

FGL2/FcγRIIB Signalling Mediates Arterial Shear Stress-Mediated Endothelial Cell Apoptosis: Implications for Coronary Artery Bypass Vein Graft Pathogenesis




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Exposure of vECs to Arterial FSS Induced a Pro-Inflammatory Phenotype and Increased Apoptosis
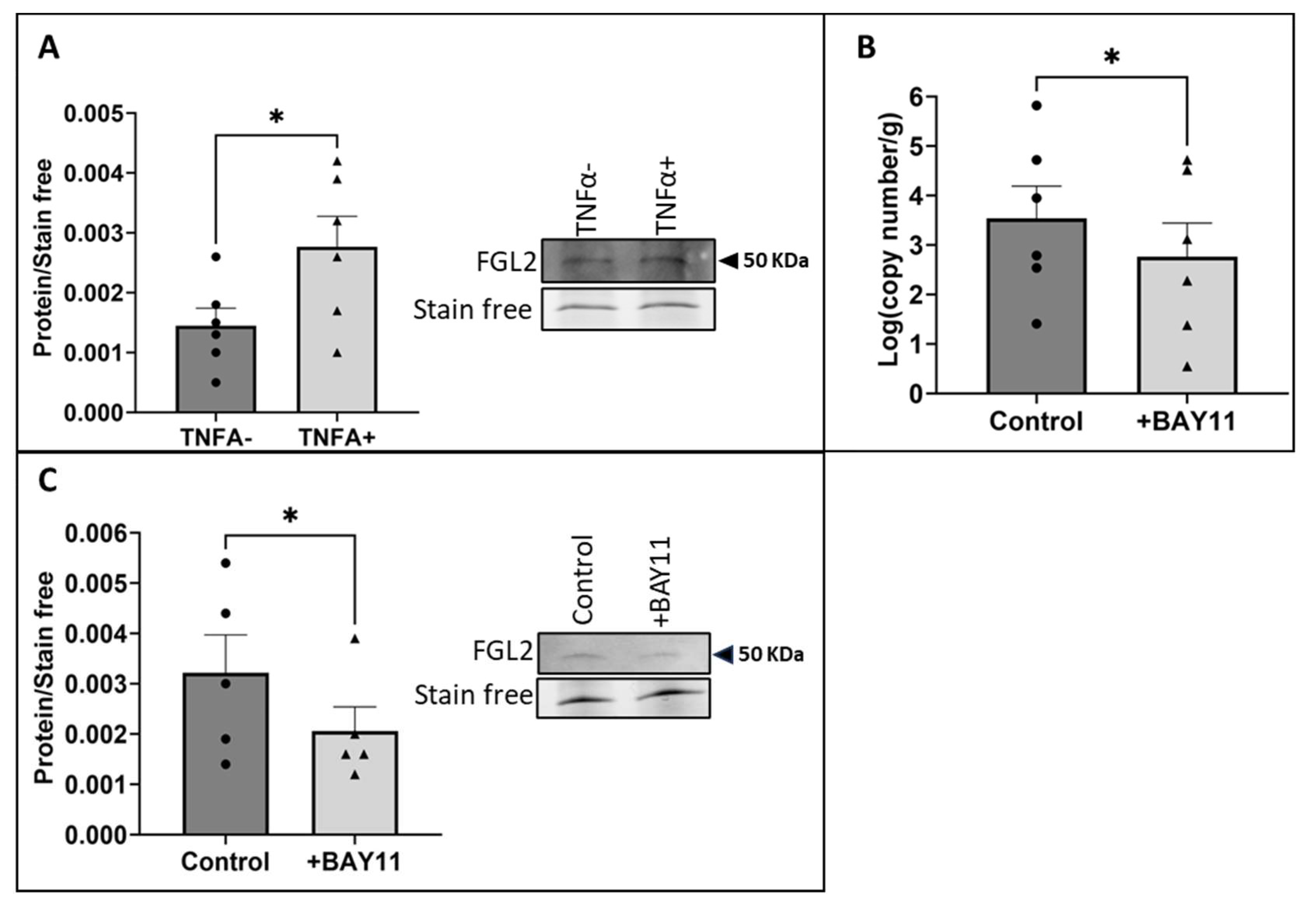
2.2. The Exposure of vECs to Arterial FSS Resulted in Increased Production and Release of FGL2
2.3. Shear Stress-Induced Production and Release of FGL2 Was Mediated by NFκB Activity
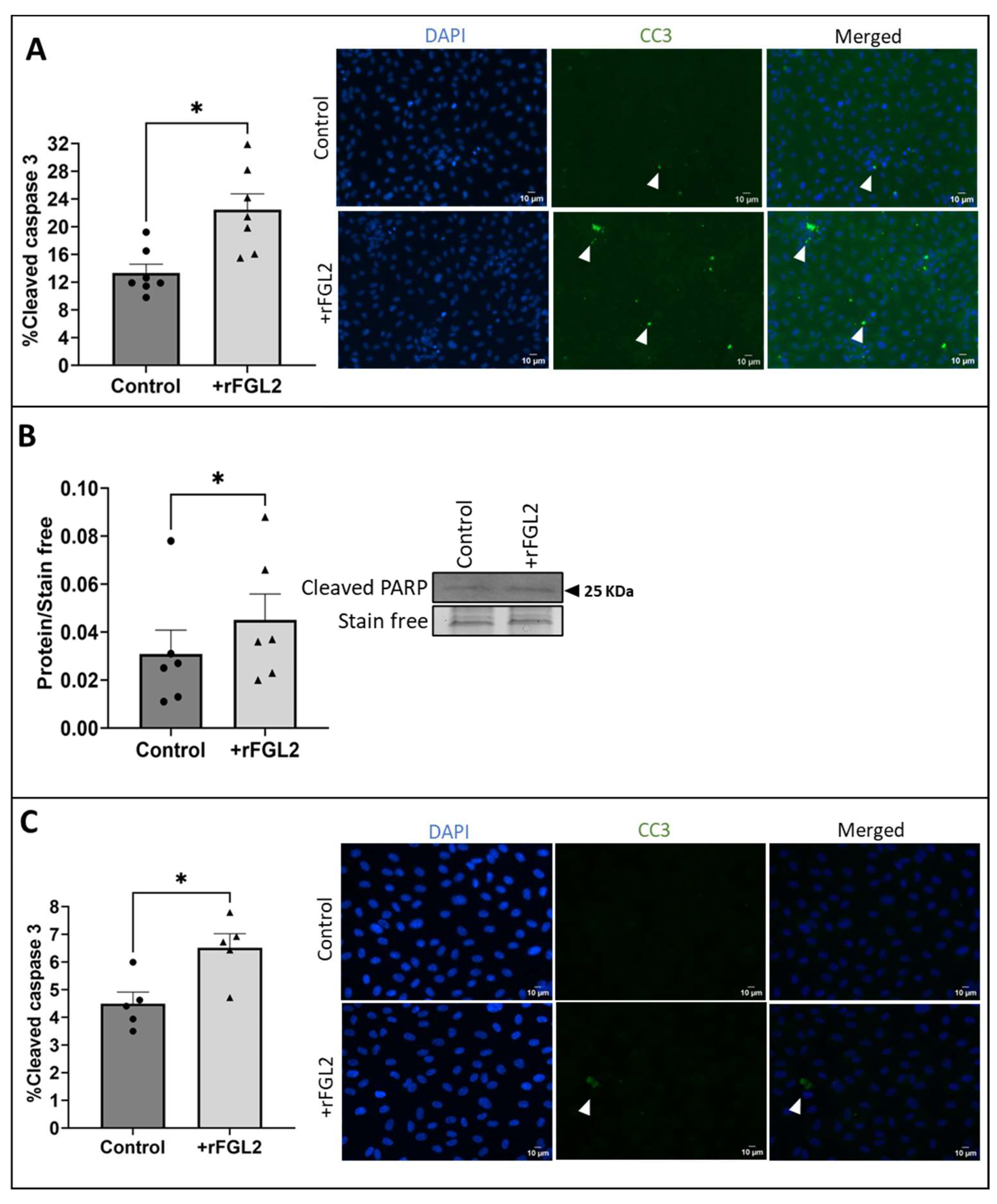
2.4. FGL2 Induced Apoptosis in vECs Cultured in Static Conditions and vECs Exposed to Venous FSS
2.5. FcγRIIB Was Detected in vECs Exposed to Venous and Arterial FSS
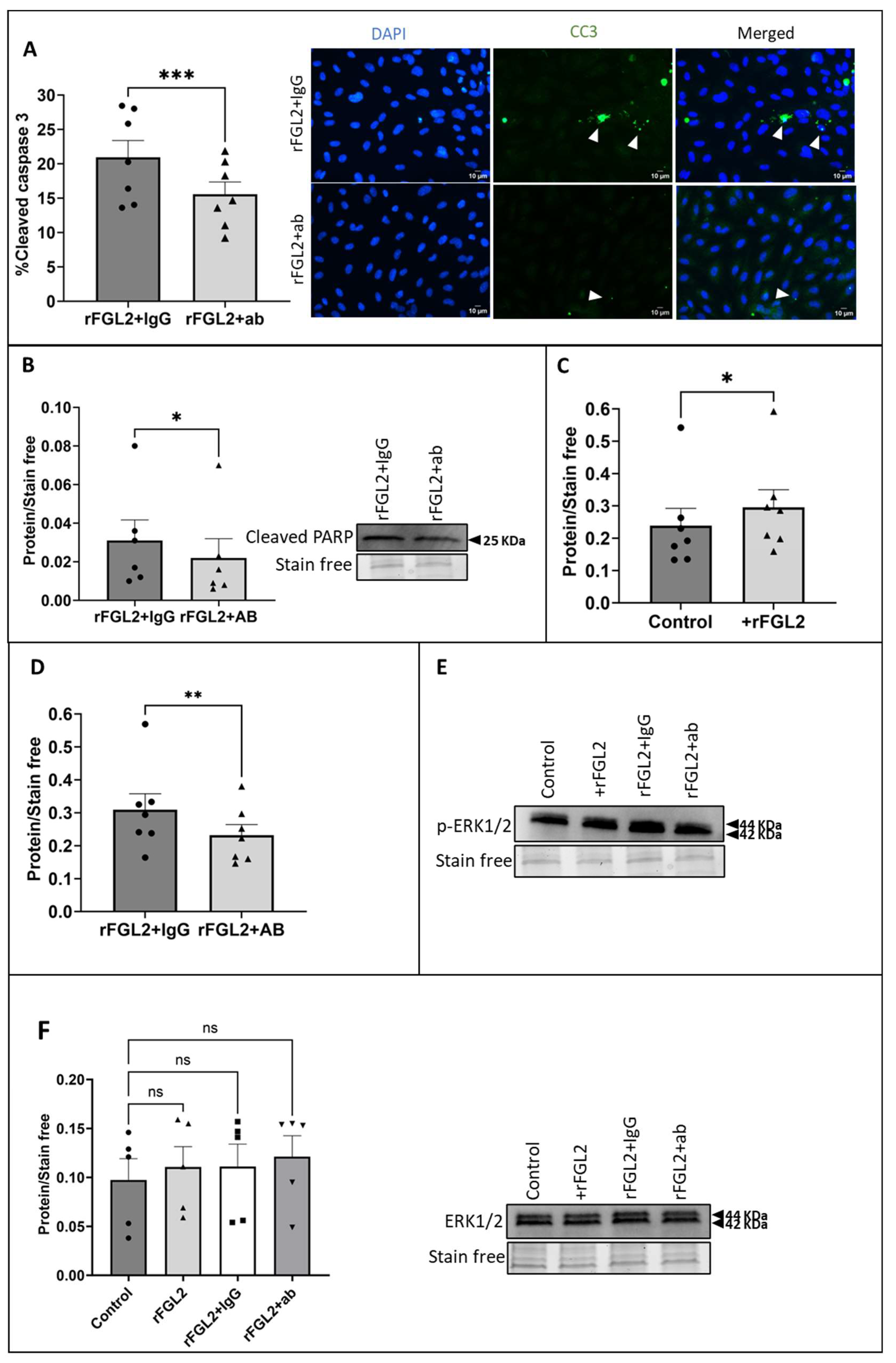
2.6. Blocking FcγRIIB Attenuated FGL2-Induced EC Apoptosis and ERK1/2 Signalling
3. Discussion
4. Materials and Methods
4.1. Culture of HUVECs
4.2. Exposure of ECs to Fluid Shear Stress
4.3. Secretome Analysis
4.4. Western Blotting
4.5. RT-qPCR
4.6. Immunocytochemistry
4.7. Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alabas, O.A.; Jernberg, T.; Pujades-Rodriguez, M.; Rutherford, M.J.; West, R.M.; Hall, M.; Timmis, A.; Lindahl, B.; Fox, K.; Hemingway, H.; et al. Statistics on mortality following acute myocardial infarction in 842 897 Europeans. Cardiovasc. Res. 2020, 116, 149–157. [Google Scholar] [CrossRef]
- Malakar, A.K.; Choudhury, D.; Halder, B.; Paul, P.; Uddin, A.; Chakraborty, S. A review on coronary artery disease, its risk factors, and therapeutics. J. Cell. Physiol. 2019, 234, 16812–16823. [Google Scholar] [CrossRef] [PubMed]
- Gaziano, T.A.; Bitton, A.; Anand, S.; Abrahams-Gessel, S.; Murphy, A. Growing epidemic of coronary heart disease in low- and middle-income countries. Curr. Probl. Cardiol. 2010, 35, 72–115. [Google Scholar] [CrossRef] [PubMed]
- Kiani, F.; Hesabi, N.; Arbabisarjou, A. Assessment of Risk Factors in Patients with Myocardial Infarction. Glob. J. Health Sci. 2015, 8, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Salehi-Abargouei, A.; Maghsoudi, Z.; Shirani, F.; Azadbakht, L. Effects of Dietary Approaches to Stop Hypertension (DASH)-style diet on fatal or nonfatal cardiovascular diseases—Incidence: A systematic review and meta-analysis on observational prospective studies. Nutrition 2013, 29, 611–618. [Google Scholar] [CrossRef]
