
Understanding Autoimmunity: Mechanisms, Predisposing Factors, and Cytokine Therapies


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
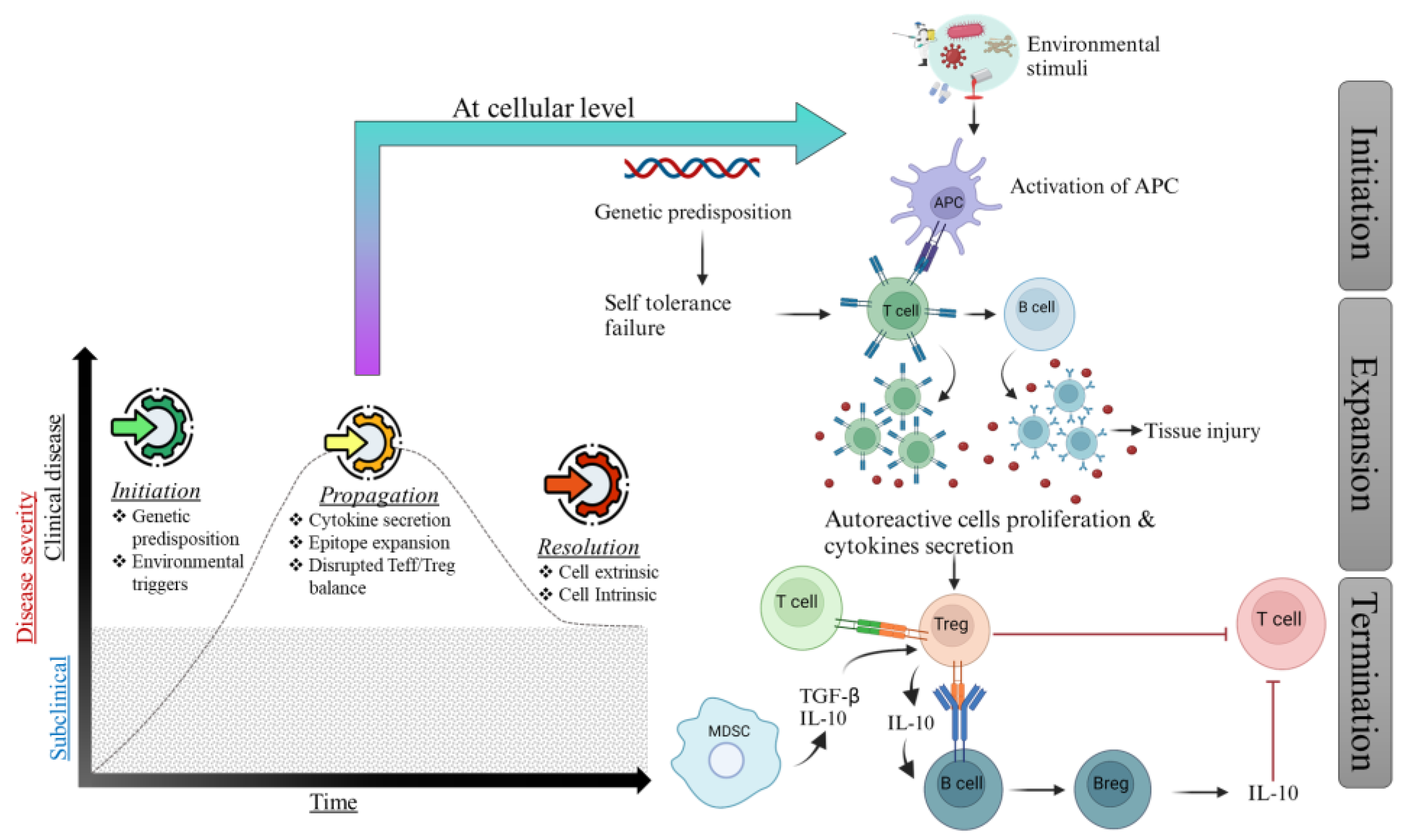
2. Mechanism of Autoimmunity
3. Immune Tolerance
3.1. Central Tolerance
3.2. Peripheral Tolerance
4. Breakdown of Tolerance
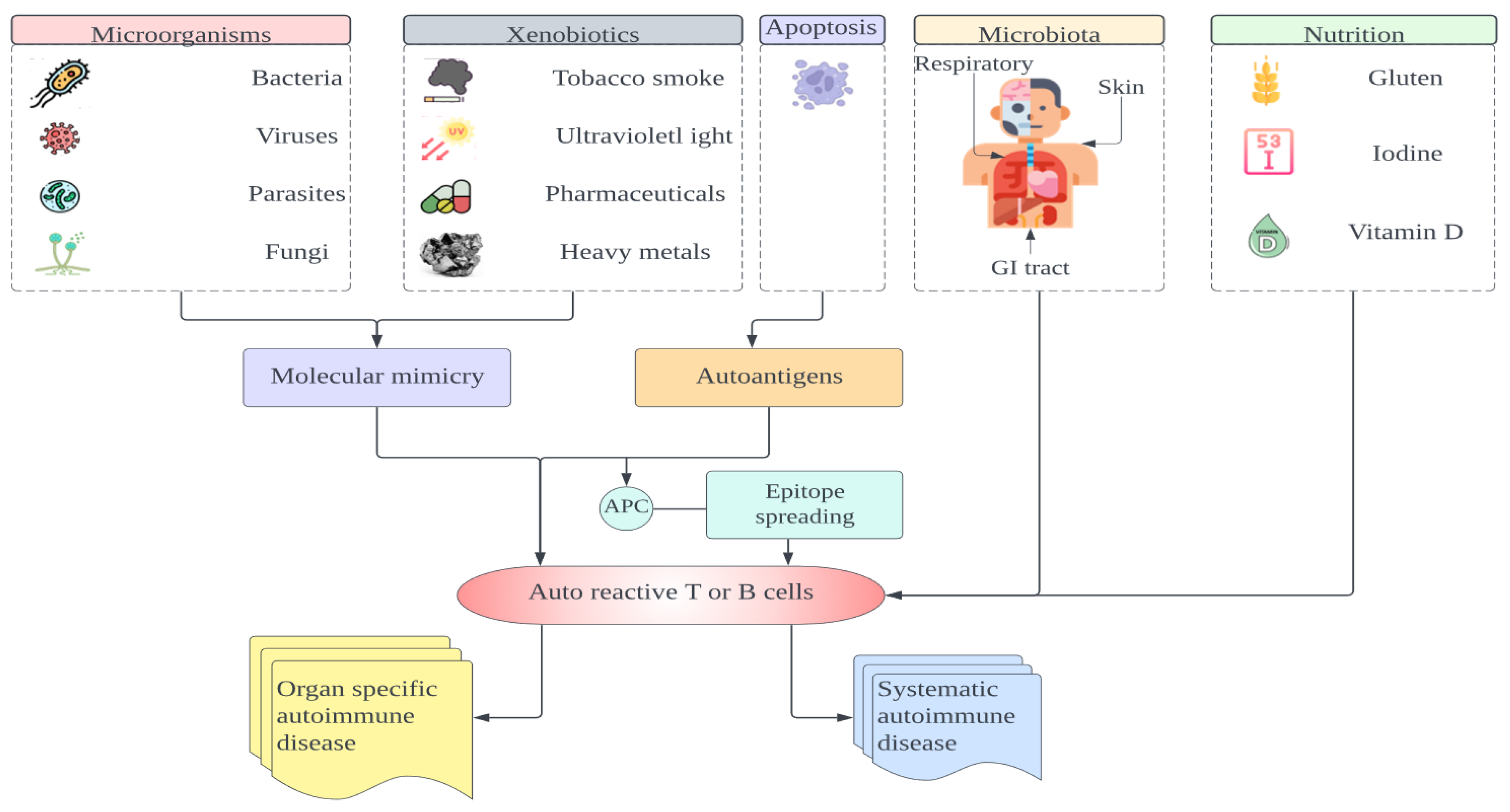
5. Predisposing Factors of Autoimmune Diseases
5.1. Gender
5.2. Environment
5.3. Genetic Risk Factors
5.4. Infections
5.5. The Gut Microbiota
5.6. UV Radiation
5.7. Vitamin D Deficiency
5.8. Alcohol
5.9. Drugs
6. Personal Care Products and Cosmetics
Smoking, Silica, Tropospheric Pollutants, and Solvents/Pesticides
7. Cytokine Therapy for Autoimmune Diseases
8. Anti-TNF Therapy
9. Anti-IL-1 Therapy
9.1. IL-6
9.2. Type 1 Anti-IFN Alpha Therapy
9.3. IL-17
9.4. IL-23
10. Alternative Therapeutic Approaches Targeting Immune Cells
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Warrington, R.; Watson, W.; Kim, H.L.; Antonetti, F.R. An introduction to immunology and immunopathology. Allergy Asthma Clin. Immunol. 2011, 7, S1. [Google Scholar] [CrossRef] [PubMed]
- Burnet, F. A Reassessment of the Forbidden Clone Hypothesis of Autoimmune Disease. Immunol. Cell Biol. 1972, 50, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Tauber, A.I.; Podolsky, S.H. Frank Macfarlane Burnet and the immune self. J. Hist. Biol. 1994, 27, 531–573. [Google Scholar] [CrossRef] [PubMed]
- Pisetsky, D.S. Pathogenesis of autoimmune disease. Nat. Rev. Nephrol. 2023, 19, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.C.; Hansoul, S.; Nicolae, D.L.; Cho, J.H.; Duerr, R.H.; Rioux, J.D.; Brant, S.R.; Silverberg, M.S.; Taylor, K.D.; Barmada, M.M.; et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat. Genet. 2008, 40, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Fraga, M.F.; Ballestar, E.; Paz, M.F.; Ropero, S.; Setien, F.; Ballestar, M.L.; Heine-Suñer, D.; Cigudosa, J.C.; Urioste, M.; Benitez, J.; et al. From The Cover: Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA 2005, 102, 10604–10609. [Google Scholar] [CrossRef] [PubMed]
- Brooks, W.H.; Le Dantec, C.; Pers, J.-O.; Youinou, P.; Renaudineau, Y. Epigenetics and autoimmunity. J. Autoimmun. 2010, 34, J207–J219. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Menezes, J.S.; Umesaki, Y.; Mazmanian, S.K. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2010, 108, 4615–4622. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Gómez, A.; Perretti, M.; Soehnlein, O. Resolution of inflammation: An integrated view. EMBO Mol. Med. 2013, 5, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.S.; A Kriegel, M. Evolving concepts of host–pathobiont interactions in autoimmunity. Curr. Opin. Immunol. 2023, 80, 102265. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, M.D.; Remedios, K.A.; Abbas, A.K. Mechanisms of human autoimmunity. J. Clin. Investig. 2015, 125, 2228–2233. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Yu, L. Understanding Islet Autoantibodies in Prediction of Type 1 Diabetes. J. Endocr. Soc. 2023, 8, bvad160. [Google Scholar] [CrossRef]
- A Piccirillo, C.; Bjur, E.; Topisirovic, I.; Sonenberg, N.; Larsson, O. Translational control of immune responses: From transcripts to translatomes. Nat. Immunol. 2014, 15, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Hogquist, K.A. T-Cell Tolerance: Central and Peripheral. Cold Spring Harb. Perspect. Biol. 2012, 4, a006957. [Google Scholar] [CrossRef]
- Gutcher, I.; Becher, B. APC-derived cytokines and T cell polarization in autoimmune inflammation. J. Clin. Investig. 2007, 117, 1119–1127. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Sung, N.; Gilman-Sachs, A.; Kwak-Kim, J. T Helper (Th) Cell Profiles in Pregnancy and Recurrent Pregnancy Losses: Th1/Th2/Th9/Th17/Th22/Tfh Cells. Front. Immunol. 2020, 11, 2025. [Google Scholar] [CrossRef] [PubMed]
- Vedeler, C. Fc receptors for immunoglobulin G—A role in the pathogenesis of Guillain–Barré syndrome and multiple sclerosis. J. Neuroimmunol. 2001, 118, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Bunte, K.; Beikler, T. Th17 Cells and the IL-23/IL-17 Axis in the Pathogenesis of Periodontitis and Immune-Mediated Inflammatory Diseases. Int. J. Mol. Sci. 2019, 20, 3394. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Pascual, V. Type I Interferon in Systemic Lupus Erythematosus and Other Autoimmune Diseases. Immunity 2006, 25, 383–392. [Google Scholar] [CrossRef]
- Leung, S.; Liu, X.; Fang, L.; Chen, X.; Guo, T.; Zhang, J. The cytokine milieu in the interplay of pathogenic Th1/Th17 cells and regulatory T cells in autoimmune disease. Cell. Mol. Immunol. 2010, 7, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Weaver, C.T.; Hatton, R.D.; Mangan, P.R.; Harrington, L.E. IL-17 Family Cytokines and the Expanding Diversity of Effector T Cell Lineages. Annu. Rev. Immunol. 2007, 25, 821–852. [Google Scholar] [CrossRef] [PubMed]
- Komiyama, Y.; Nakae, S.; Matsuki, T.; Nambu, A.; Ishigame, H.; Kakuta, S.; Sudo, K.; Iwakura, Y. IL-17 Plays an Important Role in the Development of Experimental Autoimmune Encephalomyelitis. J. Immunol. 2006, 177, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.-Y.; Kim, J.-Y.; Kim, K.-W.; Park, M.-K.; Moon, Y.; Kim, W.-U.; Kim, H.-Y. IL-17 induces production of IL-6 and IL-8 in rheumatoid arthritis synovial fibroblasts via NF-κB- and PI3-kinase/Akt-dependent pathways. Arthritis Res. Ther. 2004, 6, R120–R128. [Google Scholar] [CrossRef] [PubMed]
- Kannan, A.K.; Su, Z.; Gauvin, D.M.; Paulsboe, S.E.; Duggan, R.; Lasko, L.M.; Honore, P.; Kort, M.E.; McGaraughty, S.P.; Scott, V.E.; et al. IL-23 induces regulatory T cell plasticity with implications for inflammatory skin diseases. Sci. Rep. 2019, 9, 17675. [Google Scholar] [CrossRef] [PubMed]
- Savage, N.D.L.; de Boer, T.; Walburg, K.V.; Joosten, S.A.; van Meijgaarden, K.; Geluk, A.; Ottenhoff, T.H.M. Human Anti-Inflammatory Macrophages Induce Foxp3+GITR+CD25+ Regulatory T Cells, Which Suppress via Membrane-Bound TGFβ-1. J. Immunol. 2008, 181, 2220–2226. [Google Scholar] [CrossRef]
- Sokolov, A.V.; Shmidt, A.A.; Lomakin, Y.A. B Cell Regulation in Autoimmune Diseases. Acta Nat. 2018, 10, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wakeham, J.; Harkness, R.; Xing, Z. Macrophages Are a Significant Source of Type 1 Cytokines during Mycobacterial Infection. J. Clin. Investig. 1999, 103, 1023–1029. [Google Scholar] [CrossRef] [PubMed]
