
Adjuvant Properties of Caffeic Acid in Cancer Treatment
 , , ,
, , ,  and
and 
Abstract
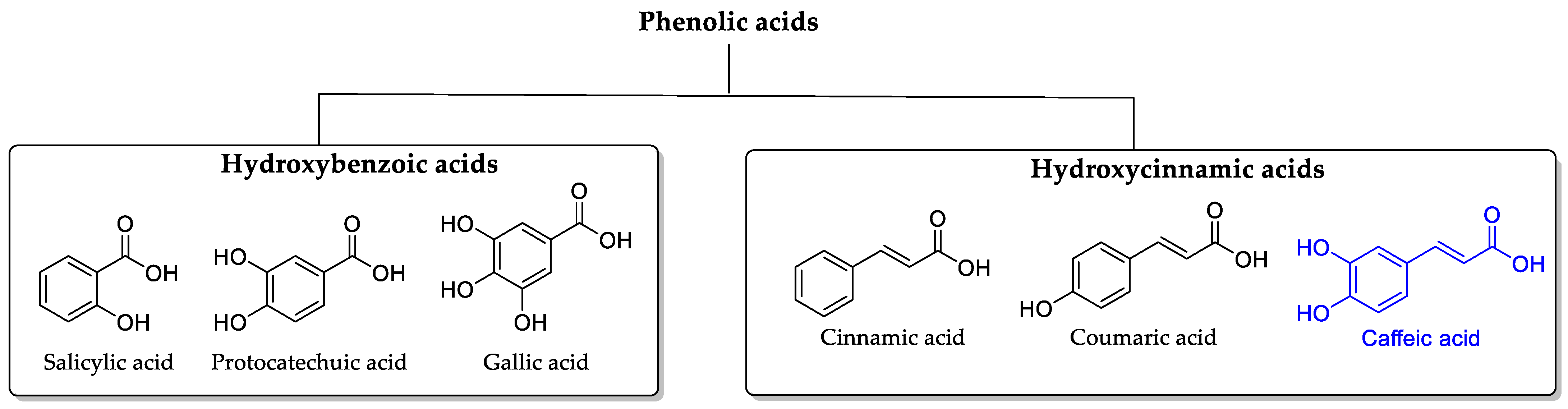
1. Introduction
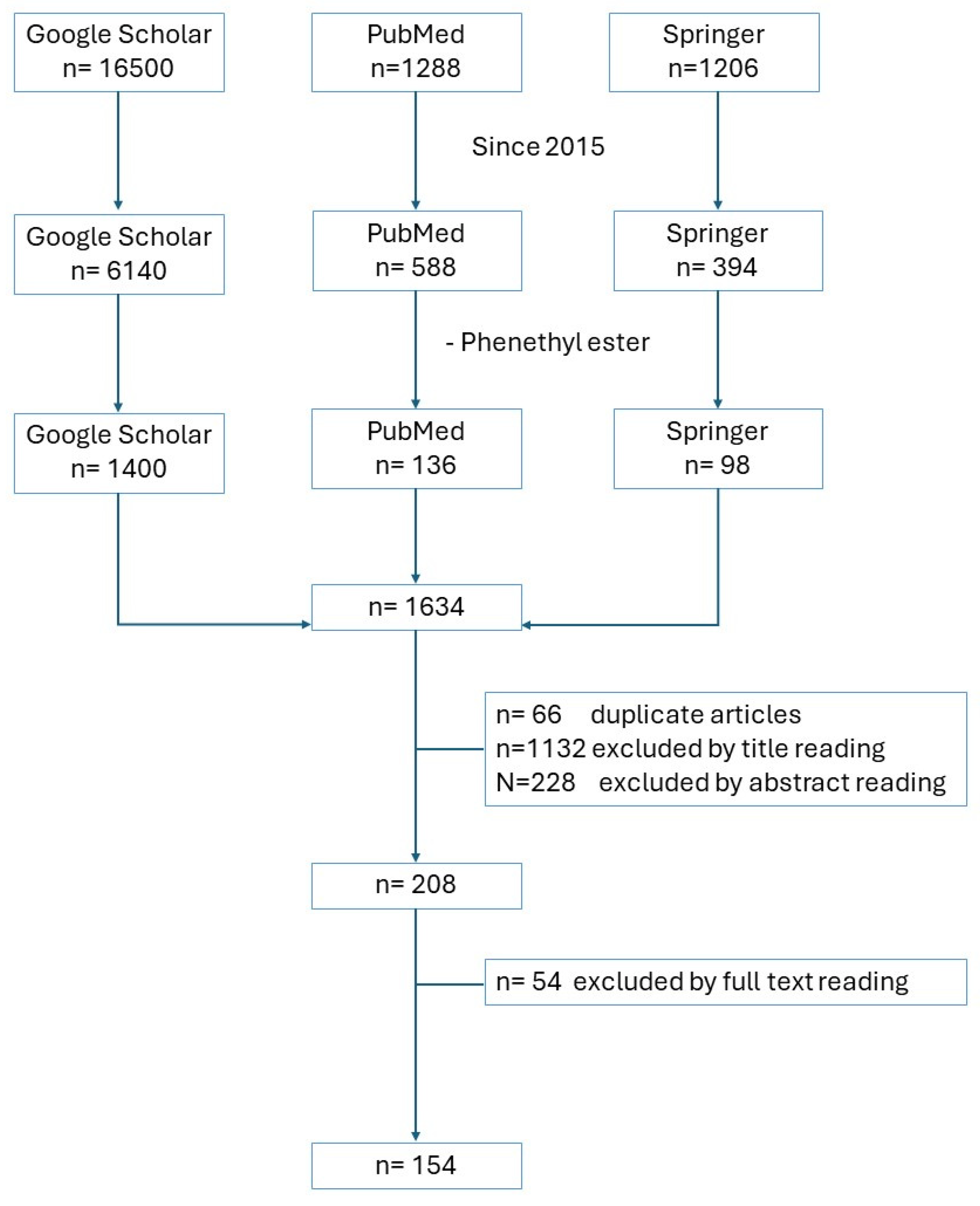
2. Methodology
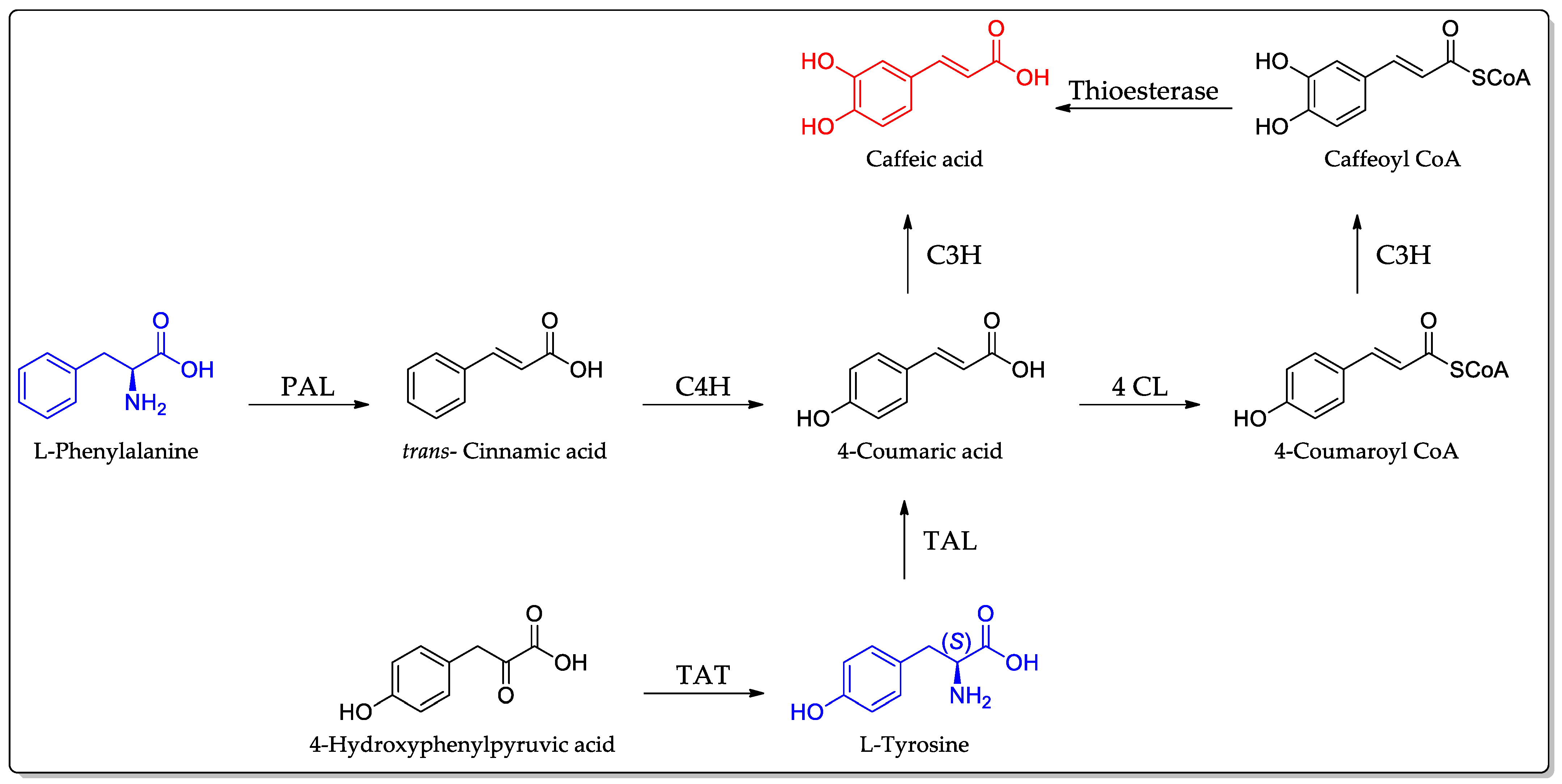
3. Biosynthesis of Caffeic Acid
4. Natural Sources with High Content of CA
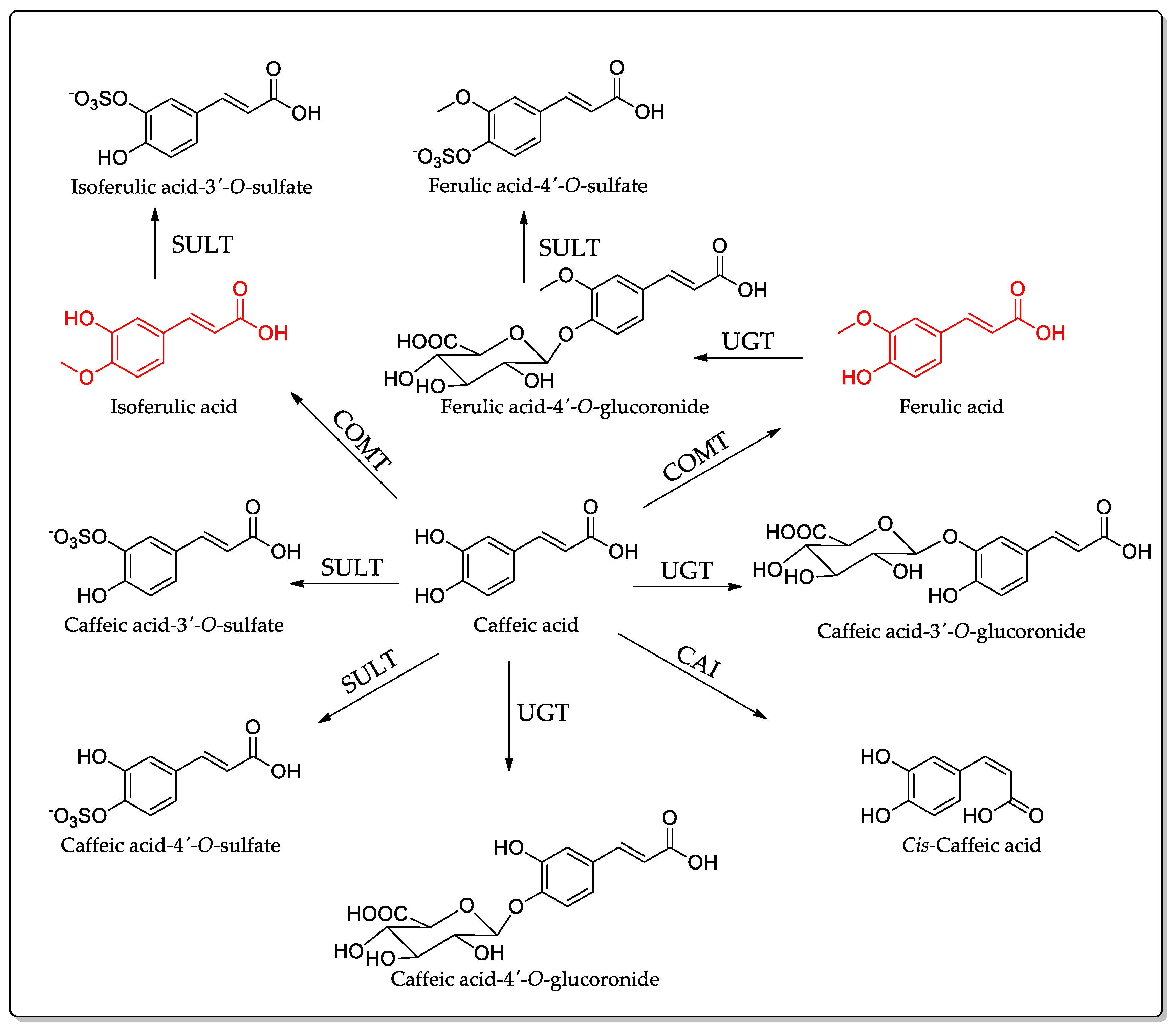
5. Absorption, Distribution, and Metabolism of CA
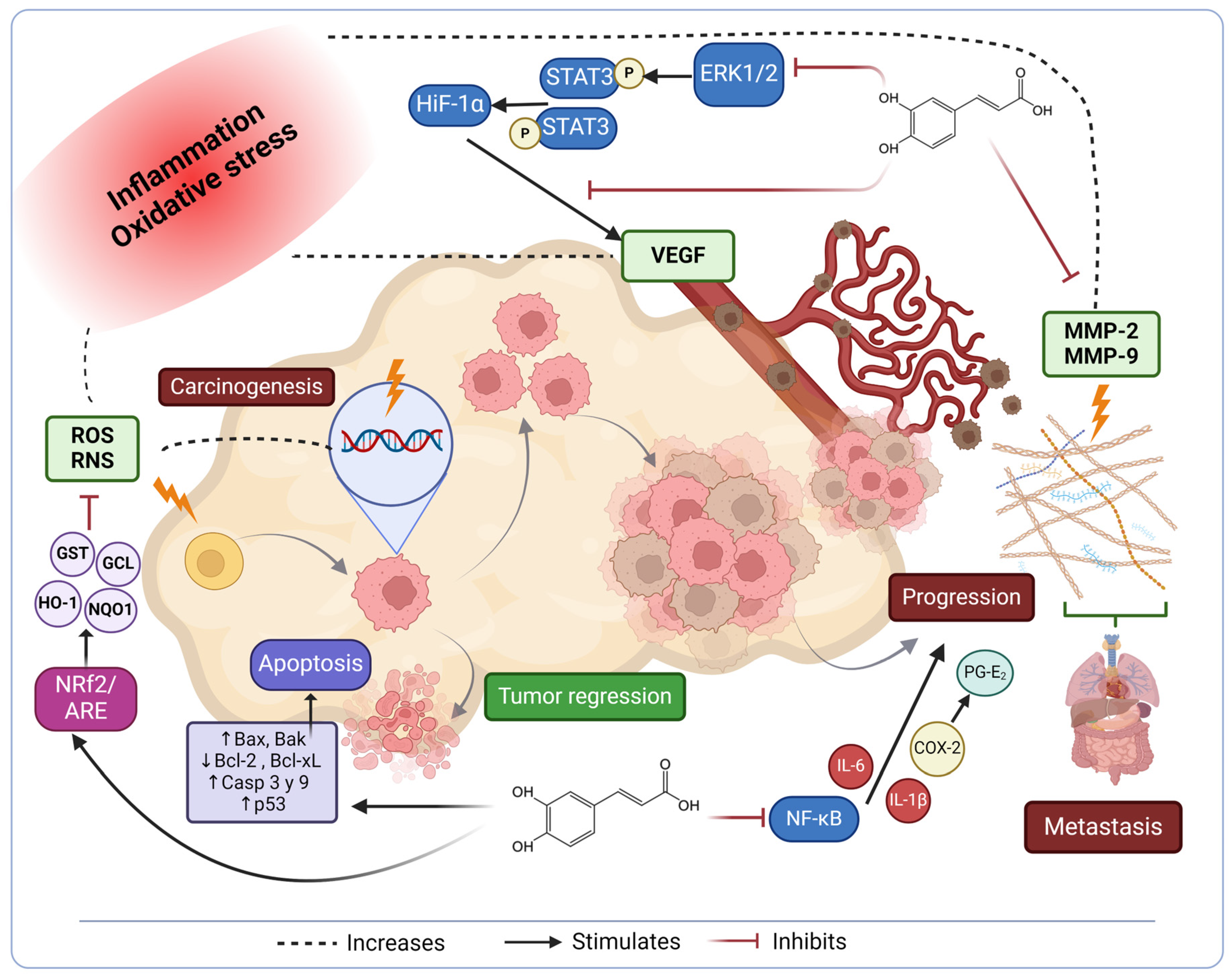
6. Signaling Pathways Affected in Cancer Progression
7. Anti-Inflammatory Activity of Caffeic Acid against Cancer
8. Caffeic Acid Properties against Cancer
9. Caffeic Acid as an Adjuvant for Chemotherapy
9.1. In Vitro and In Vivo Trials

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Aim | Cancer Type: Model | Treatment Conditions | Finding | Reference |
|---|---|---|---|---|
|
To assess the efficacy of cisplatin + CA treatment in human cervical cancer |
• In vitro: human cervical cancer cell lines: HeLa, SiHa, CaSki (HPV-positive), and C33A (HPV- negative) cells. |
CA (300 µM) and cisplatin (11 µM) for 24 h | The combination of cisplatin and CA significantly inhibited cell growth of HeLa and CaSki cell lines, with a combination index < 1, indicating a synergistic effect. The combination significantly increased the expression of caspases 3, 7, and 9, demonstrating apoptosis. | [128] |
| To assess the efficacy of the combined treatment with cisplatin + CA against ovarian carcinoma |
• In vitro: ovarian carcinoma cells A2780 and ovarian carcinoma- resistant A2780cisR cells. |
CA (10 µM) and cisplatin (5 µM) for 24 h |
The combined therapy restores the sensitivity of resistant cells to cisplatin, achieving a similar level of cell viability as that observed in sensitive cells (60% viability). When the cisplatin/caffeic acid ratio was increased to 1:10 (5:50 µM), the caspase activity rose significantly by 4.3-fold. | [130] |
| To evaluate the effects of metformin (Met) and CA on metastatic human cervical cancer |
• In vitro: metastatic human cervical HTB-34 cell line. | CA (100 µM) and Met (10 mM) for 24 h |
CA (100 µM) and Met (10 mM) activated AMPK. CA increased oxidative stress, affecting bioenergetics pathways and making HTB-34 cells more sensitive to Met. CA and Met suppressed HTB-34 cells by different mechanisms. | [133] |
|
To determine the efficacy and underlying mechanisms of CA in combination with paclitaxel for the treatment in human non-small cell lung carcinoma (NSCLC) |
• In vitro: human non-small cell lung carcinoma H1299 cells. • In vivo: mouse xenograft model by subcutaneous injections of H1299 cells. |
In vitro: 100 μM CA + 10 μM of paclitaxel for 24 h In vivo: 20 mg/kg CA and 10 mg/kg paclitaxelad ministered concomitantly for three weeks. |
In vitro, combination treatment decreased the proliferation of NSCLC H1299 cells by the MAPK pathway. CA induced sub-G1 cell cycle arrest in H1299 cells. In vivo, the combined treatment with CA and paclitaxel exerted a more effective suppressive effect on tumor growth in H1299 xenografts without causing significant adverse effects. | [134] |
|
To evaluate the synergistic antitumor activity and the physicochemical and pharmacokinetic properties of caffeic acid/5-FU-cocrystal in vitro and in vivo. |
• In vitro: human colon cancer HCT-116, breast cancer MDA-MB-231, and lung cancer A549 cell lines • In vivo: Sprague Dawley rats |
In vitro: HCT-116; MDA-MB-231 (15.19 μM); and A549 (11.57 μM) of caffeic acid/5-FU cocrystal for 48 h. In vivo: oral dose of 50 mg kg−1. |
In vitro: Cocrystallization of CA + 5-FU optimized the physicochemical properties of 5-FU and exerted a synergistic antitumor effect (CI < 1), thus enhancing the anticancer activity of 5-FU. In vivo: The aqueous solubility and permeability of 5-FU in the cocrystal increased by 1.92 and 4.22-fold, respectively, compared to the original drug 5-FU. | [131] |
|
To evaluate the effects of CA and imatinib (IM) and their synergistic effects on chronic myeloid leukemia model |
• In vitro: human myelogenous leukemia cell line K562 and (IM)-resistant cells. | Synergistic effects of CA (up to 38 µM) and IM (up to 0.15 µM) on K562 cells. | CA induced apoptosis in IM-resistant cells and reduced their proliferation. Combination treatment with CA and IM showed synergistic effects, increasing the antiproliferative activity. | [114] |
|
To assess the activity of Pancreatic Ductal Adenocarcinoma (PDAC) by treatment with CA, gemcitabine (Gem), and doxorubicin (DOX) |
• In vitro: Human epithelioid carcinoma attached cell lines Panc-1 and Mia-PaCa-2. Both have increased potential of migration and invasion, as well as Gem resistance |
Cytotoxic analysis of CA was measured at 24 and 48 h in combination with Gem and DOX. |
CA showed cytotoxic concentrations (IC50) of 37.37 µM and 15.06 µM against Panc-1 and Mia-PaCa-2, respectively. Cotreatment with a combination of CA and Gem or DOX did not show synergic activity; in contrast, it showed antagonism, suggesting that CA could display interactions with Gem or DOX. | [135] |
| To study the effect of CA and DOX on lung cancer |
• In vitro: mouse pulmonary system adenocarcinoma LA795 cell line • In vivo: Balb/c mice and Sprague Dawley (SD) rats |