- Pan, A.; Wang, Y.; Talaei, M.; Hu, F.B. Relation of Smoking with Total Mortality and Cardiovascular Events Among Patients with Diabetes Mellitus: A Meta-Analysis and Systematic Review. Circulation 2015, 132, 1795–1804. [Google Scholar] [CrossRef] [PubMed]
- Joseph, P.; Leong, D.; McKee, M.; Anand, S.S.; Schwalm, J.D.; Teo, K.; Mente, A.; Yusuf, S. Reducing the Global Burden of Cardiovascular Disease, Part 1: The Epidemiology and Risk Factors. Circ. Res. 2017, 121, 677–694. [Google Scholar] [CrossRef] [PubMed]
- Neumann, F.-J.; Sousa-Uva, M.; Ahlsson, A.; Alfonso, F.; Banning, A.P.; Benedetto, U.; Byrne, R.A.; Collet, J.-P.; Falk, V.; Head, S.J.; et al. 2018 ESC/EACTS Guidelines on myocardial revascularization. Eur. Heart J. 2019, 40, 87–165. [Google Scholar] [CrossRef]
- Guida, G.; Ward, A.O.; Bruno, V.D.; George, S.J.; Caputo, M.; Angelini, G.D.; Zakkar, M. Saphenous vein graft disease, pathophysiology, prevention, and treatment. A review of the literature. J. Card. Surg. 2020, 35, 1314–1321. [Google Scholar] [CrossRef]
- Spadaccio, C.; Antoniades, C.; Nenna, A.; Chung, C.; Will, R.; Chello, M.; Gaudino, M.F.L. Preventing treatment failures in coronary artery disease: What can we learn from the biology of in-stent restenosis, vein graft failure, and internal thoracic arteries? Cardiovasc. Res. 2020, 116, 505–519. [Google Scholar] [CrossRef]
- Goldman, S.; Zadina, K.; Moritz, T.; Ovitt, T.; Sethi, G.; Copeland, J.G.; Thottapurathu, L.; Krasnicka, B.; Ellis, N.; Anderson, R.J.; et al. Long-term patency of saphenous vein and left internal mammary artery grafts after coronary artery bypass surgery: Results from a Department of Veterans Affairs Cooperative Study. J. Am. Coll. Cardiol. 2004, 44, 2149–2156. [Google Scholar] [CrossRef] [PubMed]
- Desai, N.D.; Cohen, E.A.; Naylor, C.D.; Fremes, S.E. A Randomized Comparison of Radial-Artery and Saphenous-Vein Coronary Bypass Grafts. N. Engl. J. Med. 2004, 351, 2302–2309. [Google Scholar] [CrossRef] [PubMed]
- Caliskan, E.; de Souza, D.R.; Böning, A.; Liakopoulos, O.J.; Choi, Y.H.; Pepper, J.; Gibson, C.M.; Perrault, L.P.; Wolf, R.K.; Kim, K.B.; et al. Saphenous vein grafts in contemporary coronary artery bypass graft surgery. Nat. Rev. Cardiol. 2020, 17, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.O.; Caputo, M.; Angelini, G.D.; George, S.J.; Zakkar, M. Activation and inflammation of the venous endothelium in vein graft disease. Atherosclerosis 2017, 265, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Ladak, S.S.; McQueen, L.W.; Layton, G.R.; Aujla, H.; Adebayo, A.; Zakkar, M. The Role of Endothelial Cells in the Onset, Development and Modulation of Vein Graft Disease. Cells 2022, 11, 3066. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.F. Flow-mediated endothelial mechanotransduction. Physiol. Rev. 1995, 75, 519–560. [Google Scholar] [CrossRef]