- Elshafey, R.; Siaj, M.; Zourob, M. In Vitro Selection, Characterization, and Biosensing Application of High-Affinity Cylindrospermopsin-Targeting Aptamers. Anal. Chem. 2014, 86, 9196–9203. [Google Scholar] [CrossRef] [PubMed]
- Sundberg, E.J.; Deng, L.; Mariuzza, R.A. TCR recognition of peptide/MHC class II complexes and superantigens. Semin. Immunol. 2007, 19, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.F.A.P. The golden anniversary of the thymus. Nat. Rev. Immunol. 2011, 11, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.M. T Cell Receptor Gene Diversity and Selection. Annu. Rev. Biochem. 1990, 59, 475–496. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.P.; Ruiter, B.; Virkud, Y.V.; Tu, A.A.; Monian, B.; Moon, J.J.; Love, J.C.; Shreffler, W.G. Identification of antigen-specific TCR sequences based on biological and statistical enrichment in unselected individuals. J. Clin. Investig. 2021, 6, e140028. [Google Scholar] [CrossRef] [PubMed]
- Rashighi, M.; Harris, J.E. T Cell Receptor Sequencing in Autoimmunity. Physiol. Behav. 2017, 176, 139–148. [Google Scholar]
- Klein, L.; Kyewski, B.; Allen, P.M.; Hogquist, K.A. Positive and negative selection of the T cell repertoire: What thymocytes see (and don’t see). Nat. Rev. Immunol. 2014, 14, 377–391. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, H.; Sprent, J. Negative Selection in the Thymus Includes Semimature T Cells. J. Exp. Med. 1997, 185, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.S.; Venanzi, E.S.; Klein, L.; Chen, Z.; Berzins, S.P.; Turley, S.J.; von Boehmer, H.; Bronson, R.; Dierich, A.; Benoist, C.; et al. Projection of an Immunological Self Shadow Within the Thymus by the Aire Protein. Science 2002, 298, 1395–1401. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, C.; Trofimov, A.; Brochu, S.; Lemieux, S.; Perreault, C. Differential Features of AIRE-Induced and AIRE-Independent Promiscuous Gene Expression in Thymic Epithelial Cells. J. Immunol. 2015, 195, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Husebye, E.S.; Anderson, M.S.; Kämpe, O. Autoimmune Polyendocrine Syndromes. Ingelfinger JR, editor. N. Engl. J. Med. 2018, 378, 1132–1141. [Google Scholar] [CrossRef]
- Bluestone, J.A.; Bour-Jordan, H.; Cheng, M.; Anderson, M. T cells in the control of organ-specific autoimmunity. J. Clin. Investig. 2015, 125, 2250–2260. [Google Scholar] [CrossRef]
- Žumer, K.; Saksela, K.; Peterlin, B.M. The Mechanism of Tissue-Restricted Antigen Gene Expression by AIRE. J. Immunol. 2013, 190, 2479–2482. [Google Scholar] [CrossRef]
- Suvas, S.; Singh, V.; Sahdev, S.; Vohra, H.; Agrewala, J.N. Distinct Role of CD80 and CD86 in the Regulation of the Activation of B Cell and B Cell Lymphoma. J. Biol. Chem. 2002, 277, 7766–7775. [Google Scholar] [CrossRef] [PubMed]
- Diaz, D.; Chara, L.; Chevarria, J.; Ubeda, M.; Muñoz, L.; Barcenilla, H.; Sánchez, M.A.; Moreno, Z.; Monserrat, J.; Albillos, A.; et al. Loss of surface antigens is a conserved feature of apoptotic lymphocytes from several mammalian species. Cell. Immunol. 2011, 271, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Zhong, W.; Lu, X.; Shi, B.; Zhu, Y.; Chen, L.; Zhang, G.; Zhang, X. Association of Graves’ Disease and Prevalence of Circulating IFN-γ-producing CD28− T Cells. J. Clin. Immunol. 2008, 28, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Esensten, J.H.; Helou, Y.A.; Chopra, G.; Weiss, A.; Bluestone, J.A. CD28 Costimulation: From Mechanism to Therapy. Immunity 2016, 44, 973–988. [Google Scholar] [CrossRef] [PubMed]
- Rudd, C.E.; Taylor, A.; Schneider, H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol. Rev. 2009, 229, 12–26. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, T.C.; Davies, D.R.; Hisaminato, A.; Resnicow, D.I.; Gupta, S.; Waugh, S.M.; Nagabukuro, A.; Wadatsu, T.; Hishigaki, H.; Gawande, B.; et al. Non-helical DNA Triplex Forms a Unique Aptamer Scaffold for High Affinity Recognition of Nerve Growth Factor. Structure 2015, 23, 1293–1304. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Fife, B.T.; Guleria, I.; Bupp, M.G.; Eagar, T.N.; Tang, Q.; Bour-Jordan, H.; Yagita, H.; Azuma, M.; Sayegh, M.H.; Bluestone, J.A. Insulin-induced remission in new-onset NOD mice is maintained by the PD-1–PD-L1 pathway. J. Exp. Med. 2006, 203, 2737–2747. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Xiong, L.; Wang, Y.; Ding, L.; Hu, S.; Zhao, M.; Zhou, L. Treatment of murine lupus with PD-LIg. Clin. Immunol. 2016, 162, 1–8. [Google Scholar] [CrossRef]
- Curran, M.A.; Montalvo, W.; Yagita, H.; Allison, J.P. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 4275–4280. [Google Scholar] [CrossRef] [PubMed]
- Hui, R.; Garon, E.B.; Goldman, J.W.; Leighl, N.B.; Hellmann, M.D.; Patnaik, A.; Gandhi, L.; Eder, J.P.; Ahn, M.-J.; Horn, L.; et al. Pembrolizumab as first-line therapy for patients with PD-L1-positive advanced non-small cell lung cancer: A phase 1 trial. Ann. Oncol. 2017, 28, 874–881. [Google Scholar] [CrossRef] [PubMed]
- Schilsky, J.; Dvorai, R.H.; Yang, C.; Suo, L.; Saracino, G.; Shahbazov, R. Belatacept based immunosuppression: What and when to combine? Transpl. Immunol. 2024, 85, 102050. [Google Scholar] [CrossRef]
- Egen, J.G.; Kuhns, M.S.; Allison, J.P. CTLA-4: New insights into its biological function and use in tumor immunotherapy. Nat. Immunol. 2002, 3, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.M.R.; Muller, Y.D.; Bluestone, J.A.; Tang, Q. Next-generation regulatory T cell therapy. Nat. Rev. Drug Discov. 2019, 18, 749–769. [Google Scholar] [CrossRef] [PubMed]
- Dorsey, M.J.; Petrovic, A.; Morrow, M.R.; Dishaw, L.J.; Sleasman, J.W. FOXP3 expression following bone marrow transplantation for IPEX syndrome after reduced-intensity conditioning. Immunol. Res. 2009, 44, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Aschenbrenner, K.; D’Cruz, L.M.; Vollmann, E.H.; Hinterberger, M.; Emmerich, J.; Swee, L.K.; Rolink, A.; Klein, L. Selection of Foxp3+ regulatory T cells specific for self antigen expressed and presented by Aire+ medullary thymic epithelial cells. Nat. Immunol. 2007, 8, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Gardner, J.M.; DeVoss, J.J.; Friedman, R.S.; Wong, D.J.; Tan, Y.X.; Zhou, X.; Johannes, K.P.; Su, M.A.; Chang, H.Y.; Krummel, M.F.; et al. Deletional Tolerance Mediated by Extrathymic Aire-Expressing Cells. Science 2008, 321, 843–847. [Google Scholar] [CrossRef] [PubMed]