In vitro: Not specified In vivo: CA (5.4 mg/kg bw) + DOX (4.1 mg/kg bw) |
In vitro: CA inhibited TMEM16A with an IC50 of 29.47 ± 3.19 μM. CA regulated the proliferation, migration, and apoptosis of lung cancer cells targeting TMEM16A (binding sites: D439, E448, and R753). CA regulated the growth of lung cancer through the MAPK pathway. CA + DOX inhibited lung cancer cell growth more than a double dose of either one. In vivo: CA + DOX achieved a tumor suppression rate of 85.6% and compensated for side effects. | [132] |
| To evaluate the effects of tocotrienols and CA encapsulated in a nanoemulsion with cisplatin on lung and liver cancer |
• In vivo: human lung cancer cell A549 and liver HEP G2 cancer cells. | Not specified |
TRF, CA, and CIS synergistically enhanced late-phase apoptosis and improved cell cycle arrest in the G0/G1 phase. ROS generation was enhanced using TRF:CA:CIS by 16.9% and 30.2% for A549 and HEP G2, respectively. | [129] |
|
To evaluate the oxidative stress induced by multi-walled carbon nanotube (MWCNT) treatment on islets and β-cells. |
• In vivo: islets and β-cells |
CA significantly reduced ROS production after MWCNT treatment and increased insulin secretion together with the enzymes SOD, GSH-Px, CAT, and GSH, but it decreased the level of MDA. | [136] | |
|
To evaluate the effect of CA encapsulated in a nanoemulsion on the reduction of nephrotoxicity effects |
• In vitro: non-cancer cells of the HEK 293 kidney line | CA (0.08–1.75 μM) + CIS 0.03 μM | Improved cell viability in kidney cells from 33% to over 95%. | [129] |
|
To evaluate delivery systems with CA for the treatment of breast cancer, loaded on oxidant carbon nanotube (OCNT) and/or chitosan (CS). |
• In vitro: human breast cancer MDA-MB-231 cell line |
CA (100 µg/mL); oxidant carbon nanotube (OCNT)/CA (80 µg/mL); and chitosan (CS)/OCNT/CA (30 µg/mL) | The delivery system based on CS/OCNT/CA showed a higher cytotoxic effect on MDA-MB-231 compared to OCNT/CA and CA alone through apoptosis. | [137] |
9.2. Clinical Trials
10. Radioprotective Potential of Caffeic Acid in Chemotherapy
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kohn, M. Heinrich Hlasiwetz (1825–1875). J. Chem. Educ. 1945, 22, 55. [Google Scholar] [CrossRef]
- Kiokias, S.; Proestos, C.; Oreopoulou, V. Phenolic Acids of Plant Origin-A Review on Their Antioxidant Activity In Vitro (O/W Emulsion Systems) Along with Their in Vivo Health Biochemical Properties. Foods 2020, 9, 534. [Google Scholar] [CrossRef]
- Dias, M.C.; Pinto, D.; Silva, A.M.S. Plant Flavonoids: Chemical Characteristics and Biological Activity. Molecules 2021, 26, 5377. [Google Scholar] [CrossRef]
- Tzanova, M.; Atanasov, V.; Yaneva, Z.; Ivanova, D.; Dinev, T. Selectivity of current extraction techniques for flavonoids from plant materials. Processes 2020, 8, 1222. [Google Scholar] [CrossRef]
- Bule, M.; Khan, F.; Nisar, M.F.; Niaz, K.; Nabavi, S.; Saeedi, M.; Sanches Silva, A. Tannins (hydrolysable tannins, condensed tannins, phlorotannins, flavono-ellagitannins). In Recent Advances in Natural Products Analysis; Elsevier: Amsterdam, The Netherlands, 2020; pp. 132–146. [Google Scholar]
- Su, X.; Zhou, D.; Li, N. Bioactive stilbenes from plants. Stud. Nat. Prod. Chem. 2022, 73, 265–403. [Google Scholar]
- Wang, L.X.; Wang, H.L.; Huang, J.; Chu, T.Z.; Peng, C.; Zhang, H.; Chen, H.L.; Xiong, Y.A.; Tan, Y.Z. Review of lignans from 2019 to 2021: Newly reported compounds, diverse activities, structure-activity relationships and clinical applications. Phytochemistry 2022, 202, 113326. [Google Scholar] [CrossRef]
- Dulo, B.; Phan, K.; Githaiga, J.; Raes, K.; De Meester, S. Natural quinone dyes: A review on structure, extraction techniques, analysis and application potential. Waste Biomass Valorization 2021, 12, 6339–6374. [Google Scholar] [CrossRef]
- Gouda, M.A.; Hussein, B.H.M.; El-Demerdash, A.; Ibrahim, M.E.; Salem, M.A.; Helal, M.H.; Hamama, W.S. A review: Synthesis and medicinal importance of coumarins and their analogues (Part II). Curr. Bioact. Compd. 2020, 16, 993–1008. [Google Scholar] [CrossRef]
- Lončar, M.; Jakovljević, M.; Šubarić, D.; Pavlić, M.; Buzjak Služek, V.; Cindrić, I.; Molnar, M. Coumarins in Food and Methods of Their Determination. Foods 2020, 9, 645. [Google Scholar] [CrossRef]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef]
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules 2020, 25, 5474. [Google Scholar] [CrossRef]
- Yang, S.Y.; Pyo, M.C.; Nam, M.H.; Lee, K.W. ERK/Nrf2 pathway activation by caffeic acid in HepG2 cells alleviates its hepatocellular damage caused by t-butylhydroperoxide-induced oxidative stress. BMC Complement. Altern. Med. 2019, 19, 139. [Google Scholar] [CrossRef]
- Colonnello, A.; Aguilera-Portillo, G.; Rubio-López, L.C.; Robles-Bañuelos, B.; Rangel-López, E.; Cortez-Núñez, S.; Evaristo-Priego, Y.; Silva-Palacios, A.; Galván-Arzate, S.; García-Contreras, R.; et al. Comparing the Neuroprotective Effects of Caffeic Acid in Rat Cortical Slices and Caenorhabditis elegans: Involvement of Nrf2 and SKN-1 Signaling Pathways. Neurotox. Res. 2020, 37, 326–337. [Google Scholar] [CrossRef]
- Guerriero, E.; Sorice, A.; Capone, F.; Costantini, S.; Palladino, P.; D’Ischia, M.; Castello, G. Effects of lipoic acid, caffeic acid and a synthesized lipoyl-caffeic conjugate on human hepatoma cell lines. Molecules 2011, 16, 6365–6377. [Google Scholar] [CrossRef]