- Roux, E.; Bougaran, P.; Dufourcq, P.; Couffinhal, T. Fluid Shear Stress Sensing by the Endothelial Layer. Front. Physiol. 2020, 11, 861. [Google Scholar] [CrossRef] [PubMed]
- Baeyens, N.; Bandyopadhyay, C.; Coon, B.G.; Yun, S.; Schwartz, M.A. Endothelial fluid shear stress sensing in vascular health and disease. J. Clin. Investig. 2016, 126, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Gooch, K.J.; Firstenberg, M.S.; Shrefler, B.S.; Scandling, B.W. Biomechanics and Mechanobiology of Saphenous Vein Grafts. J. Biomech. Eng. 2018, 140, 020804. [Google Scholar] [CrossRef]
- Lilly, B. We Have Contact: Endothelial Cell-Smooth Muscle Cell Interactions. Physiology 2014, 29, 234–241. [Google Scholar] [CrossRef]
- Lee, D.D.; Schwarz, M.A. Cell-Cell Communication Breakdown and Endothelial Dysfunction. Crit. Care Clin. 2020, 36, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.O.; Angelini, G.D.; Caputo, M.; Evans, P.C.; Johnson, J.L.; Suleiman, M.S.; Tulloh, R.M.; George, S.J.; Zakkar, M. NF-κB inhibition prevents acute shear stress-induced inflammation in the saphenous vein graft endothelium. Sci. Rep. 2020, 10, 15133. [Google Scholar] [CrossRef] [PubMed]
- Bryan, A.J.; Angelini, G.D. The biology of saphenous vein graft occlusion: Etiology and strategies for prevention. Curr. Opin. Cardiol. 1994, 9, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-Z.; Yang, Y.; Liu, K.; Long, R.; Jin, N.; Huang, S.-Y.; You, Y.; Dai, J.; Fan, C.; Wang, J.; et al. FGL2 prothrombinase contributes to the early stage of coronary microvascular obstruction through a fibrin-dependent pathway. Int. J. Cardiol. 2019, 274, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Ghanekar, A.; Mendicino, M.; Liu, H.; He, W.; Liu, M.; Zhong, R.; Phillips, M.J.; Levy, G.A.; Grant, D.R. Endothelial induction of fgl2 contributes to thrombosis during acute vascular xenograft rejection. J. Immunol. 2004, 172, 5693–5701. [Google Scholar] [CrossRef]