- Abbas, A.K.; Benoist, C.; A Bluestone, J.; Campbell, D.J.; Ghosh, S.; Hori, S.; Jiang, S.; Kuchroo, V.K.; Mathis, D.; Roncarolo, M.G.; et al. Regulatory T cells: Recommendations to simplify the nomenclature. Nat. Immunol. 2013, 14, 307–308. [Google Scholar] [CrossRef]
- Round, J.L.; Mazmanian, S.K. The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 2009, 9, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Ander, S.E.; Diamond, M.S.; Coyne, C.B. Immune responses at the maternal-fetal interface. Sci. Immunol. 2019, 4, eaat6114. [Google Scholar] [CrossRef] [PubMed]
- Rosser, E.C.; Mauri, C. Regulatory B Cells: Origin, Phenotype, and Function. Immunity 2015, 42, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Talmadge, J.E.; Gabrilovich, D.I. History of myeloid-derived suppressor cells. Nat. Rev. Cancer 2013, 13, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Belkaid, Y.; Hand, T.W. Role of the Microbiota in Immunity and Inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Rasouli-Saravani, A.; Jahankhani, K.; Moradi, S.; Gorgani, M.; Shafaghat, Z.; Mirsanei, Z.; Mehmandar, A.; Mirzaei, R. Role of microbiota short-chain fatty acids in the pathogenesis of autoimmune diseases. Biomed. Pharmacother. 2023, 162, 114620. [Google Scholar] [CrossRef] [PubMed]
- Allard, B.; Allard, D.; Buisseret, L.; Stagg, J. The adenosine pathway in immuno-oncology. Nat. Rev. Clin. Oncol. 2020, 17, 611–629. [Google Scholar] [CrossRef] [PubMed]
- Tsilifis, C.; Slatter, M.A.; Gennery, A.R. Too much of a good thing: A review of primary immune regulatory disorders. Front. Immunol. 2023, 14, 1279201. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J.; on behalf of the HLH Across Speciality Collaboration, UK. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef] [PubMed]
- Lein, L.U.K.; Lugmann, M.A.K.; Ave, K.L.R.N.; Uohy, V.I.K.T.; Yewski, B.R.K. Shaping of the Autoreactive T-Cell Repertoire by a Splice Variant of Self Protein Expressed in Thymic Epithelial Cells. Nat. Med. 2000, 6, 56–61. [Google Scholar]
- Rao, E.; Weiss, B.; Fukami, M.; Rump, A.; Niesler, B.; Mertz, A. The Insulin Gene Is Tran Scribed in the Human Thymus and Transcription Levels Correlated with Allelic Variation at the INS VNTR-IDDM2 Susceptibility Locus for Type 1 Diabetes. Nat. Genet. 1997, 15, 57–61. [Google Scholar]
- Goldacre, M.J.; Wotton, C.J.; Seagroatt, V.; Yeates, D. Multiple sclerosis after infectious mononucleosis: Record linkage study. J. Epidemiol. Community Health 2004, 58, 1032–1035. [Google Scholar] [CrossRef] [PubMed]
- Smatti, M.K.; Cyprian, F.S.; Nasrallah, G.K.; Al Thani, A.A.; Almishal, R.O.; Yassine, H.M. Viruses and Autoimmunity: A Review on the Potential Interaction and Molecular Mechanisms. Viruses 2019, 11, 762. [Google Scholar] [CrossRef] [PubMed]
- Tacken, P.J.; de Vries, I.J.M.; Torensma, R.; Figdor, C.G. Dendritic-cell immunotherapy: From ex vivo loading to in vivo targeting. Nat. Rev. Immunol. 2007, 7, 790–802. [Google Scholar] [CrossRef]
- Vreugdenhil, G.R.; Schloot, N.C.; Hoorens, A.; Rongen, C.; Pipeleers, D.G.; Melchers, W.J.G.; Roep, B.O.; Galama, J.M.D. Acute Onset of Type I Diabetes Mellitus after Severe Echovirus 9 Infection: Putative Pathogenic Pathways. Clin. Infect. Dis. 2000, 31, 1025–1031. [Google Scholar] [CrossRef] [PubMed]
- Clare, E.; Clare, E.; Pa-C, E. Acute disseminated encephalomyelitis. J. Am. Acad. Physician Assist. 2022, 35, 32–35. [Google Scholar] [CrossRef] [PubMed]
- Knapp, S. Diabetes and Infection: Is There a Link?—A Mini-Review. Gerontology 2012, 59, 99–104. [Google Scholar] [CrossRef] [PubMed]
- King, A.J.F. Animal Models of Diabetes; Springer Science and Business Media LLC: Dordrecht, The Netherlands, 2020. [Google Scholar]
- A Lyons, P.; Hancock, W.W.; Denny, P.; Lord, C.J.; Hill, N.J.; Armitage, N.; Siegmund, T.; A Todd, J.; Phillips, M.S.; Hess, J.F.; et al. The NOD Idd9 Genetic Interval Influences the Pathogenicity of Insulitis and Contains Molecular Variants of Cd30, Tnfr2, and Cd137. Immunity 2000, 13, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Janssens, I.; Cools, N. Regulating the regulators: Is introduction of an antigen-specific approach in regulatory T cells the next step to treat autoimmunity? Cell. Immunol. 2020, 358, 104236. [Google Scholar] [CrossRef]
- Tedeschi, S.K.; Bermas, B.; Costenbader, K.H. Sexual disparities in the incidence and course of SLE and RA. Clin. Immunol. 2013, 149, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Markle, J.; Fish, E. SeXX matters in immunity. Trends Immunol. 2014, 35, 97–104. [Google Scholar] [CrossRef]
- ter Horst, R.; Jaeger, M.; Smeekens, S.P.; Oosting, M.; Swertz, M.A.; Li, Y.; Kumar, V.; Diavatopoulos, D.A.; Jansen, A.F.; Lemmers, H.; et al. Host and Environmental Factors Influencing Individual Human Cytokine Responses. Cell 2016, 167, 1111–1124.e13. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, C.M.; Jeganathan, V.; Diamond, B. Hormonal Regulation of B Cell Development: 17β-Estradiol Impairs Negative Selection of High-Affinity DNA-Reactive B Cells at More Than One Developmental Checkpoint. J. Immunol. 2006, 176, 2703–2710. [Google Scholar] [CrossRef] [PubMed]
- Dragin, N.; Bismuth, J.; Cizeron-Clairac, G.; Biferi, M.G.; Berthault, C.; Serraf, A.; Nottin, R.; Klatzmann, D.; Cumano, A.; Barkats, M.; et al. Estrogen-mediated downregulation of AIRE influences sexual dimorphism in autoimmune diseases. J. Clin. Investig. 2016, 126, 1525–1537. [Google Scholar] [CrossRef]
- Kivity, S.; Arango, M.T.; Ehrenfeld, M.; Tehori, O.; Shoenfeld, Y.; Anaya, J.-M.; Agmon-Levin, N. Infection and autoimmunity in Sjogren’s syndrome: A clinical study and comprehensive review. J. Autoimmun. 2014, 51, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Narasimhan, S.; Marchesi, J.R.; Benson, A.; Wong, F.S.; Wen, L. Long term effect of gut microbiota transfer on diabetes development. J. Autoimmun. 2014, 53, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Tsokos, G.C.; Lo, M.S.; Reis, P.C.; Sullivan, K.E. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat. Rev. Rheumatol. 2016, 12, 716–730. [Google Scholar] [CrossRef] [PubMed]
- Ivanovski, T.; Miralles, F. Lambert-Eaton Myasthenic syndrome: Early diagnosis is key. Degener. Neurol. Neuromuscul. Dis. 2019, 9, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Loehrer, P.A.; Zieger, L.; Simon, O.J. Update on Paraneoplastic Cerebellar Degeneration. Brain Sci. 2021, 11, 1414. [Google Scholar] [CrossRef] [PubMed]