- Jung, J.E.; Kim, H.S.; Lee, C.S.; Park, D.H.; Kim, Y.N.; Lee, M.J.; Lee, J.W.; Park, J.W.; Kim, M.S.; Ye, S.K.; et al. Caffeic acid and its synthetic derivative CADPE suppress tumor angiogenesis by blocking STAT3-mediated VEGF expression in human renal carcinoma cells. Carcinogenesis 2007, 28, 1780–1787. [Google Scholar] [CrossRef]
- Yang, G.; Fu, Y.; Malakhova, M.; Kurinov, I.; Zhu, F.; Yao, K.; Li, H.; Chen, H.; Li, W.; Lim, D.Y.; et al. Caffeic acid directly targets ERK1/2 to attenuate solar UV-induced skin carcinogenesis. Cancer Prev. Res. 2014, 7, 1056–1566. [Google Scholar] [CrossRef]
- Espíndola, K.M.M.; Ferreira, R.G.; Narvaez, L.E.M.; Silva Rosario, A.C.R.; Da Silva, A.H.M.; Silva, A.G.B.; Vieira, A.P.O.; Monteiro, M.C. Chemical and pharmacological aspects of caffeic acid and its activity in hepatocarcinoma. Front. Oncol. 2019, 9, 541. [Google Scholar] [CrossRef]
- Ramirez-Ahumada Mdel, C.; Timmermann, B.N.; Gang, D.R. Biosynthesis of curcuminoids and gingerols in turmeric (Curcuma longa) and ginger (Zingiber officinale): Identification of curcuminoid synthase and hydroxycinnamoyl-CoA thioesterases. Phytochemistry 2006, 67, 2017–2029. [Google Scholar] [CrossRef]
- Fraser, C.M.; Chapple, C. The phenylpropanoid pathway in Arabidopsis. Arab. Book 2011, 9, e0152. [Google Scholar] [CrossRef]
- Vogt, T. Phenylpropanoid biosynthesis. Mol. Plant 2010, 3, 2–20. [Google Scholar] [CrossRef]
- Fujioka, K.; Shibamoto, T. Quantitation of volatiles and nonvolatile acids in an extract from coffee beverages: Correlation with antioxidant activity. J. Agric. Food Chem. 2006, 54, 6054–6058. [Google Scholar] [CrossRef] [PubMed]
- Mattila, P.; Kumpulainen, J. Determination of free and total phenolic acids in plant-derived foods by HPLC with diode-array detection. J. Agric. Food Chem. 2002, 50, 3660–3667. [Google Scholar] [CrossRef] [PubMed]
- Al-Farsi, M.; Alasalvar, C.; Morris, A.; Baron, M.; Shahidi, F. Comparison of antioxidant activity, anthocyanins, carotenoids, and phenolics of three native fresh and sun-dried date (Phoenix dactylifera L.) varieties grown in Oman. J. Agric. Food Chem. 2005, 53, 7592–7599. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, N.; Kayano, S.; Kikuzaki, H.; Sumino, K.; Katagiri, K.; Mitani, T. Identification, quantitative determination, and antioxidative activities of chlorogenic acid isomers in prune (Prunus domestica L.). J. Agric. Food Chem. 2000, 48, 5512–5516. [Google Scholar] [CrossRef] [PubMed]
- Bianco, A.; Buiarelli, F.; Cartoni, G.; Coccioli, F.; Jasionowska, R.; Margherita, P. Analysis by liquid chromatography-tandem mass spectrometry of biophenolic compounds in olives and vegetation waters, Part I. J. Sep. Sci. 2003, 26, 409–416. [Google Scholar] [CrossRef]
- Rossetto, M.; Lante, A.; Vanzani, P.; Spettoli, P.; Scarpa, M.; Rigo, A. Red chicories as potent scavengers of highly reactive radicals: A study on their phenolic composition and peroxyl radical trapping capacity and efficiency. J. Agric. Food Chem. 2005, 53, 8169–8175. [Google Scholar] [CrossRef]
- Lee, J. Caffeic acid derivatives in dried Lamiaceae and Echinacea purpurea products. J. Funct. Foods 2010, 2, 158–162. [Google Scholar] [CrossRef]
- Meinhart, A.D.; Damin, F.M.; Caldeirão, L.; de Jesus Filho, M.; da Silva, L.C.; da Silva Constant, L.; Teixeira Filho, J.; Wagner, R.; Teixeira Godoy, H. Study of new sources of six chlorogenic acids and caffeic acid. J. Food Compos. Anal. 2019, 82, 103244. [Google Scholar] [CrossRef]
- Zielinski, H.; Kozlowska, H.; Lewczuk, B. Bioactive compounds in the cereal grains before and after hydrothermal processing. Innov. Food Sci. Emerg. Technol. 2001, 2, 159–169. [Google Scholar] [CrossRef]
- Weidner, S.; Amarowicz, R.; Karamać, M.; Dąbrowski, G. Phenolic acids in caryopses of two cultivars of wheat, rye and triticale that display different resistance to pre-harvest sprouting. Eur. Food Res. Technol. 1999, 210, 109–113. [Google Scholar] [CrossRef]
- Çayan, F.; Deveci, E.; Tel-Çayan, G.; Duru, M.E. Identification and quantification of phenolic acid compounds of twenty-six mushrooms by HPLC–DAD. J. Food Meas. Charact. 2020, 14, 1690–1698. [Google Scholar] [CrossRef]
- Pérez-García, F.; Adzet, T.; Cañigueral, S. Activity of artichoke leaf extract on reactive oxygen species in human leukocytes. Free Radic. Res. 2000, 33, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Llorach, R.; Espín, J.C.; Tomás-Barberán, F.A.; Ferreres, F. Artichoke (Cynara scolymus L.) byproducts as a potential source of health-promoting antioxidant phenolics. J. Agric. Food Chem. 2002, 50, 3458–3464. [Google Scholar] [CrossRef] [PubMed]
- Chaowuttikul, C.; Palanuvej, C.; Ruangrungsi, N. Quantification of chlorogenic acid, rosmarinic acid, and caffeic acid contents in selected Thai medicinal plants using RP-HPLC-DAD. Braz. J. Pharm. Sci. 2020, 56, e17547. [Google Scholar] [CrossRef]
- Vieira, M.A.; Maraschin, M.; Pagliosa, C.M.; Podestá, R.; de Simas, K.N.; Rockenbach, I.I.; Amboni, R.D.; Amante, E.R. Phenolic acids and methylxanthines composition and antioxidant properties of mate (Ilex paraguariensis) residue. J Food Sci 2010, 75, C280–C285. [Google Scholar] [CrossRef] [PubMed]