- Fan, C.; Wang, J.; Mao, C.; Li, W.; Liu, K.; Wang, Z. The FGL2 prothrombinase contributes to the pathological process of experimental pulmonary hypertension. J. Appl. Physiol. 2019, 127, 1677–1687. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Yan, J.; Rao, G.; Latha, K.; Overwijk, W.W.; Heimberger, A.B.; Li, S. The Duality of Fgl2–Secreted Immune Checkpoint Regulator Versus Membrane-Associated Procoagulant: Therapeutic Potential and Implications. Int. Rev. Immunol. 2016, 35, 325–339. [Google Scholar] [CrossRef]
- Selzner, N.; Liu, H.; Boehnert, M.U.; Adeyi, O.A.; Shalev, I.; Bartczak, A.M.; Xue-Zhong, M.; Manuel, J.; Rotstein, O.D.; McGilvray, I.D.; et al. FGL2/fibroleukin mediates hepatic reperfusion injury by induction of sinusoidal endothelial cell and hepatocyte apoptosis in mice. J. Hepatol. 2012, 56, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Yang, C.; Wang, L.; Li, L.; Zhao, T.; Hu, L.; Rong, R.; Xu, M.; Zhu, T. The regulatory T cell effector soluble fibrinogen-like protein 2 induces tubular epithelial cell apoptosis in renal transplantation. Exp. Biol. Med. 2014, 239, 193–201. [Google Scholar] [CrossRef]
- Yanaba, K.; Asano, Y.; Noda, S.; Akamata, K.; Aozasa, N.; Taniguchi, T.; Takahashi, T.; Ichimura, Y.; Toyama, T.; Sumida, H.; et al. Increased circulating fibrinogen-like protein 2 in patients with systemic sclerosis. Clin. Rheumatol. 2013, 32, 43–47. [Google Scholar] [CrossRef]
- Liu, M.; Mendicino, M.; Ning, Q.; Ghanekar, A.; He, W.; McGilvray, I.; Shalev, I.; Pivato, D.; Clark, D.A.; Phillips, M.J.; et al. Cytokine-induced hepatic apoptosis is dependent on FGL2/fibroleukin: The role of Sp1/Sp3 and STAT1/PU.1 composite cis elements. J. Immunol. 2006, 176, 7028–7038. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Yan, W.; Guo, W.; Xi, D.; Zhou, Y.; Li, W.; Gao, S.; Liu, M.; Levy, G.; Luo, X. Hepatitis B virus-induced hFGL2 transcription is dependent on c-Ets-2 and MAPK signal pathway. J. Biol. Chem. 2008, 283, 32715–32729. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xu, L.; Zeng, Q.; Wang, J.; Wang, M.; Xi, D.; Wang, X.; Yang, D.; Luo, X.; Ning, Q. Downregulation of FGL2/prothrombinase delays HCCLM6 xenograft tumour growth and decreases tumour angiogenesis. Liver Int. 2012, 32, 1585–1595. [Google Scholar] [CrossRef] [PubMed]
- Dora, K.A. Cell-cell communication in the vessel wall. Vasc. Med. 2001, 6, 43–50. [Google Scholar] [CrossRef]