- Frommer, L.; Kahaly, G.J. Type 1 Diabetes and Autoimmune Thyroid Disease—The Genetic Link. Front. Endocrinol. 2021, 12, 618213. [Google Scholar] [CrossRef]
- Matzaraki, V.; Kumar, V.; Wijmenga, C.; Zhernakova, A. The MHC locus and genetic susceptibility to autoimmune and infectious diseases. Genome Biol. 2017, 18, 76. [Google Scholar] [CrossRef] [PubMed]
- Chentoufi, A.A.; Gaudreau, S.; Nguyen, A.; Sabha, M.; Amrani, A.; ElGhazali, G. Type I Diabetes-Associated Tolerogenic Properties of Interleukin-2. J. Immunol. Res. 2011, 2011, 289343. [Google Scholar] [CrossRef] [PubMed]
- Lettre, G.; Rioux, J.D. Autoimmune diseases: Insights from genome-wide association studies. Hum. Mol. Genet. 2008, 17, R116–R121. [Google Scholar] [CrossRef]
- Ahmad, R.; Ahsan, H. Dual autoimmune diseases: Rheumatoid arthritis with systemic lupus erythematosus and Type 1 diabetes mellitus with multiple sclerosis. Rheumatol. Autoimmun. 2022, 2, 120–128. [Google Scholar] [CrossRef]
- Iwamoto, T.; Niewold, T.B. Genetics of human lupus nephritis. Clin. Immunol. 2016, 185, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Coenen, M.J.; Trynka, G.; Heskamp, S.; Franke, B.; van Diemen, C.C.; Smolonska, J.; van Leeuwen, M.; Brouwer, E.; Boezen, M.H.; Postma, D.S.; et al. Common and different genetic background for rheumatoid arthritis and coeliac disease. Hum. Mol. Genet. 2009, 18, 4195–4203. [Google Scholar] [CrossRef] [PubMed]
- International Consortium for Systemic Lupus Erythematosus Genetics (SLEGEN); Harley, J.B.; Alarcón-Riquelme, M.E.; Criswell, L.A.; Jacob, C.O.; Kimberly, R.P.; Moser, K.L.; Tsao, B.P.; Vyse, T.J.; Langefeld, C.D.; et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat. Genet. 2008, 40, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Esplin, E.D.; Oei, L.; Snyder, M.P. Pharmacogenomics Personalized Sequencing and the Future Of Personalized sequencing and the future of medicine: Discovery, diagnosis and defeat of disease. Pharmacogenomics 2014, 15, 1771–1790. [Google Scholar] [CrossRef] [PubMed]
- Bogdanos, D.P.; Smyk, D.S.; Invernizzi, P.; Rigopoulou, E.I.; Blank, M.; Pouria, S.; Shoenfeld, Y. Infectome: A platform to trace infectious triggers of autoimmunity. Autoimmun. Rev. 2013, 12, 726–740. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, M.W. Rheumatic Fever, Autoimmunity, and Molecular Mimicry: The Streptococcal Connection. Int. Rev. Immunol. 2014, 33, 314–329. [Google Scholar] [CrossRef]
- Guilherme, L.; Oshiro, S.E.; Faé, K.C.; Cunha-Neto, E.; Renesto, G.; Goldberg, A.C.; Tanaka, A.C.; A Pomerantzeff, P.M.; Kiss, M.H.; Silva, C.; et al. T-Cell Reactivity against Streptococcal Antigens in the Periphery Mirrors Reactivity of Heart-Infiltrating T Lymphocytes in Rheumatic Heart Disease Patients. Infect. Immun. 2001, 69, 5345–5351. [Google Scholar] [CrossRef] [PubMed]
- Ilonen, J.; Lempainen, J.; Veijola, R. The heterogeneous pathogenesis of type 1 diabetes mellitus. Nat. Rev. Endocrinol. 2019, 15, 635–650. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhu, G.; Charlesworth, J.C.; Simpson, S.; Rubicz, R.; Göring, H.H.; A Patsopoulos, N.; Laverty, C.; Wu, F.; Henders, A.; et al. Genetic loci for Epstein-Barr virus nuclear antigen-1 are associated with risk of multiple sclerosis. Mult. Scler. J. 2016, 22, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Kremer, J.M.; Westhovens, R.; Leon, M.; Di Giorgio, E.; Alten, R.; Steinfeld, S.; Russell, A.; Dougados, M.; Emery, P.; Nuamah, I.F.; et al. Treatment of Rheumatoid Arthritis by Selective Inhibition of T-Cell Activation with Fusion Protein CTLA4Ig. New Engl. J. Med. 2003, 349, 1907–1915. [Google Scholar] [CrossRef]
- Strachan, D.P. Hay Fever, Hygiene, and Household Size. Br. Med. J. 1989, 299, 1259–1260. [Google Scholar] [CrossRef] [PubMed]
- Vieira, S.; Pagovich, O.; Kriegel, M. Diet, microbiota and autoimmune diseases. Lupus 2014, 23, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Van Praet, J.T.; Donovan, E.; Vanassche, I.; Drennan, M.B.; Windels, F.; Dendooven, A.; Allais, L.; A Cuvelier, C.; van de Loo, F.; Norris, P.S.; et al. Commensal microbiota influence systemic autoimmune responses. EMBO J. 2015, 34, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Kostic, A.D.; Gevers, D.; Siljander, H.; Vatanen, T.; Hyötyläinen, T.; Hämäläinen, A.-M.; Peet, A.; Tillmann, V.; Pöhö, P.; Mattila, I.; et al. The Dynamics of the Human Infant Gut Microbiome in Development and in Progression toward Type 1 Diabetes. Cell Host Microbe 2015, 17, 260–273. [Google Scholar] [CrossRef]
- Aota, N.; Shiohara, T. Viral connection between drug rashes and autoimmune diseases: How autoimmune responses are generated after resolution of drug rashes. Autoimmun. Rev. 2009, 8, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Foxman, E.F.; Iwasaki, A. Genome–virome interactions: Examining the role of common viral infections in complex disease. Nat. Rev. Microbiol. 2011, 9, 254–264. [Google Scholar] [CrossRef]
- Moon, C.; Stappenbeck, T.S. Viral interactions with the host and microbiota in the intestine. Curr. Opin. Immunol. 2012, 24, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Kau, A.L.; Ahern, P.P.; Griffin, N.W.; Goodman, A.L.; Gordon, J.I. Human nutrition, the gut microbiome and the immune system. Nature 2011, 474, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Greer, R.L.; Morgun, A.; Shulzhenko, N. Bridging immunity and lipid metabolism by gut microbiota. J. Allergy Clin. Immunol. 2013, 132, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Greiner, T.U.; Hyötyläinen, T.; Knip, M.; Bäckhed, F.; Orešič, M. The Gut Microbiota Modulates Glycaemic Control and Serum Metabolite Profiles in Non-Obese Diabetic Mice. PLoS ONE 2014, 9, e110359. [Google Scholar] [CrossRef] [PubMed]
- Rogier, R.; Koenders, M.I.; Abdollahi-Roodsaz, S. Toll-Like Receptor Mediated Modulation of T Cell Response by Commensal Intestinal Microbiota as a Trigger for Autoimmune Arthritis. J. Immunol. Res. 2015, 2015, 527696. [Google Scholar] [CrossRef] [PubMed]
- Coleman, D.J.; Garcia, G.; Hyter, S.; Jang, H.S.; Chagani, S.; Liang, X.; Larue, L.; Ganguli-Indra, G.; Indra, A.K. Retinoid-X-Receptors (α/β) in Melanocytes Modulate Innate Immune Responses and Differentially Regulate Cell Survival following UV Irradiation. PLoS Genet. 2014, 10, e1004321. [Google Scholar] [CrossRef] [PubMed]