- Berté, K.A.; Beux, M.R.; Spada, P.K.; Salvador, M.; Hoffmann-Ribani, R. Chemical composition and antioxidant activity of yerba-mate (Ilex paraguariensis A.St.-Hil., Aquifoliaceae) extract as obtained by spray drying. J. Agric. Food Chem. 2011, 59, 5523–5527. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Oki, T.; Kobayashi, T.; Kai, Y.; Okuno, S. Single-laboratory validation for the determination of caffeic acid and seven caffeoylquinic acids in sweet potato leaves. Biosci. Biotechnol. Biochem. 2014, 78, 2073–2080. [Google Scholar] [CrossRef]
- Zheng, W.; Wang, S.Y. Antioxidant activity and phenolic compounds in selected herbs. J. Agric. Food Chem. 2001, 49, 5165–5170. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-S.; Lee, H.-A.; Hong, C.-O.; Yang, S.-Y.; Hong, S.-Y.; Park, S.-Y.; Lee, H.-J.; Lee, K.-W. Quantification of caffeic acid and rosmarinic acid and antioxidant activities of hot-water extracts from leaves of Perilla frutescens. Korean J. Food Sci. Technol. 2009, 41, 302–306. [Google Scholar]
- Exarchou, V.; Troganis, A.; Gerothanassis, I.P.; Tsimidou, M.; Boskou, D. Identification and quantification of caffeic and rosmarinic acid in complex plant extracts by the use of variable-temperature two-dimensional nuclear magnetic resonance spectroscopy. J. Agric. Food Chem. 2001, 49, 2–8. [Google Scholar] [CrossRef]
- Kowalski, R.; Kowalska, G. Phenolic acid contents in fruits of aubergine (Solanum melongena L.). Pol. J. Food Nutr. Sci. 2005, 14, 37–41. [Google Scholar]
- Meinhart, A.D.; Damin, F.M.; Caldeirão, L.; de Jesus Filho, M.; da Silva, L.C.; da Silva Constant, L.; Filho, J.T.; Wagner, R.; Godoy, H.T. Chlorogenic and caffeic acids in 64 fruits consumed in Brazil. Food Chem. 2019, 286, 51–63. [Google Scholar] [CrossRef]
- Grabska-Kobylecka, I.; Kaczmarek-Bak, J.; Figlus, M.; Prymont-Przyminska, A.; Zwolinska, A.; Sarniak, A.; Wlodarczyk, A.; Glabinski, A.; Nowak, D. The Presence of Caffeic Acid in Cerebrospinal Fluid: Evidence That Dietary Polyphenols Can Cross the Blood-Brain Barrier in Humans. Nutrients 2020, 12, 1531. [Google Scholar] [CrossRef]
- Omar, M.H.; Mullen, W.; Stalmach, A.; Auger, C.; Rouanet, J.M.; Teissedre, P.L.; Caldwell, S.T.; Hartley, R.C.; Crozier, A. Absorption, disposition, metabolism, and excretion of [3-(14)C]caffeic acid in rats. J. Agric. Food Chem. 2012, 60, 5205–5214. [Google Scholar] [CrossRef]
- Kishida, K.; Matsumoto, H. Urinary excretion rate and bioavailability of chlorogenic acid, caffeic acid, p-coumaric acid, and ferulic acid in non-fasted rats maintained under physiological conditions. Heliyon 2019, 5, e02708. [Google Scholar] [CrossRef]
- World Health Organization. WHO MORTALITY DATABASE. Interactive Platform Visualizing Mortality Data. Noncommunicable Diseases. Available online: https://platform.who.int/mortality/themes/theme-details/mdb/noncommunicable-diseases (accessed on 5 April 2024).
- Helfinger, V.; Schröder, K. Redox control in cancer development and progression. Mol. Asp. Med. 2018, 63, 88–98. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Gào, X.; Schöttker, B. Reduction-oxidation pathways involved in cancer development: A systematic review of literature reviews. Oncotarget 2017, 8, 51888–51906. [Google Scholar] [CrossRef]
- Shifera, A.S. The zinc finger domain of IKKγ (NEMO) protein in health and disease. J. Cell. Mol. Med. 2010, 14, 2404–2414. [Google Scholar] [CrossRef] [PubMed]
- Barczewski, A.H.; Ragusa, M.J.; Mierke, D.F.; Pellegrini, M. The IKK-binding domain of NEMO is an irregular coiled coil with a dynamic binding interface. Sci. Rep. 2019, 9, 2950. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. Nuclear factor-κB in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Sarmiento-Salinas, F.L.; Perez-Gonzalez, A.; Acosta-Casique, A.; Ix-Ballote, A.; Diaz, A.; Treviño, S.; Rosas-Murrieta, N.H.; Millán-Perez-Peña, L.; Maycotte, P. Reactive oxygen species: Role in carcinogenesis, cancer cell signaling and tumor progression. Life Sci. 2021, 284, 119942. [Google Scholar] [CrossRef] [PubMed]
- Giridharan, S.; Srinivasan, M. Mechanisms of NF-κB p65 and strategies for therapeutic manipulation. J. Inflamm. Res. 2018, 11, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, H.; Motohashi, H. NRF2 addiction in cancer cells. Cancer Sci. 2018, 109, 900–911. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.; Kim, E.H.; Lee, J.; Roh, J.L. Nrf2 inhibition reverses resistance to GPX4 inhibitor-induced ferroptosis in head and neck cancer. Free Radic. Biol. Med. 2018, 129, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Best, S.A.; Sutherland, K.D. “Keaping” a lid on lung cancer: The Keap1-Nrf2 pathway. Cell Cycle 2018, 17, 1696–1707. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Spentzos, D.; Fountzilas, E.; Francoeur, N.; Sanisetty, S.; Grammatikos, A.P.; Hecht, J.L.; Cannistra, S.A. Keap1 mutations and Nrf2 pathway activation in epithelial ovarian cancer. Cancer Res. 2011, 71, 5081–5089. [Google Scholar] [CrossRef] [PubMed]
- da Costa, D.R.C. NRF2 and Keap1 Genetic Polymorphisms in Breast Cancer. 2022. Available online: https://ubibliorum.ubi.pt/bitstream/10400.6/12896/1/9344_19837.pdf (accessed on 10 June 2024).