- Marsden, P.A.; Ning, Q.; Fung, L.S.; Luo, X.; Chen, Y.; Mendicino, M.; Ghanekar, A.; Scott, J.A.; Miller, T.; Chan, C.W. The Fgl2/fibroleukin prothrombinase contributes to immunologically mediated thrombosis in experimental and human viral hepatitis. J. Clin. Investig. 2003, 112, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Su, K.; Chen, F.; Yan, W.-M.; Zeng, Q.-L.; Xu, L.; Xi, D.; Pi, B.; Luo, X.-P.; Ning, Q. Fibrinogen-like protein 2/fibroleukin prothrombinase contributes to tumor hypercoagulability via IL-2 and IFN-γ. World J. Gastroenterol. 2008, 14, 5980. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Liu, K.; Wang, Y.; Su, G.; Deng, H.; Zeng, Q.; Liao, Y.; Wang, Z. Expression and significance of fgl2 prothrombinase in cardiac microvascular endothelial cells of rats with type 2 diabetes. J. Huazhong Univ. Sci. Technol. Med. Sci. 2010, 30, 575–581. [Google Scholar] [CrossRef]
- Ning, Q.; Sun, Y.; Han, M.; Zhang, L.; Zhu, C.; Zhang, W.; Guo, H.; Li, J.; Yan, W.; Gong, F.; et al. Role of Fibrinogen-Like Protein 2 Prothrombinase/Fibroleukin in Experimental and Human Allograft Rejection1. J. Immunol. 2005, 174, 7403–7411. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.F.; Kreisle, R.A.; Shi, Y.D. Detection of Fcgamma receptors on human endothelial cells stimulated with cytokines tumour necrosis factor-alpha (TNF-alpha) and interferon-gamma (IFN-gamma). Clin. Exp. Immunol. 1998, 112, 533–538. [Google Scholar] [CrossRef]
- Tanigaki, K.; Sundgren, N.; Khera, A.; Vongpatanasin, W.; Mineo, C.; Shaul, P.W. Fcγ receptors and ligands and cardiovascular disease. Circ. Res. 2015, 116, 368–384. [Google Scholar] [CrossRef]
- Song, X.; Zou, X.; Ge, W.; Hou, C.; Cao, Z.; Zhao, H.; Zhang, T.; Jin, L.; Fu, Y.; Kong, W.; et al. Blocking FcγRIIB in Smooth Muscle Cells Reduces Hypertension. Circ. Res. 2021, 129, 308–325. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Kassab, G.S. Role of shear stress and stretch in vascular mechanobiology. J. R. Soc. Interface 2011, 8, 1379–1385. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Wu, D.; Zhang, P.; Li, W.; Wang, J.; Su, G.; Liao, Y.; Wang, Z.; Liu, K. The Significance of Microthrombosis and fgl2 in No-Reflow Phenomenon of Rats with Acute Myocardial Ischemia/Reperfusion. Clin. Appl. Thromb./Hemost. 2013, 19, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Motwani, J.G.; Topol, E.J. Aortocoronary saphenous vein graft disease: Pathogenesis, predisposition, and prevention. Circulation 1998, 97, 916–931. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.; Angelini, G.; George, S.; Zakkar, M. Contribution of the classical NF-kappaB pathway to venous endothelial inflammation following acute increases in shear stress: Implications for vein graft failure. Atherosclerosis 2017, 263, e135. [Google Scholar] [CrossRef]