- Metwally, D.; Sayed, K.; Hay, R.A.; Rashed, L. Reduction in tissue plasmin: A new mechanism of action of narrowband ultraviolet B in psoriasis. Clin. Exp. Dermatol. 2014, 40, 416–420. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-Y.; Leung, P.S.C.; Adamopoulos, I.E.; Gershwin, M.E. The Implication of Vitamin D and Autoimmunity: A Comprehensive Review. Clin. Rev. Allergy Immunol. 2013, 45, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Gröber, U.; Spitz, J.; Reichrath, J.; Kisters, K.; Holick, M.F. Vitamin D. Derm. Endocrinol. 2013, 5, 331–347. [Google Scholar] [CrossRef]
- Gorman, S.; Hart, P.H. The current state of play of rodent models to study the role of vitamin D in UV-induced immunomodulation. Photochem. Photobiol. Sci. 2012, 11, 1788–1796. [Google Scholar] [CrossRef] [PubMed]
- D’Aurizio, F.; Villalta, D.; Metus, P.; Doretto, P.; Tozzoli, R. Is vitamin D a player or not in the pathophysiology of autoimmune thyroid diseases? Autoimmun. Rev. 2015, 14, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Wilkes, N.C. Holick Vitamin D Deficiency. In Essence of Anesthesia Practice, 3rd ed.; Saunders: Philadelphia, PA, USA, 2007; pp. 382–383. [Google Scholar] [CrossRef]
- Jaakkola, J.J.; Gissler, M. Maternal smoking in pregnancy as a determinant of rheumatoid arthritis and other inflammatory polyarthropathies during the first 7 years of life. Leuk. Res. 2005, 34, 664–671. [Google Scholar] [CrossRef]
- van der Helm-van Mil, A.H.M.; Verpoort, K.N.; le Cessie, S.; Huizinga, T.W.; de Vries, R.R.; Toes, R.E. The HLA–DRB1 shared epitope alleles differ in the interaction with smoking and predisposition to antibodies to cyclic citrullinated peptide. Arthritis Rheum. 2007, 56, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Somers, E.; Richardson, B. Environmental exposures, epigenetic changes and the risk of lupus. Lupus 2014, 23, 568–576. [Google Scholar] [CrossRef]
- Gilbert, K.M.; Reisfeld, B.; Zurlinden, T.J.; Kreps, M.N.; Erickson, S.W.; Blossom, S.J. Modeling toxicodynamic effects of trichloroethylene on liver in mouse model of autoimmune hepatitis. Toxicol. Appl. Pharmacol. 2014, 279, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Lerner, A.; Matthias, T. Changes in intestinal tight junction permeability associated with industrial food additives explain the rising incidence of autoimmune disease. Autoimmun. Rev. 2015, 14, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Goldman, W.; Seltzer, R.; Reuman, P. Association between treatment with central nervous system stimulants and Raynaud’s syndrome in children: A retrospective case–control study of rheumatology patients. Arthritis Rheum. 2008, 58, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Steenland, K.; Goldsmith, D.F. Silica exposure and autoimmune diseases. Am. J. Ind. Med. 1995, 28, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Caslin, B.; Mohler, K.; Thiagarajan, S.; Melamed, E. Alcohol as friend or foe in autoimmune diseases: A role for gut microbiome? Gut Microbes 2021, 13, 1916278. [Google Scholar] [CrossRef] [PubMed]
- Adamzik, K.; McAleer, M.A.; Kirby, B. Alcohol and psoriasis: Sobering thoughts. Clin. Exp. Dermatol. 2013, 38, 819–822. [Google Scholar] [CrossRef] [PubMed]
- Nath, B.; Szabo, G. Alcohol-induced Modulation of Signaling Pathways in Liver Parenchymal and Nonparenchymal Cells: Implications for Immunity. Semin. Liver Dis. 2009, 29, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Warrington, R.; Silviu-Dan, F.; Wong, T. Drug allergy. Allergy Asthma Clin. Immunol. 2018, 14, 60. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Gershwin, M.E. Drug-Induced Lupus Erythematosus. Drug Saf. 2011, 34, 357–374. [Google Scholar] [CrossRef] [PubMed]
- Araújo-Fernández, S.; Ahijón-Lana, M.; Isenberg, D. Drug-induced lupus: Including anti-tumour necrosis factor and interferon induced. Lupus 2014, 23, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.S.; Wither, J.; Bernatsky, S.; Claudio, J.O.; Clarke, A.; Rioux, J.D.; Fortin, P.R. Occupational and Environmental Exposures and Risk of Systemic Lupus Erythematosus: Silica, Sunlight, Solvents. Rheumatology 2010, 49, 2172–2180. [Google Scholar] [CrossRef] [PubMed]
- Amano, K.; Leung, P.S.C.; Rieger, R.; Quan, C.; Wang, X.; Marik, J.; Suen, Y.F.; Kurth, M.J.; Nantz, M.H.; Ansari, A.A.; et al. Chemical Xenobiotics and Mitochondrial Autoantigens in Primary Biliary Cirrhosis: Identification of Antibodies against a Common Environmental, Cosmetic, and Food Additive, 2-Octynoic Acid. J. Immunol. 2005, 174, 5874–5883. [Google Scholar] [CrossRef] [PubMed]
- Leung, P.S.; Wang, J.; Naiyanetr, P.; Kenny, T.P.; Lam, K.S.; Kurth, M.J.; Gershwin, M.E. Environment and primary biliary cirrhosis: Electrophilic drugs and the induction of AMA. J. Autoimmun. 2013, 41, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Al-Mousa, H.; Al-Saud, B. Primary Immunodeficiency Diseases in Highly Consanguineous Populations from Middle East and North Africa: Epidemiology, Diagnosis, and Care. Front. Immunol. 2017, 8, 678. [Google Scholar] [CrossRef] [PubMed]
- Notarangelo, L.D. Primary immunodeficiencies. J. Allergy Clin. Immunol. 2009, 125, S182–S194. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.S.; Bynum, M.L.; Somers, E.C. Recent insights in the epidemiology of autoimmune diseases: Improved prevalence estimates and understanding of clustering of diseases. J. Autoimmun. 2009, 33, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Zarrin, A.A.; Bao, K.; Lupardus, P.; Vucic, D. Kinase inhibition in autoimmunity and inflammation. Nat. Rev. Drug Discov. 2020, 20, 39–63. [Google Scholar] [CrossRef] [PubMed]
- Attwood, M.M.; Fabbro, D.; Sokolov, A.V.; Knapp, S.; Schiöth, H.B. Trends in kinase drug discovery: Targets, indications and inhibitor design. Nat. Rev. Drug Discov. 2021, 20, 839–861. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Schett, G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat. Rev. Immunol. 2007, 7, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Italiani, P.; Manca, M.L.; Angelotti, F.; Melillo, D.; Pratesi, F.; Puxeddu, I.; Boraschi, D.; Migliorini, P. IL-1 family cytokines and soluble receptors in systemic lupus erythematosus. Arthritis Res. Ther. 2018, 20, 27. [Google Scholar] [CrossRef] [PubMed]
- Kruglov, A.A.; Lampropoulou, V.; Fillatreau, S.; Nedospasov, S.A. Pathogenic and Protective Functions of TNF in Neuroinflammation Are Defined by Its Expression in T Lymphocytes and Myeloid Cells. J. Immunol. 2011, 187, 5660–5670. [Google Scholar] [CrossRef] [PubMed]