- Sánchez-Ortega, M.; Carrera, A.C.; Garrido, A. Role of NRF2 in lung cancer. Cells 2021, 10, 1879. [Google Scholar] [CrossRef] [PubMed]
- Tossetta, G.; Fantone, S.; Montanari, E.; Marzioni, D.; Goteri, G. Role of NRF2 in Ovarian Cancer. Antioxidants 2022, 11, 663. [Google Scholar] [CrossRef] [PubMed]
- Alves, F.M.; dos Santos Jaques, H.; Orrutéa, J.F.G.; de Oliveira Silva, A.G.; Machado, M.G.; Smaniotto, L.L.; Federige, A.C.L.; Colleto, M.I.O.; de Souza, J.A.O.; Rech, D.; et al. Changes in systemic oxidative stress correlate to chemoresistance and poor prognosis features in women with breast cancer. Rev. Senol. Patol. Mamar. 2024, 37, 100598. [Google Scholar] [CrossRef]
- Taguchi, K.; Yamamoto, M. The KEAP1-NRF2 System in Cancer. Front. Oncol. 2017, 7, 85. [Google Scholar] [CrossRef]
- Shurin, M.R.; Umansky, V. Cross-talk between HIF and PD-1/PD-L1 pathways in carcinogenesis and therapy. J. Clin. Investig. 2022, 132, e159473. [Google Scholar] [CrossRef]
- Jin, X.; Dai, L.; Ma, Y.; Wang, J.; Liu, Z. Implications of HIF-1α in the tumorigenesis and progression of pancreatic cancer. Cancer Cell Int. 2020, 20, 273. [Google Scholar] [CrossRef]
- Ucaryilmaz Metin, C.; Ozcan, G. The HIF-1α as a potent inducer of the hallmarks in gastric cancer. Cancers 2022, 14, 2711. [Google Scholar] [CrossRef]
- Li, M.; Li, G.; Yang, X.; Yin, W.; Lv, G.; Wang, S. HIF in Gastric Cancer: Regulation and Therapeutic Target. Molecules 2022, 27, 4893. [Google Scholar] [CrossRef]
- Mohamed, O.A.A.; Tesen, H.S.; Hany, M.; Sherif, A.; Abdelwahab, M.M.; Elnaggar, M.H. The role of hypoxia on prostate cancer progression and metastasis. Mol. Biol. Rep. 2023, 50, 3873–3884. [Google Scholar] [CrossRef]
- Elzakra, N.; Kim, Y. HIF-1α Metabolic Pathways in Human Cancer. Adv. Exp. Med. Biol. 2021, 1280, 243–260. [Google Scholar]
- Green, K.J.; Niessen, C.M.; Rübsam, M.; Perez White, B.E.; Broussard, J.A. The Desmosome-Keratin Scaffold Integrates ErbB Family and Mechanical Signaling to Polarize Epidermal Structure and Function. Front. Cell Dev. Biol. 2022, 10, 903696. [Google Scholar] [CrossRef]
- Kumagai, S.; Koyama, S.; Nishikawa, H. Antitumour immunity regulated by aberrant ERBB family signalling. Nat. Rev. Cancer 2021, 21, 181–197. [Google Scholar] [CrossRef]
- Chu, E.C.; Tarnawski, A.S. PTEN regulatory functions in tumor suppression and cell biology. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2004, 10, Ra235–Ra241. [Google Scholar]
- Choudhury, A.D. PTEN-PI3K pathway alterations in advanced prostate cancer and clinical implications. Prostate 2022, 82 (Suppl. S1), S60–S72. [Google Scholar] [CrossRef] [PubMed]
- Tortorella, E.; Giantulli, S.; Sciarra, A.; Silvestri, I. AR and PI3K/AKT in Prostate Cancer: A Tale of Two Interconnected Pathways. Int. J. Mol. Sci. 2023, 24, 2046. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.C. Targeting the PI3K/Akt/mTOR pathway in non-small cell lung cancer (NSCLC). Thorac. Cancer 2020, 11, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Wu, Y.; He, P.; Fan, Y.; Zhong, X.; Zheng, H.; Luo, T. PI3K/AKT/mTOR-Targeted Therapy for Breast Cancer. Cells 2022, 11, 2508. [Google Scholar] [CrossRef] [PubMed]
- NCBI PTEN Phosphatase and Tensin Homolog [Homo sapiens (Human)]. Available online: https://www.ncbi.nlm.nih.gov/gene?Db=gene&Cmd=ShowDetailView&TermToSearch=5728 (accessed on 10 June 2024).
- Xiao, J.; Hu, C.P.; He, B.X.; Chen, X.; Lu, X.X.; Xie, M.X.; Li, W.; He, S.Y.; You, S.J.; Chen, Q. PTEN expression is a prognostic marker for patients with non-small cell lung cancer: A systematic review and meta-analysis of the literature. Oncotarget 2016, 7, 57832–57840. [Google Scholar] [CrossRef] [PubMed]
- Elwy, F.; Assem, M.M.; Hassan, N.H.A.; Helwa, R. PTEN mutations prevalence in HER2-positive breast cancer patients. Rev. Senol. Patol. Mamar. 2023, 36, 100410. [Google Scholar] [CrossRef]
- Patnam, S.; Samal, R.; Koyyada, R.; Joshi, P.; Singh, A.D.; Nagalla, B.; Soma, M.R.; Sannareddy, R.R.; Ippili, K.; Raju, S. Exosomal PTEN as a predictive marker of aggressive gliomas. Neurol. India 2022, 70, 215–222. [Google Scholar] [PubMed]
- Benitez, J.A.; Finlay, D.; Castanza, A.; Parisian, A.D.; Ma, J.; Longobardi, C.; Campos, A.; Vadla, R.; Izurieta, A.; Scerra, G.; et al. PTEN deficiency leads to proteasome addiction: A novel vulnerability in glioblastoma. Neuro-Oncology 2021, 23, 1072–1086. [Google Scholar] [CrossRef]
- Nóbrega, M.; Cilião, H.L.; Souza, M.F.; Souza, M.R.; Serpeloni, J.M.; Fuganti, P.E.; Cólus, I.M.S. Association of polymorphisms of PTEN, AKT1, PI3K, AR, and AMACR genes in patients with prostate cancer. Genet. Mol. Biol. 2020, 43, e20180329. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhuang, Y.; Shao, S.J.; Trivedi, P.; Zheng, B.; Huang, G.L.; He, Z.; Zhang, X. Essential contribution of the JAK/STAT pathway to carcinogenesis, lytic infection of herpesviruses and pathogenesis of COVID-19 (Review). Mol. Med. Rep. 2024, 29, 39. [Google Scholar] [CrossRef]
- Smirnova, O.V.; Ostroukhova, T.Y.; Bogorad, R.L. JAK-STAT pathway in carcinogenesis: Is it relevant to cholangiocarcinoma progression? World J. Gastroenterol. 2007, 13, 6478–6491. [Google Scholar] [CrossRef]
- Hassin, O.; Oren, M. Drugging p53 in cancer: One protein, many targets. Nat. Rev. Drug Discov. 2023, 22, 127–144. [Google Scholar] [CrossRef] [PubMed]
- Marei, H.E.; Althani, A.; Afifi, N.; Hasan, A.; Caceci, T.; Pozzoli, G.; Morrione, A.; Giordano, A.; Cenciarelli, C. p53 signaling in cancer progression and therapy. Cancer Cell Int. 2021, 21, 703. [Google Scholar] [CrossRef]
- Zhang, T.; Ma, C.; Zhang, Z.; Zhang, H.; Hu, H. NF-κB signaling in inflammation and cancer. MedComm 2021, 2, 618–653. [Google Scholar] [CrossRef]
- Hibino, S.; Kawazoe, T.; Kasahara, H.; Itoh, S.; Ishimoto, T.; Sakata-Yanagimoto, M.; Taniguchi, K. Inflammation-Induced Tumorigenesis and Metastasis. Int. J. Mol. Sci. 2021, 22, 5421. [Google Scholar] [CrossRef] [PubMed]
- Piotrowski, I.; Kulcenty, K.; Suchorska, W. Interplay between inflammation and cancer. Rep. Pract. Oncol. Radiother. 2020, 25, 422–427. [Google Scholar] [CrossRef]
- Kruk, J.; Aboul-Enein, H.Y. Reactive Oxygen and Nitrogen Species in Carcinogenesis: Implications of Oxidative Stress on the Progression and Development of Several Cancer Types. Mini-Rev. Med. Chem. 2017, 17, 904–919. [Google Scholar] [CrossRef] [PubMed]
- Moldogazieva, N.T.; Lutsenko, S.V.; Terentiev, A.A. Reactive Oxygen and Nitrogen Species-Induced Protein Modifications: Implication in Carcinogenesis and Anticancer Therapy. Cancer Res. 2018, 78, 6040–6047. [Google Scholar] [CrossRef]
- Lambrou, G.I.; Hatziagapiou, K.; Vlahopoulos, S. Inflammation and tissue homeostasis: The NF-κB system in physiology and malignant progression. Mol. Biol. Rep. 2020, 47, 4047–4063. [Google Scholar] [CrossRef]
- Kim, S.R.; Jung, Y.R.; Kim, D.H.; An, H.J.; Kim, M.K.; Kim, N.D.; Chung, H.Y. Caffeic acid regulates LPS-induced NF-κB activation through NIK/IKK and c-Src/ERK signaling pathways in endothelial cells. Arch. Pharmacal. Res. 2014, 37, 539–547. [Google Scholar] [CrossRef]
- Liu, M.; Fang, G.; Yin, S.; Zhao, X.; Zhang, C.; Li, J.; Liu, Z. Caffeic Acid Prevented LPS-Induced Injury of Primary Bovine Mammary Epithelial Cells through Inhibiting NF-κB and MAPK Activation. Mediat. Inflamm. 2019, 2019, 1897820. [Google Scholar] [CrossRef]
