L. Regulation of CXCR4 signaling. Biochim. Et Biophys. Acta 2007, 1768, 952–963. [Google Scholar] [CrossRef] [PubMed]
- Busillo, J.M.; Armando, S.; Sengupta, R.; Meucci, O.; Bouvier, M.; Benovic, J.L. Site-specific phosphorylation of CXCR4 is dynamically regulated by multiple kinases and results in differential modulation of CXCR4 signaling. J. Biol. Chem. 2010, 285, 7805–7817. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.S.; Chang, B.Y.; Chang, S.; Tong, T.; Ham, S.; Sherry, B.; Burger, J.A.; Rai, K.R.; Chiorazzi, N. BTK inhibition results in impaired CXCR4 chemokine receptor surface expression, signaling and function in chronic lymphocytic leukemia. Leukemia 2016, 30, 833–843. [Google Scholar] [CrossRef]
- Hernandez, P.A.; Gorlin, R.J.; Lukens, J.N.; Taniuchi, S.; Bohinjec, J.; Francois, F.; Klotman, M.E.; Diaz, G.A. Mutations in the chemokine receptor gene CXCR4 are associated with WHIM syndrome, a combined immunodeficiency disease. Nat. Genet. 2003, 34, 70–74. [Google Scholar] [CrossRef]
- Kawai, T.; Choi, U.; Whiting-Theobald, N.L.; Linton, G.F.; Brenner, S.; Sechler, J.M.G.; Murphy, P.M.; Malech, H.L. Enhanced function with decreased internalization of carboxy-terminus truncated CXCR4 responsible for WHIM syndrome. Exp. Hematol. 2005, 33, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xie, J.; Wang, D.; Han, X.; Chen, M.; Shi, G.; Jiang, L.; Zhao, M. CXCR4high megakaryocytes regulate host-defense immunity against bacterial pathogens. eLife 2022, 11, e78662. [Google Scholar] [CrossRef] [PubMed]
- Heib, T.; Gross, C.; Müller, M.L.; Stegner, D.; Pleines, I. Isolation of murine bone marrow by centrifugation or flushing for the analysis of hematopoietic cells—A comparative study. Platelets 2021, 32, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Frydman, G.H.; Ellett, F.; Jorgensen, J.; Marand, A.L.; Zukerberg, L.; Selig, M.K.; Tessier, S.N.; Wong, K.H.K.; Olaleye, D.; Vanderburg, C.R.; et al. Megakaryocytes respond during sepsis and display innate immune cell behaviors. Front. Immunol. 2023, 14, 1083339. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Marin, F.; Gutti, R.; Liu, Z.J.; Sola-Visner, M. MiR-9 contributes to the developmental differences in CXCR-4 expression in human megakaryocytes. J. Thromb. Haemost. 2014, 12, 282–285. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Kaiser, W.J.; Holbrook, L.M.; Tucker, K.L.; Stanley, R.G.; Gibbins, J.M. A functional proteomic method for the enrichment of peripheral membrane proteins reveals the collagen binding protein Hsp47 is exposed on the surface of activated human platelets. J. Proteome Res. 2009, 8, 2903–2914. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nock, S.H.; Blanco-Lopez, M.R.; Stephenson-Deakin, C.; Jones, S.; Unsworth, A.J. Pim Kinase Inhibition Disrupts CXCR4 Signalling in Megakaryocytes and Platelets by Reducing Receptor Availability at the Surface. Int. J. Mol. Sci. 2024, 25, 7606. https://doi.org/10.3390/ijms25147606
Nock SH, Blanco-Lopez MR, Stephenson-Deakin C, Jones S, Unsworth AJ. Pim Kinase Inhibition Disrupts CXCR4 Signalling in Megakaryocytes and Platelets by Reducing Receptor Availability at the Surface. International Journal of Molecular Sciences. 2024; 25(14):7606. https://doi.org/10.3390/ijms25147606
Chicago/Turabian StyleNock, Sophie H., Maria R. Blanco-Lopez, Chloe Stephenson-Deakin, Sarah Jones, and Amanda J. Unsworth. 2024. "Pim Kinase Inhibition Disrupts CXCR4 Signalling in Megakaryocytes and Platelets by Reducing Receptor Availability at the Surface" International Journal of Molecular Sciences 25, no. 14: 7606. https://doi.org/10.3390/ijms25147606
APA StyleNock, S. H., Blanco-Lopez, M. R., Stephenson-Deakin, C., Jones, S., & Unsworth, A. J. (2024). Pim Kinase Inhibition Disrupts CXCR4 Signalling in Megakaryocytes and Platelets by Reducing Receptor Availability at the Surface. International Journal of Molecular Sciences, 25(14), 7606. https://doi.org/10.3390/ijms25147606