- Locksley, R.M.; Killeen, N.; Lenardo, M.J. The TNF and TNF Receptor Superfamilies. Cell 2001, 104, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B. Signalling pathways of the TNF superfamily: A double-edged sword. Nat. Rev. Immunol. 2003, 3, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Collart, M.A.; Baeuerle, P.; Vassalli, P. Regulation of Tumor Necrosis Factor Alpha Transcription in Macrophages: Involvement of Four κΒ-Like Motifs and of Constitutive and Inducible Forms of NF-κB. Mol. Cell. Biol. 1990, 10. [Google Scholar] [CrossRef] [PubMed]
- Camussi, G.; Albano, E.; Tetta, C.; Bussolino, F. The molecular action of tumor necrosis factor-α. Eur. J. Biochem. 1991, 202, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Cordingley, F.; Hoffbrand, A.; Heslop, H.; Turner, M.; Bianchi, A.; Reittie, J.; Vyakarnam, A.; Meager, A.; Brenner, M. Tumour Necrosis Factor as an Autocrine Tumour Growth Factor for Chronic B-Cell Malignancies. Lancet 1988, 331, 969–971. [Google Scholar] [CrossRef] [PubMed]
- Keffer, J.; Probert, L.; Cazlaris, H.; Georgopoulos, S.; Kaslaris, E.; Kioussis, D.; Kollias, G. Transgenic mice expressing human tumour necrosis factor: A predictive genetic model of arthritis. EMBO J. 1991, 10, 4025–4031. [Google Scholar] [CrossRef]
- Feldmann, M. Translating Molecular Insights in Autoimmunity into Effective Therapy. Annu. Rev. Immunol. 2009, 27, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Campanati, A.; Paolinelli, M.; Diotallevi, F.; Martina, E.; Molinelli, E.; Offidani, A. Pharmacodynamics OF TNF α inhibitors for the treatment of psoriasis. Expert Opin. Drug Metab. Toxicol. 2019, 15, 913–925. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Kontermann, R.E.; Pfizenmaier, K. Selective Targeting of TNF Receptors as a Novel Therapeutic Approach. Front. Cell Dev. Biol. 2020, 8, 401. [Google Scholar] [CrossRef] [PubMed]
- Atretkhany, K.-S.N.; Gogoleva, V.S.; Drutskaya, M.S.; Nedospasov, S.A. Distinct modes of TNF signaling through its two receptors in health and disease. J. Leukoc. Biol. 2020, 107, 893–905. [Google Scholar] [CrossRef] [PubMed]
- Talotta, R.; Berzi, A.; Atzeni, F.; Batticciotto, A.; Clerici, M.; Sarzi-Puttini, P.; Trabattoni, D. Paradoxical Expansion of Th1 and Th17 Lymphocytes in Rheumatoid Arthritis Following Infliximab Treatment: A Possible Explanation for a Lack of Clinical Response. J. Clin. Immunol. 2015, 35, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Conrad, C.; Di Domizio, J.; Mylonas, A.; Belkhodja, C.; Demaria, O.; Navarini, A.A.; Lapointe, A.-K.; French, L.E.; Vernez, M.; Gilliet, M. TNF blockade induces a dysregulated type I interferon response without autoimmunity in paradoxical psoriasis. Nat. Commun. 2018, 9, 25. [Google Scholar] [CrossRef] [PubMed]
- Salomon, B.L. Insights into the biology and therapeutic implications of TNF and regulatory T cells. Nat. Rev. Rheumatol. 2021, 17, 487–504. [Google Scholar] [CrossRef]
- Dinarello, C.A. Immunological and Inflammatory Functions of the Interleukin-1 Family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef] [PubMed]
- Mizel, S.B. Biochemical and Biological Characterization of Lymphocyte-Activating Factor (Laf) Produced by the Murine Macrophage Cell Line, P388d. Ann. N. Y. Acad. Sci. 1979, 332, 539–549. [Google Scholar] [CrossRef]
- di Giovine, F.S.; Duff, G.W. Interleukin 1: The first interleukin. Immunol. Today 1990, 11, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. The IL-1 family of cytokines and receptors in rheumatic diseases. Nat. Rev. Rheumatol. 2019, 15, 612–632. [Google Scholar] [CrossRef] [PubMed]
- von Moltke, J.; Ayres, J.S.; Kofoed, E.M.; Chavarría-Smith, J.; Vance, R.E. Recognition of Bacteria by Inflammasomes. Annu. Rev. Immunol. 2013, 31, 73–106. [Google Scholar] [CrossRef] [PubMed]
- Pirzada, R.H.; Javaid, N.; Choi, S. The Roles of the NLRP3 Inflammasome in Neurodegenerative and Metabolic Diseases and in Relevant Advanced Therapeutic Interventions. Genes 2020, 11, 131. [Google Scholar] [CrossRef]
- Jung, S.M.; Kim, W.-U. Targeted Immunotherapy for Autoimmune Disease. Immune Netw. 2022, 22, e9. [Google Scholar] [CrossRef] [PubMed]
- Skendros, P.; Papagoras, C.; Mitroulis, I.; Ritis, K. Autoinflammation: Lessons from the study of familial Mediterranean fever. J. Autoimmun. 2019, 104, 102305. [Google Scholar] [CrossRef]
- Broderick, L.; Hoffman, H.M. IL-1 and autoinflammatory disease: Biology, pathogenesis and therapeutic targeting. Nat. Rev. Rheumatol. 2022, 18, 448–463. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Dinarello, C.A.; Molgora, M.; Garlanda, C. Interleukin-1 and Related Cytokines in the Regulation of Inflammation and Immunity. Immunity 2019, 50, 778–795. [Google Scholar] [CrossRef] [PubMed]
- Geng, J.; Wang, F.; Huang, Z.; Chen, X.; Wang, Y. Perspectives on anti-IL-1 inhibitors as potential therapeutic interventions for severe COVID-19. Cytokine 2021, 143, 155544. [Google Scholar] [CrossRef] [PubMed]
- Baskar, S.; Klein, A.L.; Zeft, A. The Use of IL-1 Receptor Antagonist (Anakinra) in Idiopathic Recurrent Pericarditis: A Narrative Review. Cardiol. Res. Pract. 2016, 2016, 7840724. [Google Scholar] [CrossRef] [PubMed]
- Grom, A.A.; Horne, A.; De Benedetti, F. Macrophage activation syndrome in the era of biologic therapy. Nat. Rev. Rheumatol. 2016, 12, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A.; van der Meer, J.W. Treating inflammation by blocking interleukin-1 in humans. Semin. Immunol. 2013, 25, 469–484. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A.; Thompson, R.C. Blocking IL-1: Interleukin 1 receptor antagonist in vivo and in vitro. Immunol. Today 1991, 12, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Choy, E.H.; De Benedetti, F.; Takeuchi, T.; Hashizume, M.; John, M.R.; Kishimoto, T. Translating IL-6 biology into effective treatments. Nat. Rev. Rheumatol. 2020, 16, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Legouffe, E.; Liautard, J.; Gaillard, J.P.; Rossi, J.; Wijdenes, J.; Bataille, R.; Klein, B.; Brochier, J. Human anti-mouse antibody response to the injection of murine monoclonal antibodies against IL-6. Clin. Exp. Immunol. 1994, 98, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Gabay, C.; Emery, P.; van Vollenhoven, R.; Dikranian, A.; Alten, R.; Pavelka, K.; Klearman, M.; Musselman, D.; Agarwal, S.; Green, J.; et al. Tocilizumab monotherapy versus adalimumab monotherapy for treatment of rheumatoid arthritis (ADACTA): A randomised, double-blind, controlled phase 4 trial. Lancet 2013, 381, 1541–1550. [Google Scholar] [CrossRef] [PubMed]