- Sandra, F.; Ketherin, K. Caffeic acid inhibits RANKL and TNF-α-induced phosphorylation of p38 mitogen-activated protein kinase in RAW-D cells. Indones. Biomed. J. 2018, 10, 140–143. [Google Scholar] [CrossRef]
- Zhang, M.; Zhou, J.; Wang, L.; Li, B.; Guo, J.; Guan, X.; Han, Q.; Zhang, H. Caffeic acid reduces cutaneous tumor necrosis factor alpha (TNF-α), IL-6 and IL-1β levels and ameliorates skin edema in acute and chronic model of cutaneous inflammation in mice. Biol. Pharm. Bull. 2014, 37, 347–354. [Google Scholar] [CrossRef]
- Wang, L.; Lu, M.; Yi, M.; Chen, L.; Shen, J.; Li, Z.; Li, L.; Yang, Y.; Zhang, J.; Li, Y. Caffeic acid attenuates the autocrine IL-6 in hepatocellular carcinoma via the epigenetic silencing of the NF-κB-IL-6-STAT-3 feedback loop. RSC Adv. 2015, 5, 52952–52957. [Google Scholar] [CrossRef]
- Zielińska, D.; Zieliński, H.; Laparra-Llopis, J.M.; Szawara-Nowak, D.; Honke, J.; Giménez-Bastida, J.A. Caffeic Acid Modulates Processes Associated with Intestinal Inflammation. Nutrients 2021, 13, 554. [Google Scholar] [CrossRef]
- Huang, Y.K.; Tseng, K.F.; Tsai, P.H.; Wang, J.S.; Lee, C.Y.; Shen, M.Y. IL-8 as a Potential Therapeutic Target for Periodontitis and Its Inhibition by Caffeic Acid Phenethyl Ester In Vitro. Int. J. Mol. Sci. 2021, 22, 3641. [Google Scholar] [CrossRef]
- Alam, M.; Ahmed, S.; Elasbali, A.M.; Adnan, M.; Alam, S.; Hassan, M.I.; Pasupuleti, V.R. Therapeutic implications of caffeic acid in cancer and neurological diseases. Front. Oncol. 2022, 12, 860508. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, B.J.; Kim, J.H.; Yu, Y.S.; Kim, K.W. Anti-angiogenic effect of caffeic acid on retinal neovascularization. Vasc. Pharmacol. 2009, 51, 262–267. [Google Scholar] [CrossRef]
- Gu, W.; Yang, Y.; Zhang, C.; Zhang, Y.; Chen, L.; Shen, J.; Li, G.; Li, Z.; Li, L.; Li, Y.; et al. Caffeic acid attenuates the angiogenic function of hepatocellular carcinoma cells via reduction in JNK-1-mediated HIF-1α stabilization in hypoxia. RSC Adv. 2016, 6, 82774–82782. [Google Scholar] [CrossRef]
- Guo, Z.; Yang, Y.; Li, L.; Zhao, Q.; Li, Y.; Liu, Z.; Hao, L.; Guo, B.; Diao, A. The novel prolyl hydroxylase-2 inhibitor caffeic acid upregulates hypoxia inducible factor and protects against hypoxia. Eur. J. Pharmacol. 2022, 934, 175307. [Google Scholar] [CrossRef]
- Istyastono, E.P.; Yuniarti, N.; Prasasty, V.D.; Mungkasi, S.; Waskitha, S.S.W.; Yanuar, M.R.S.; Riswanto, F.D.O. Caffeic Acid in Spent Coffee Grounds as a Dual Inhibitor for MMP-9 and DPP-4 Enzymes. Molecules 2023, 28, 7182. [Google Scholar] [CrossRef]
- Chung, T.W.; Moon, S.K.; Chang, Y.C.; Ko, J.H.; Lee, Y.C.; Cho, G.; Kim, S.H.; Kim, J.G.; Kim, C.H. Novel and therapeutic effect of caffeic acid and caffeic acid phenyl ester on hepatocarcinoma cells: Complete regression of hepatoma growth and metastasis by dual mechanism. FASEB J. 2004, 18, 1670–1681. [Google Scholar] [CrossRef]
- Chang, W.C.; Hsieh, C.H.; Hsiao, M.W.; Lin, W.C.; Hung, Y.C.; Ye, J.C. Caffeic acid induces apoptosis in human cervical cancer cells through the mitochondrial pathway. Taiwan. J. Obstet. Gynecol. 2010, 49, 419–424. [Google Scholar] [CrossRef]
- Sandra, F.; Rizal, M.I.; Wahid, A.H.A.; Andajana, M.; Celinna, M. Caffeic acid induces intrinsic apoptotic pathway in mg-63 osteosarcoma cells through bid truncation and cytochrome c release. Indones. Biomed. J. 2022, 14, 323–328. [Google Scholar] [CrossRef]
- Sandra, F.; Hudono, K.F.; Putri, A.A.; Putri, C.A.P. Caspase inhibitor diminishes caffeic acid-induced apoptosis in osteosarcoma cells. Indones. Biomed. J. 2017, 9, 160–164. [Google Scholar] [CrossRef]
- Rezaei-Seresht, H.; Cheshomi, H.; Falanji, F.; Movahedi-Motlagh, F.; Hashemian, M.; Mireskandari, E. Cytotoxic activity of caffeic acid and gallic acid against MCF-7 human breast cancer cells: An in silico and in vitro study. Avicenna J. Phytomed. 2019, 9, 574–586. [Google Scholar]
- Dziedzic, A.; Kubina, R.; Kabała-Dzik, A.; Tanasiewicz, M. Induction of Cell Cycle Arrest and Apoptotic Response of Head and Neck Squamous Carcinoma Cells (Detroit 562) by Caffeic Acid and Caffeic Acid Phenethyl Ester Derivative. Evid. Based Complement. Altern. Med. 2017, 2017, 6793456. [Google Scholar] [CrossRef]
- Kabała-Dzik, A.; Rzepecka-Stojko, A.; Kubina, R.; Wojtyczka, R.D.; Buszman, E.; Stojko, J. Caffeic Acid Versus Caffeic Acid Phenethyl Ester in the Treatment of Breast Cancer MCF-7 Cells: Migration Rate Inhibition. Integr. Cancer Ther. 2018, 17, 1247–1259. [Google Scholar] [CrossRef]
- Sun, L.; Ren, J.; Feng, X.; Li, S.; Wang, Y.; Jiang, Y.; Zheng, C. Caffeic Acid Markedly Induced Apoptosis of Human Multiple Myeloma Cells through the Caspase-dependent Pathway. Pharmacogn. Mag. 2023, 19, 720–726. [Google Scholar] [CrossRef]
- Feriotto, G.; Tagliati, F.; Giriolo, R.; Casciano, F.; Tabolacci, C.; Beninati, S.; Khan, M.T.H.; Mischiati, C. Caffeic Acid Enhances the Anti-Leukemic Effect of Imatinib on Chronic Myeloid Leukemia Cells and Triggers Apoptosis in Cells Sensitive and Resistant to Imatinib. Int. J. Mol. Sci. 2021, 22, 1644. [Google Scholar] [CrossRef]
- Sandra, F.; Rizal, M.I.; Aliwarga, C.C.; Hadimartana, J.C.; Celinna, M. Caffeic Acid Induces Apoptosis in MG-63 Osteosarcoma Cells via Protein Kinase C Delta (PKCδ) Translocation and Mitochondrial Membrane Potential Reduction. Indones. Biomed. J. 2022, 14, 358–364. [Google Scholar] [CrossRef]
- Pelinson, L.P.; Assmann, C.E.; Palma, T.V.; da Cruz, I.B.M.; Pillat, M.M.; Mânica, A.; Stefanello, N.; Weis, G.C.C.; de Oliveira Alves, A.; de Andrade, C.M.; et al. Antiproliferative and apoptotic effects of caffeic acid on SK-Mel-28 human melanoma cancer cells. Mol. Biol. Rep. 2019, 46, 2085–2092. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.T.; Chen, I.L.; Chou, C.T.; Liang, W.Z.; Kuo, D.H.; Shieh, P.; Jan, C.R. Effect of caffeic acid on Ca(2+) homeostasis and apoptosis in SCM1 human gastric cancer cells. Arch. Toxicol. 2013, 87, 2141–2150. [Google Scholar] [CrossRef] [PubMed]
- Abdelwahab, T.S.; Abdelhamed, R.E.; Ali, E.N.; Mansour, N.A.; Abdalla, M.S. Evaluation of Silver Nanoparticles Caffeic Acid Complex Compound as New Potential Therapeutic Agent against Cancer Incidence in Mice. Asian Pac. J. Cancer Prev. APJCP 2021, 22, 3189–3201. [Google Scholar] [CrossRef] [PubMed]
- Robati, A.K.; Shahsavari, Z.; Vaezi, M.A.; Safizadeh, B.; Shirian, F.I.; Tavakoli-Yaraki, M. Caffeic acid stimulates breast cancer death through Reactive oxygen species (ROS) formation, Caspase activation and mitochondrial membrane potential depletion. Acta Biochim. Iran. 2024, 1, 176–182. [Google Scholar] [CrossRef]
- Amorim, R.; Magalhães, C.C.; Benfeito, S.; Cagide, F.; Tavares, L.C.; Santos, K.; Sardão, V.A.; Datta, S.; Cortopassi, G.A.; Baldeiras, I.; et al. Mitochondria dysfunction induced by decyl-TPP mitochondriotropic antioxidant based on caffeic acid AntiOxCIN(6) sensitizes cisplatin lung anticancer therapy due to a remodeling of energy metabolism. Biochem. Pharmacol. 2024, 219, 115953. [Google Scholar] [CrossRef]
- Mirzaei, S.; Gholami, M.H.; Zabolian, A.; Saleki, H.; Farahani, M.V.; Hamzehlou, S.; Far, F.B.; Sharifzadeh, S.O.; Samarghandian, S.; Khan, H.; et al. Caffeic acid and its derivatives as potential modulators of oncogenic molecular pathways: New hope in the fight against cancer. Pharmacol. Res. 2021, 171, 105759. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Li, J.; Hao, Q.; Vadgama, J.V.; Wu, Y. AMP-activated protein kinase: A potential therapeutic target for triple-negative breast cancer. Breast Cancer Res. BCR 2019, 21, 29. [Google Scholar] [CrossRef]