- Liu, M.; Leibowitz, J.L.; Clark, D.A.; Mendicino, M.; Ning, Q.; Ding, J.W.; D’Abreo, C.; Fung, L.; Marsden, P.A.; Levy, G.A. Gene transcription of fgl2 in endothelial cells is controlled by Ets-1 and Oct-1 and requires the presence of both Sp1 and Sp3. Eur. J. Biochem. 2003, 270, 2274–2286. [Google Scholar] [CrossRef] [PubMed]
- Samano, N.; Geijer, H.; Liden, M.; Fremes, S.; Bodin, L.; Souza, D. The no-touch saphenous vein for coronary artery bypass grafting maintains a patency, after 16 years, comparable to the left internal thoracic artery: A randomized trial. J. Thorac. Cardiovasc. Surg. 2015, 150, 880–888. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.Y.; Bobryshev, Y.V.; Cherian, S.M.; Liang, H.; Tran, D.; Inder, S.J.; Lord, R.S.A.; Ashwell, K.W.S.; Farnsworth, A.E. Expression of Apoptosis-Related Proteins and Structural Features of Cell Death in Explanted Aortocoronary Saphenous Vein Bypass Grafts. Cardiovasc. Surg. 2001, 9, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Tricot, O.; Mallat, Z.; Heymes, C.; Belmin, J.; Lesèche, G.; Tedgui, A. Relation Between Endothelial Cell Apoptosis and Blood Flow Direction in Human Atherosclerotic Plaques. Circulation 2000, 101, 2450–2453. [Google Scholar] [CrossRef]
- Cornelissen, J.; Armstrong, J.; Holt, C.M. Mechanical Stretch Induces Phosphorylation of p38-MAPK and Apoptosis in Human Saphenous Vein. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 451–456. [Google Scholar] [CrossRef]
- Ji, Q.; Wang, Y.L.; Xia, L.M.; Yang, Y.; Wang, C.S.; Mei, Y.Q. High shear stress suppresses proliferation and migration but promotes apoptosis of endothelial cells co-cultured with vascular smooth muscle cells via down-regulating MAPK pathway. J. Cardiothorac. Surg. 2019, 14, 216. [Google Scholar] [CrossRef]
- Liu, H.; Shalev, I.; Manuel, J.; He, W.; Leung, E.; Crookshank, J.; Liu, M.F.; Diao, J.; Cattral, M.; Clark, D.A.; et al. The FGL2-FcgammaRIIB pathway: A novel mechanism leading to immunosuppression. Eur. J. Immunol. 2008, 38, 3114–3126. [Google Scholar] [CrossRef]
- Morris, A.B.; Farley, C.R.; Pinelli, D.F.; Adams, L.E.; Cragg, M.S.; Boss, J.M.; Scharer, C.D.; Fribourg, M.; Cravedi, P.; Heeger, P.S.; et al. Signaling through the Inhibitory Fc Receptor FcγRIIB Induces CD8(+) T Cell Apoptosis to Limit T Cell Immunity. Immunity 2020, 52, 136–150.e136. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Fcγ receptors as regulators of immune responses. Nat. Rev. Immunol. 2008, 8, 34–47. [Google Scholar] [CrossRef]