- Khanna, D.; Lin, C.J.F.; Furst, D.E.; Goldin, J.; Kim, G.; Kuwana, M.; Allanore, Y.; Matucci-Cerinic, M.; Distler, O.; Shima, Y.; et al. Tocilizumab in systemic sclerosis: A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Respir. Med. 2020, 8, 963–974. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhang, M.; Qiu, W.; Ma, H.; Zhang, X.; Zhu, Z.; Yang, C.-S.; Jia, D.; Zhang, T.-X.; Yuan, M.; et al. Safety and efficacy of tocilizumab versus azathioprine in highly relapsing neuromyelitis optica spectrum disorder (TANGO): An open-label, multicentre, randomised, phase 2 trial. Lancet Neurol. 2020, 19, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Arras, D.; Rose-John, S. IL-6 pathway in the liver: From physiopathology to therapy. J. Hepatol. 2016, 64, 1403–1415. [Google Scholar] [CrossRef] [PubMed]
- Yuk, C.M.; Park, H.J.; Kwon, B.-I.; Lah, S.J.; Chang, J.; Kim, J.-Y.; Lee, K.-M.; Park, S.-H.; Hong, S.; Lee, S.-H. Basophil-derived IL-6 regulates TH17 cell differentiation and CD4 T cell immunity. Sci. Rep. 2017, 7, 41744. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef]
- Garbers, C.; Heink, S.; Korn, T.; Rose-John, S. Interleukin-6: Designing specific therapeutics for a complex cytokine. Nat. Rev. Drug Discov. 2018, 17, 395–412. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Wu, L.-W.; Grivennikov, S.I.; de Jong, P.R.; Lian, I.; Yu, F.-X.; Wang, K.; Ho, S.B.; Boland, B.S.; Chang, J.T.; et al. A gp130–Src–YAP module links inflammation to epithelial regeneration. Nature 2015, 519, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, P.C.; Behrmann, I.; Müller-Newen, G.; Schaper, F.; Graeve, L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem. J. 1998, 334, 297–314. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Gadina, M.; Schreiber, R.D. Cytokine Signaling in 2002. Cell 2002, 109, S121–S131. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Taga, T.; Saito, M.; Suematsu, S.; Kumanogoh, A.; Tanaka, T.; Fujiwara, H.; Hirata, M.; Yamagami, T.; Nakahata, T.; et al. Targeted disruption of gp130, a common signal transducer for the interleukin 6 family of cytokines, leads to myocardial and hematological disorders. Proc. Natl. Acad. Sci. USA 1996, 93, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Zhao, M.; Chang, C.; Wu, H.; Lu, Q. Type I Interferons in the Pathogenesis and Treatment of Autoimmune Diseases. Clin. Rev. Allergy Immunol. 2020, 59, 248–272. [Google Scholar] [CrossRef]
- Yu, R.; Zhu, B.; Chen, D. Type I interferon-mediated tumor immunity and its role in immunotherapy. Cell. Mol. Life Sci. 2022, 79, 191. [Google Scholar] [CrossRef] [PubMed]
- A de Weerd, N.; Vivian, J.P.; Nguyen, T.K.; E Mangan, N.; A Gould, J.; Braniff, S.-J.; Zaker-Tabrizi, L.; Fung, K.Y.; Forster, S.C.; Beddoe, T.; et al. Structural basis of a unique interferon-β signaling axis mediated via the receptor IFNAR1. Nat. Immunol. 2013, 14, 901–907. [Google Scholar] [CrossRef] [PubMed]
- Piehler, J.; Thomas, C.; Garcia, K.; Schreiber, G. Structural and dynamic determinants of type I interferon receptor assembly and their functional interpretation. Immunol. Rev. 2012, 250, 317–334. [Google Scholar] [CrossRef] [PubMed]
- Thorlacius, G.E.; Wahren-Herlenius, M.; Rönnblom, L. An update on the role of type I interferons in systemic lupus erythematosus and Sjögren’s syndrome. Curr. Opin. Rheumatol. 2018, 30, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Swiecki, M.; Colonna, M. The multifaceted biology of plasmacytoid dendritic cells. Nat. Rev. Immunol. 2015, 15, 471–485. [Google Scholar] [CrossRef] [PubMed]
- Chasset, F.; Arnaud, L. Targeting interferons and their pathways in systemic lupus erythematosus. Autoimmun. Rev. 2018, 17, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Kalunian, K.C.; Merrill, J.T.; Maciuca, R.; McBride, J.M.; Townsend, M.J.; Wei, X.; Davis, J.C.; Kennedy, W.P. A Phase II study of the efficacy and safety of rontalizumab (rhuMAb interferon-α) in patients with systemic lupus erythematosus (ROSE). Ann. Rheum. Dis. 2015, 75, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Khamashta, M.; Merrill, J.T.; Werth, V.P.; Furie, R.; Kalunian, K.; Illei, G.G.; Drappa, J.; Wang, L.; Greth, W. Sifalimumab, an anti-interferon-α monoclonal antibody, in moderate to severe systemic lupus erythematosus: A randomised, double-blind, placebo-controlled study. Ann. Rheum. Dis. 2016, 75, 1909–1916. [Google Scholar] [CrossRef] [PubMed]
- Morand, E.F.; Furie, R.; Tanaka, Y.; Bruce, I.N.; Askanase, A.D.; Richez, C.; Bae, S.-C.; Brohawn, P.Z.; Pineda, L.; Berglind, A.; et al. Trial of Anifrolumab in Active Systemic Lupus Erythematosus. N. Engl. J. Med. 2020, 382, 211–221. [Google Scholar] [CrossRef]
- Moseley, T.; Haudenschild, D.; Rose, L.; Reddi, A. Interleukin-17 family and IL-17 receptors. Cytokine Growth Factor Rev. 2003, 14, 155–174. [Google Scholar] [CrossRef]
- Cua, D.J.; Tato, C.M. Innate IL-17-producing cells: The sentinels of the immune system. Nat. Rev. Immunol. 2010, 10, 479–489. [Google Scholar] [CrossRef] [PubMed]
- McGeachy, M.J.; Cua, D.J.; Gaffen, S.L. The IL-17 Family of Cytokines in Health and Disease. Immunity 2019, 50, 892–906. [Google Scholar] [CrossRef] [PubMed]
- Veldhoen, M. Interleukin 17 is a chief orchestrator of immunity. Nat. Immunol. 2017, 18, 612–621. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, S.; Sakuraba, A. Distinct roles of interleukin-17 and T helper 17 cells among autoimmune diseases. J. Transl. Autoimmun. 2021, 4, 100104. [Google Scholar] [CrossRef] [PubMed]
- Hirahara, K.; Poholek, A.; Vahedi, G.; Laurence, A.; Kanno, Y.; Milner, J.D.; O’shea, J.J. Mechanisms underlying helper T-cell plasticity: Implications for immune-mediated disease. J. Allergy Clin. Immunol. 2013, 131, 1276–1287. [Google Scholar] [CrossRef] [PubMed]
- Langrish, C.L.; Chen, Y.; Blumenschein, W.M.; Mattson, J.; Basham, B.; Sedgwick, J.D.; McClanahan, T.; Kastelein, R.A.; Cua, D.J. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 2005, 201, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Liu, C.; Hartupee, J.; Altuntas, C.Z.; Gulen, M.F.; Jane-Wit, D.; Xiao, J.; Lu, Y.; Giltiay, N.; Liu, J.; et al. The adaptor Act1 is required for interleukin 17–dependent signaling associated with autoimmune and inflammatory disease. Nat. Immunol. 2007, 8, 247–256. [Google Scholar] [CrossRef]