- Park, S.R.; Kim, S.R.; Hong, I.S.; Lee, H.Y. A Novel Therapeutic Approach for Colorectal Cancer Stem Cells: Blocking the PI3K/Akt Signaling Axis With Caffeic Acid. Front. Cell Dev. Biol. 2020, 8, 585987. [Google Scholar] [CrossRef]
- Kimsa-Dudek, M.; Synowiec-Wojtarowicz, A.; Krawczyk, A. Phenolic acids and a static magnetic field change the expression of transforming growth factor β isoforms in amelanotic melanoma cells. Mol. Biol. Rep. 2023, 50, 4207–4216. [Google Scholar] [CrossRef]
- Yang, Y.; Jin, M.; Meng, Y.; Dai, Y.; Chen, S.; Zhou, Y.; Li, Y.; Tang, L. Involvement and targeted intervention of benzo(a)pyrene-regulated apoptosis related proteome modification and muti-drug resistance in hepatocellular carcinoma. Cell Death Dis. 2023, 14, 265. [Google Scholar] [CrossRef]
- Gatti, L.; Zunino, F. Overview of tumor cell chemoresistance mechanisms. Methods Mol. Med. 2005, 111, 127–148. [Google Scholar] [PubMed]
- Teng, Y.N.; Wang, C.C.N.; Liao, W.C.; Lan, Y.H.; Hung, C.C. Caffeic Acid Attenuates Multi-Drug Resistance in Cancer Cells by Inhibiting Efflux Function of Human P-glycoprotein. Molecules 2020, 25, 247. [Google Scholar] [CrossRef]
- Koraneekit, A.; Limpaiboon, T.; Sangka, A.; Boonsiri, P.; Daduang, S.; Daduang, J. Synergistic effects of cisplatin-caffeic acid induces apoptosis in human cervical cancer cells via the mitochondrial pathways. Oncol. Lett. 2018, 15, 7397–7402. [Google Scholar] [CrossRef]
- Raviadaran, R.; Ng, M.H.; Chandran, D.; Ooi, K.K.; Manickam, S. Stable W/O/W multiple nanoemulsion encapsulating natural tocotrienols and caffeic acid with cisplatin synergistically treated cancer cell lines (A549 and HEP G2) and reduced toxicity on normal cell line (HEK 293). Mater. Sci. Eng. C Mater. Biol. Appl. 2021, 121, 111808. [Google Scholar] [CrossRef]
- Sirota, R.; Gibson, D.; Kohen, R. The timing of caffeic acid treatment with cisplatin determines sensitization or resistance of ovarian carcinoma cell lines. Redox Biol. 2017, 11, 170–175. [Google Scholar] [CrossRef]
- Yu, Y.-M.; Wang, L.-Y.; Bu, F.-Z.; Wang, L.-L.; Li, Y.-T.; Wang, C.; Wu, Z.-Y. The supramolecular self-assembly of 5-fluorouracil and caffeic acid through cocrystallization strategy opens up a new way for the development of synergistic antitumor pharmaceutical cocrystal. CrystEngComm 2020, 22, 7992–8006. [Google Scholar] [CrossRef]
- Bai, X.; Li, S.; Liu, X.; An, H.; Kang, X.; Guo, S. Caffeic Acid, an Active Ingredient in Coffee, Combines with DOX for Multitarget Combination Therapy of Lung Cancer. J. Agric. Food Chem. 2022, 70, 8326–8337. [Google Scholar] [CrossRef]
- Tyszka-Czochara, M.; Konieczny, P.; Majka, M. Caffeic Acid Expands Anti-Tumor Effect of Metformin in Human Metastatic Cervical Carcinoma HTB-34 Cells: Implications of AMPK Activation and Impairment of Fatty Acids De Novo Biosynthesis. Int. J. Mol. Sci. 2017, 18, 462. [Google Scholar] [CrossRef] [PubMed]
- Min, J.; Shen, H.; Xi, W.; Wang, Q.; Yin, L.; Zhang, Y.; Yu, Y.; Yang, Q.; Wang, Z.N. Synergistic Anticancer Activity of Combined Use of Caffeic Acid with Paclitaxel Enhances Apoptosis of Non-Small-Cell Lung Cancer H1299 Cells in Vivo and in Vitro. Cell. Physiol. Biochem. 2018, 48, 1433–1442. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Tak, H.; Rathore, K.; Banavath, H.N.; Tejavath, K.K. Caffeic acid, a dietary polyphenol pre-sensitizes PDAC to chemotherapeutic drug. J. Biomol. Struct. Dyn. 2023; ahead of print. [Google Scholar]
- Ahangarpour, A.; Alboghobeish, S.; Oroojan, A.A.; Dehghani, M.A. Caffeic acid protects mice pancreatic islets from oxidative stress induced by multi-walled carbon nanotubes (MWCNTs). Vet. Res. Forum 2021, 12, 77–85. [Google Scholar]
- Rahimi, H.; Mohammadgholi, A.; Koosha, M. Cytotoxic Effect of Caffeic Acid Loaded in Chitosan-Grafted Carbon Nanotubes on MDA-MB-231 Breast Cancer Cell Lines. Iran. J. Sci. 2023, 47, 631–640. [Google Scholar] [CrossRef]
- Qin, P.; Wei, Y.; Hou, M.; Zhao, C.; Shen, Z. A multicenter clinical trial of caffeic acid tablet in treatment of 103 primary immune thrombocytopenia patients. Zhonghua Xue Ye Xue Za Zhi 2015, 36, 103–106. [Google Scholar]
- Yu, H.; Chen, R.; Zhou, Z.; Liu, R.; Wen, J. Efficacy and safety of caffeic acid tablets in the treatment of thrombocytopenia: A systematic review and meta-analysis. Medicine 2023, 102, e35353. [Google Scholar] [CrossRef]
- Schaue, D.; McBride, W.H. Opportunities and challenges of radiotherapy for treating cancer. Nat. Rev. Clin. Oncol. 2015, 12, 527–540. [Google Scholar] [CrossRef]
- De Ruysscher, D.; Niedermann, G.; Burnet, N.G.; Siva, S.; Lee, A.W.M.; Hegi-Johnson, F. Radiotherapy toxicity. Nat. Rev. Dis. Primers 2019, 5, 13. [Google Scholar] [CrossRef]
- Hall, E.J. Radiation, the two-edged sword: Cancer risks at high and low doses. Cancer J. 2000, 6, 343–350. [Google Scholar]
- Borrelli, M.R.; Shen, A.H.; Lee, G.K.; Momeni, A.; Longaker, M.T.; Wan, D.C. Radiation-Induced Skin Fibrosis: Pathogenesis, Current Treatment Options, and Emerging Therapeutics. Ann. Plast. Surg. 2019, 83 (4S Suppl. 1), S59–S64. [Google Scholar] [CrossRef]
- Rakici, S.Y.; Guzel, A.I.; Tumkaya, L.; Sevim Nalkiran, H.; Mercantepe, T. Pelvic radiation-induced testicular damage: An experimental study at 1 Gray. Syst. Biol. Reprod. Med. 2020, 66, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Delanian, S.; Lefaix, J.L.; Pradat, P.F. Radiation-induced neuropathy in cancer survivors. Radiother. Oncol. 2012, 105, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Kuderer, N.M.; Desai, A.; Lustberg, M.B.; Lyman, G.H. Mitigating acute chemotherapy-associated adverse events in patients with cancer. Nat. Rev. Clin. Oncol. 2022, 19, 681–697. [Google Scholar] [CrossRef]
- van den Boogaard, W.M.C.; Komninos, D.S.J.; Vermeij, W.P. Chemotherapy Side-Effects: Not All DNA Damage Is Equal. Cancers 2022, 14, 627. [Google Scholar] [CrossRef]
- Devipriya, N.; Sudheer, A.R.; Menon, V.P. Caffeic acid protects human peripheral blood lymphocytes against gamma radiation-induced cellular damage. J. Biochem. Mol. Toxicol. 2008, 22, 175–186. [Google Scholar] [CrossRef]
- Lukmanul Hakkim, F.; Miura, M.; Matsuda, N.; Alharassi, A.S.; Guillemin, G.; Yamauchi, M.; Arivazhagan, G.; Song, H. An in vitro evidence for caffeic acid, rosmarinic acid and trans cinnamic acid as a skin protectant against γ-radiation. Int. J. Low Radiat. 2014, 9, 305–316. [Google Scholar] [CrossRef]
- Sayed, H.M.; Said, M.M.; Morcos, N.Y.S.; El Gawish, M.A.; Ismail, A.F.M. Antitumor and Radiosensitizing Effects of Zinc Oxide-Caffeic Acid Nanoparticles against Solid Ehrlich Carcinoma in Female Mice. Integr. Cancer Ther. 2021, 20, 15347354211021920. [Google Scholar] [CrossRef]
- Hausser, J.; Alon, U. Tumour heterogeneity and the evolutionary trade-offs of cancer. Nat. Rev. Cancer 2020, 20, 247–257. [Google Scholar] [CrossRef]
- Andrade, S.; Pereira, M.C.; Loureiro, J.A. Caffeic acid loaded into engineered lipid nanoparticles for Alzheimer’s disease therapy. Colloids Surf. B Biointerfaces 2023, 225, 113270. [Google Scholar] [CrossRef]
- Kamath, P.P.; Rajeevan, R.; Maity, S.; Nayak, Y.; Narayan, R.; Mehta, C.H.; Velagacherla, V.; Konuri, A.; Nayak, U.Y. Development of nanostructured lipid carriers loaded caffeic acid topical cream for prevention of inflammation in wistar rat model. J. Appl. Pharm. Sci. 2023, 13, 064–075. [Google Scholar] [CrossRef]