- Mendez-Fernandez, Y.V.; Stevenson, B.G.; Diehl, C.J.; Braun, N.A.; Wade, N.S.; Covarrubias, R.; van Leuven, S.; Witztum, J.L.; Major, A.S. The inhibitory FcγRIIb modulates the inflammatory response and influences atherosclerosis in male apoE−/− mice. Atherosclerosis 2011, 214, 73–80. [Google Scholar] [CrossRef]
- Zhao, M.; Wigren, M.; Dunér, P.; Kolbus, D.; Olofsson, K.E.; Björkbacka, H.; Nilsson, J.; Fredrikson, G.N. FcγRIIB inhibits the development of atherosclerosis in low-density lipoprotein receptor-deficient mice. J. Immunol. 2010, 184, 2253–2260. [Google Scholar] [CrossRef]
- Hage, F. C-reactive protein and hypertension. J. Hum. Hypertens. 2014, 28, 410–415. [Google Scholar] [CrossRef]
- Sundgren, N.C.; Zhu, W.; Yuhanna, I.S.; Chambliss, K.L.; Ahmed, M.; Tanigaki, K.; Umetani, M.; Mineo, C.; Shaul, P.W. Coupling of Fcγ receptor I to Fcγ receptor IIb by SRC kinase mediates C-reactive protein impairment of endothelial function. Circ. Res. 2011, 109, 1132–1140. [Google Scholar] [CrossRef]
- Zeng, M.; Li, Q.; Chen, J.; Huang, W.; Liu, J.; Wang, C.; Huang, M.; Li, H.; Zhou, S.; Xie, M.; et al. The Fgl2 interaction with Tyrobp promotes the proliferation of cutaneous squamous cell carcinoma by regulating ERK-dependent autophagy. Int. J. Med. Sci. 2022, 19, 195–204. [Google Scholar] [CrossRef]
- Rabizadeh, E.; Cherny, I.; Lederfein, D.; Sherman, S.; Binkovsky, N.; Rosenblat, Y.; Inbal, A. The cell-membrane prothrombinase, fibrinogen-like protein 2, promotes angiogenesis and tumor development. Thromb. Res. 2015, 136, 118–124. [Google Scholar] [CrossRef]
- Hong, S.K.; Wu, P.K.; Park, J.I. A cellular threshold for active ERK1/2 levels determines Raf/MEK/ERK-mediated growth arrest versus death responses. Cell. Signal. 2018, 42, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, S.; Yan, Y.; Daubert, R.A.; Han, J.; Schnellmann, R.G. ERK promotes hydrogen peroxide-induced apoptosis through caspase-3 activation and inhibition of Akt in renal epithelial cells. Am. J. Physiol. Ren. Physiol. 2007, 292, F440–F447. [Google Scholar] [CrossRef] [PubMed]
- Cagnol, S.; Chambard, J.-C. ERK and cell death: Mechanisms of ERK-induced cell death–apoptosis, autophagy and senescence. FEBS J. 2010, 277, 2–21. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, K.; Nishida, E. Regulation of MAP kinases by MAP kinase phosphatases. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2007, 1773, 1227–1237. [Google Scholar] [CrossRef] [PubMed]