- Maitra, A.; Shen, F.; Hanel, W.; Mossman, K.; Tocker, J.; Swart, D.; Gaffen, S.L. Distinct functional motifs within the IL-17 receptor regulate signal transduction and target gene expression. Proc. Natl. Acad. Sci. USA 2007, 104, 7506–7511. [Google Scholar] [CrossRef] [PubMed]
- Sieper, J.; Poddubnyy, D.; Miossec, P. The IL-23–IL-17 pathway as a therapeutic target in axial spondyloarthritis. Nat. Rev. Rheumatol. 2019, 15, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, J.E.; Yan, B.Y.; Chan, T.C.; Krueger, J.G. Discovery of the IL-23/IL-17 Signaling Pathway and the Treatment of Psoriasis. J. Immunol. 2018, 201, 1605–1613. [Google Scholar] [CrossRef] [PubMed]
- Genovese, M.C.; Greenwald, M.; Cho, C.; Berman, A.; Jin, L.; Cameron, G.S.; Benichou, O.; Xie, L.; Braun, D.; Berclaz, P.; et al. A Phase II Randomized Study of Subcutaneous Ixekizumab, an Anti–Interleukin-17 Monoclonal Antibody, in Rheumatoid Arthritis Patients Who Were Naive to Biologic Agents or Had an Inadequate Response to Tumor Necrosis Factor Inhibitors. Arthritis Rheumatol. 2014, 66, 1693–1704. [Google Scholar] [CrossRef]
- Oppmann, B.; Lesley, R.; Blom, B.; Timans, J.C.; Xu, Y.; Hunte, B.; Vega, F.; Yu, N.; Wang, J.; Singh, K.; et al. Novel p19 Protein Engages IL-12p40 to Form a Cytokine, IL-23, with Biological Activities Similar as Well as Distinct from IL-12. Immunity 2000, 13, 715–725. [Google Scholar] [CrossRef]
- Parham, C.; Chirica, M.; Timans, J.; Vaisberg, E.; Travis, M.; Cheung, J.; Pflanz, S.; Zhang, R.; Singh, K.P.; Vega, F.; et al. A Receptor for the Heterodimeric Cytokine IL-23 Is Composed of IL-12Rβ1 and a Novel Cytokine Receptor Subunit, IL-23R. J. Immunol. 2002, 168, 5699–5708. [Google Scholar] [CrossRef]
- Xiong, D.-K.; Shi, X.; Han, M.-M.; Zhang, X.-M.; Wu, N.-N.; Sheng, X.-Y.; Wang, J.-N. The regulatory mechanism and potential application of IL-23 in autoimmune diseases. Front. Pharmacol. 2022, 13, 982238. [Google Scholar] [CrossRef]
- Floss, D.M.; Schröder, J.; Franke, M.; Scheller, J. Insights into IL-23 biology: From structure to function. Cytokine Growth Factor Rev. 2015, 26, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef] [PubMed]
- de Morales, J.M.G.R.; Puig, L.; Daudén, E.; Cañete, J.D.; Pablos, J.L.; Martín, A.O.; Juanatey, C.G.; Adán, A.; Montalbán, X.; Borruel, N.; et al. Critical role of interleukin (IL)-17 in inflammatory and immune disorders: An updated review of the evidence focusing in controversies. Autoimmun. Rev. 2019, 19, 102429. [Google Scholar] [CrossRef] [PubMed]
- Musette, P.; Bouaziz, J.D. B Cell Modulation Strategies in Autoimmune Diseases: New Concepts. Front. Immunol. 2018, 9, 622. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.S.W.; Rojas, O.L.; Gommerman, J.L. B cell depletion therapies in autoimmune disease: Advances and mechanistic insights. Nat. Rev. Drug Discov. 2020, 20, 179–199. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, R.J.; Vanhoorelbeke, K.; Leypoldt, F.; Kaya, Z.; Bieber, K.; McLachlan, S.M.; Komorowski, L.; Luo, J.; Cabral-Marques, O.; Hammers, C.M.; et al. Mechanisms of Autoantibody-Induced Pathology. Front. Immunol. 2017, 8, 603. [Google Scholar] [CrossRef] [PubMed]
- Anolik, J.H.; Barnard, J.; Owen, T.; Zheng, B.; Kemshetti, S.; Looney, R.J.; Sanz, I. Delayed memory B cell recovery in peripheral blood and lymphoid tissue in systemic lupus erythematosus after B cell depletion therapy. Arthritis Rheum. 2007, 56, 3044–3056. [Google Scholar] [CrossRef] [PubMed]
- Palanichamy, A.; Barnard, J.; Zheng, B.; Owen, T.; Quach, T.; Wei, C.; Looney, R.J.; Sanz, I.; Anolik, J.H. Novel Human Transitional B Cell Populations Revealed by B Cell Depletion Therapy. J. Immunol. 2009, 182, 5982–5993. [Google Scholar] [CrossRef] [PubMed]
- Stathopoulos, P.; Dalakas, M.C. Evolution of Anti-B Cell Therapeutics in Autoimmune Neurological Diseases. Neurotherapeutics 2022, 19, 691–710. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.; Chan, A.C. Pathogenic Roles of B Cells in Human Autoimmunity. Immunity 2004, 20, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Nikolopoulos, D.; Fotis, L.; Gioti, O.; Fanouriakis, A. Tailored treatment strategies and future directions in systemic lupus erythematosus. Rheumatol. Int. 2022, 42, 1307–1319. [Google Scholar] [CrossRef] [PubMed]
- Sharkey, P.; Thomas, R. Immune tolerance therapies for autoimmune diseases: Shifting the goalpost to cure. Curr. Opin. Pharmacol. 2022, 65, 102242. [Google Scholar] [CrossRef] [PubMed]
- Schett, G.; Mackensen, A.; Mougiakakos, D. CAR T-cell therapy in autoimmune diseases. Lancet 2023, 402, 2034–2044. [Google Scholar] [CrossRef] [PubMed]
- Mackensen, A.; Mackensen, A.; Müller, F.; Müller, F.; Mougiakakos, D.; Mougiakakos, D.; Böltz, S.; Böltz, S.; Wilhelm, A.; Wilhelm, A.; et al. Anti-CD19 CAR T cell therapy for refractory systemic lupus erythematosus. Nat. Med. 2022, 28, 2124–2132. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric Antigen Receptor–Modified T Cells in Chronic Lymphoid Leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef]
- Larson, S.M.; Truscott, L.C.; Chiou, T.-T.; Patel, A.; Kao, R.; Tu, A.; Tyagi, T.; Lu, X.; Elashoff, D.; De Oliveira, S.N. Pre-clinical development of gene modification of haematopoietic stem cells with chimeric antigen receptors for cancer immunotherapy. Hum. Vaccines Immunother. 2017, 13, 1094–1104. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yasmeen, F.; Pirzada, R.H.; Ahmad, B.; Choi, B.; Choi, S. Understanding Autoimmunity: Mechanisms, Predisposing Factors, and Cytokine Therapies. Int. J. Mol. Sci. 2024, 25, 7666. https://doi.org/10.3390/ijms25147666
Yasmeen F, Pirzada RH, Ahmad B, Choi B, Choi S. Understanding Autoimmunity: Mechanisms, Predisposing Factors, and Cytokine Therapies. International Journal of Molecular Sciences. 2024; 25(14):7666. https://doi.org/10.3390/ijms25147666
Chicago/Turabian StyleYasmeen, Farzana, Rameez Hassan Pirzada, Bilal Ahmad, Bogeum Choi, and Sangdun Choi. 2024. "Understanding Autoimmunity: Mechanisms, Predisposing Factors, and Cytokine Therapies" International Journal of Molecular Sciences 25, no. 14: 7666. https://doi.org/10.3390/ijms25147666
APA StyleYasmeen, F., Pirzada, R. H., Ahmad, B., Choi, B., & Choi, S. (2024). Understanding Autoimmunity: Mechanisms, Predisposing Factors, and Cytokine Therapies. International Journal of Molecular Sciences, 25(14), 7666. https://doi.org/10.3390/ijms25147666