- Stanciauskaite, M.; Poskute, M.; Kurapkiene, V.; Marksa, M.; Jakstas, V.; Ivanauskas, L.; Kersiene, M.; Leskauskaite, D.; Ramanauskiene, K. Optimization of Delivery and Bioavailability of Encapsulated Caffeic Acid. Foods 2023, 12, 1993. [Google Scholar] [CrossRef]

| Edible Vegetables or Fruits | Culinary Plants or Herbal Infusion | ||||
|---|---|---|---|---|---|
| Scientific Name/ Common Name | Caffeic Acid mg/kg | References | Scientific Name/ Common Name | Caffeic Acid mg/kg | References |
| Allium sativum (Garlic) | 50 | [35] | Andrographis paniculata (Green chiretta) | 450 | [35] |
| Apium graveolens (Celery) | 880–1120 | [35] | Anethum graveolens (Dill) | 1840 | [35] |
| Averrhoa carambola (Carambola) | 90 | [35] | Artemisia dracunculus (Tarragon) | 620 | [35] |
| Brassica juncea (Brown mustard) | 830 | [35] | Artemisia pallens (Davana) | 110 | [35] |
| Capsicum annuum (Red Pepper) | 250–850 | [35] | Artemisia vulgaris (Mugwort) | 25.4 | [28] |
| Carica papaya (Papaya) | 5080 | [35] | Camellia sinensis (Tea plant) | 510 | [35] |
| Coriandrum sativum (Coriander) | 240 | [35] | Centella asiatica (Indian pennywort) | 860 | [35] |
| Eryngium foetidum (Cilantro) | 1600 | [35] | Chromolaena odorata (Devil weed) | 6210 | [35] |
| Daucus carota (Carrot) | 850 | [35] | Ilex paraguariensis (Yerba mate) | 63.5 | [36,37] |
| Helianthus annuus (Sunflower) | 30–1100 | [35] | Clerodendrum indicum (Bharangi) | 110 | [35] |
| Ipomoea aquatica (Water spinach) | 1130 | [35] | Clerodendrum thomsoniae | 770 | [35] |
| Ipomoea batatas (Sweet Potato) | 125 | [38] | Coffea canephora (Coffea) | 12,330 | [35] |
| Lactuca sativa (Lettuce) | 2580 | [35] | Ginkgo biloba (Ginkgo) | 398 | [39] |
| Morus alba (Mulberry) | 250 | [35] | Cymbopogon citratus (Lemon grass) | 730 | [35] |
| Morinda citrifolia (Noni) | 100 | [35] | Echinacea purpurea (Purple coneflower) | 115.9 | [28] |
| Persea americana (Avocado) | 80 | [35] | Euphorbia hirta (Asthma plant) | 210 | [35] |
| Persicaria odorata (Vietnamese coriander) | 910 | [35] | Eucommia ulmoides (Hardy rubber tree) | 190 | [35] |
| Spinacia oleracea (Spinach) | 2.4–5.3 * 370 ** | [29] [35] | Gnaphalium polycaulon (Western cudweed) | 2360 | [35] |
| Psidium guajava (Guava) | 220 | [35] | Hibiscus sabdariffa (Roselle) | 3510 | [35] |
| Punica granatum (Pomegranate) | 3050–3630 | [35] | Hyptis suaveolens (Bamburral) | 1110 | [35] |
| Raphanus sativus (Radish) | 330 | [35] | Leonotis nepetifolia (Klip dagga) | 4180 | [35] |
| Petroselinum crispum (Parsley) | 480 | [35] | Leonurus sibiricus (Motherwort) | 120 | [35] |
| Phyllanthus emblica (Gooseberry) | 290 | [35] | Salvia officinalis (Sage) | 1660 ** 74.2 * | [35] [39] |
| Physalis angulata (Poppers) | 120 | [35] | Perilla frutescens (Purple mint) | 1890 870 | [35] [40] |
| Physalis peruviana (Goldenberry) | 860 | [35] | Melissa officinalis (Lemon balm) | 26.8 1740 | [35] [28] |
| Satureja hortensis (Summer savory) | 248 | [41] | Mentha arvensis (Pudina) | 1080 | [35] |
| Solanum melongena (Eggplant) | 3.8 | [42] | Mentha cordifolia | 1000 | [35] |
| Vaccinium myrtillus (Blueberry) | 59.66 | [43] | Mentha piperita (Peppermint) | 57.6 | [28] |
| Moringa oleifera (Moringa) | 40–300 | [35] | Rosmarinus officinalis (Rosemary) | 1460 ** 43.6 ** | [35] [28] |
| Thymus vulgaris (Thyme) | 117 * 1550 ** 69.4 ** | [39] [35] [28] | Origanum majorana (Marjoram) | 104 * 67 ** | [39] [28] |
| Ocimum basilicum (Basil) | 16.6–41.1 * 54.4 ** 3110 ** | [29] [28] [35] | Origanum vulgare (Oregano) | 4100 ** 140 ** 41.4 ** | [35] [41] [28] |
| Sample | 1 h | 3 h | 6 h | 12 h | 24 h | 48 h | 72 h |
|---|---|---|---|---|---|---|---|
| Stomach | 62 | 7.1 | 0.2 | 0.4 | 0.1 | 0.2 | 0.1 |
| Duodenum | 7.9 | 8.1 | 0.2 | 0.3 | 0.1 | 0.1 | <0.1 |
| Jejunum/ileum | 9.1 | 16 | 1.1 | 1.4 | 1.1 | 0.7 | <0.1 |
| Cecum | <0.1 | 7.4 | 6.6 | 3.9 | 1.9 | 0.3 | <0.1 |
| Colon | <0.1 | 7.4 | 6.6 | 3.9 | 1.9 | 0.3 | <0.1 |
| Aim | Model | Treatment Conditions | Finding | Reference |
|---|---|---|---|---|
|
To investigate the radioprotective potential of CA against γ radiation-induced cellular changes |
In vitro: human peripheral blood lymphocytes | CA 66 µM for 30 min before γ radiation (1, 2, 3 y, and 4 Gy) |
Pre-treatment with CA before γ radiation treatment showed significant cell protection (around 80–85%). Overall, CA protects lymphocytes by decreasing (p < 0.01) DNA damage in micronucleus frequencies (MNs) by comet assay, decreasing the level of lipid peroxidation index by TBARS, and improving the antioxidant activity by increasing GSH, SOD, CAT, and GPx levels. | [148] |
|
To investigate the protective role of CA in human epidermal keratinocytes and carcinogenesis induced by cancer treatments with ionizing radiation (γ or X-rays) |
In vitro: human epidermal keratinocyte line HaCaT cells | 0.1 µg/mL of CA for 24 h prior to γ radiation at 4 Gy (1 Gy/min) for 10 min. | Pre-treatment with CA increased the cell survival significantly (p < 0.05) by about 40% at 8 Gy level and reduced ROS production by 38% (p < 0.05), which was induced radiation. CA pre-treatment considerably reduced the number of foci of DNA strand breaks at each time point compared to the control. | [149] |
| To assess the activity of zinc oxide–caffeic acid nanoparticles (ZnO-CA NPs) against cancer cell lines and evaluated on Ehrlich carcinoma treated with γ radiation. |
In vitro: human breast cancer MCF-7 cell line and human liver cancer cell line HepG2 In vivo: Ehrlich carcinoma bearing mice (EC mice) |
In vitro: Not specified. In vivo: Animals were treated with γ radiation at a dose rate of 0.45 Gy/min in a treatment of 2 Gy/week for 3 successive doses. Animals were injected IP with ZnO-CA NPs (5 mg/100 g) in different experiments. |
In vitro: ZnO-CA NPs showed antiproliferative activity against cancer cell lines. The IC50 values of ZnO-CA NPs were 9.22 and 11.53 µg/mL for MCF7 and HepG2, respectively. In vivo: ZnO-CA NPs increased the antitumor activity in mice treated with γ radiation. The LD50 for ZnO-CA NPs was determined in 50.0 mg/100 g bw. The tumor weight decreased from 56.1% (ZnO-CA NPs) to 71.9% in the combination treatment with γ radiation after 4 weeks compared to untreated solid EC tumor. | [150] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cortez, N.; Villegas, C.; Burgos, V.; Cabrera-Pardo, J.R.; Ortiz, L.; González-Chavarría, I.; Nchiozem-Ngnitedem, V.-A.; Paz, C. Adjuvant Properties of Caffeic Acid in Cancer Treatment. Int. J. Mol. Sci. 2024, 25, 7631. https://doi.org/10.3390/ijms25147631
Cortez N, Villegas C, Burgos V, Cabrera-Pardo JR, Ortiz L, González-Chavarría I, Nchiozem-Ngnitedem V-A, Paz C. Adjuvant Properties of Caffeic Acid in Cancer Treatment. International Journal of Molecular Sciences. 2024; 25(14):7631. https://doi.org/10.3390/ijms25147631
Chicago/Turabian StyleCortez, Nicole, Cecilia Villegas, Viviana Burgos, Jaime R. Cabrera-Pardo, Leandro Ortiz, Iván González-Chavarría, Vaderament-A. Nchiozem-Ngnitedem, and Cristian Paz. 2024. "Adjuvant Properties of Caffeic Acid in Cancer Treatment" International Journal of Molecular Sciences 25, no. 14: 7631. https://doi.org/10.3390/ijms25147631
APA StyleCortez, N., Villegas, C., Burgos, V., Cabrera-Pardo, J. R., Ortiz, L., González-Chavarría, I., Nchiozem-Ngnitedem, V.-A., & Paz, C. (2024). Adjuvant Properties of Caffeic Acid in Cancer Treatment. International Journal of Molecular Sciences, 25(14), 7631. https://doi.org/10.3390/ijms25147631