- Barlev, A.N.; Malkiel, S.; Kurata-Sato, I.; Dorjée, A.L.; Suurmond, J.; Diamond, B. FcγRIIB regulates autoantibody responses by limiting marginal zone B cell activation. J. Clin. Investig. 2022, 132, 157–250. [Google Scholar] [CrossRef]
- Tridandapani, S.; Phee, H.; Shivakumar, L.; Kelley, T.W.; Coggeshall, K.M. Role of ship in Fcγ RIIb-mediated inhibition of Ras activation in B cellsfn3fn3This work was supported by National Institutes of Health grants CA64268 and AI41447 and by grant P30CA16058, National Cancer Institute. Dr Coggeshall is a Scholar of the Leukemia Society of America. Mol. Immunol. 1998, 35, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Ravetch, J.V.; Bolland, S. Igg fc receptors. Annu. Rev. Immunol. 2001, 19, 275–290. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; García-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2021, 50, D543–D552. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jackson, M.L.; Bond, A.R.; Ascione, R.; Johnson, J.L.; George, S.J. FGL2/FcγRIIB Signalling Mediates Arterial Shear Stress-Mediated Endothelial Cell Apoptosis: Implications for Coronary Artery Bypass Vein Graft Pathogenesis. Int. J. Mol. Sci. 2024, 25, 7638. https://doi.org/10.3390/ijms25147638
Jackson ML, Bond AR, Ascione R, Johnson JL, George SJ. FGL2/FcγRIIB Signalling Mediates Arterial Shear Stress-Mediated Endothelial Cell Apoptosis: Implications for Coronary Artery Bypass Vein Graft Pathogenesis. International Journal of Molecular Sciences. 2024; 25(14):7638. https://doi.org/10.3390/ijms25147638
Chicago/Turabian StyleJackson, Molly L., Andrew R. Bond, Raimondo Ascione, Jason L. Johnson, and Sarah J. George. 2024. "FGL2/FcγRIIB Signalling Mediates Arterial Shear Stress-Mediated Endothelial Cell Apoptosis: Implications for Coronary Artery Bypass Vein Graft Pathogenesis" International Journal of Molecular Sciences 25, no. 14: 7638. https://doi.org/10.3390/ijms25147638
APA StyleJackson, M. L., Bond, A. R., Ascione, R., Johnson, J. L., & George, S. J. (2024). FGL2/FcγRIIB Signalling Mediates Arterial Shear Stress-Mediated Endothelial Cell Apoptosis: Implications for Coronary Artery Bypass Vein Graft Pathogenesis. International Journal of Molecular Sciences, 25(14), 7638. https://doi.org/10.3390/ijms25147638