
Comprehensive Analysis of Placental DNA Methylation Changes and Fetal Birth Weight in Pigs
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Morphology and Gene Expression Changes between the HBW and LBW Placentas
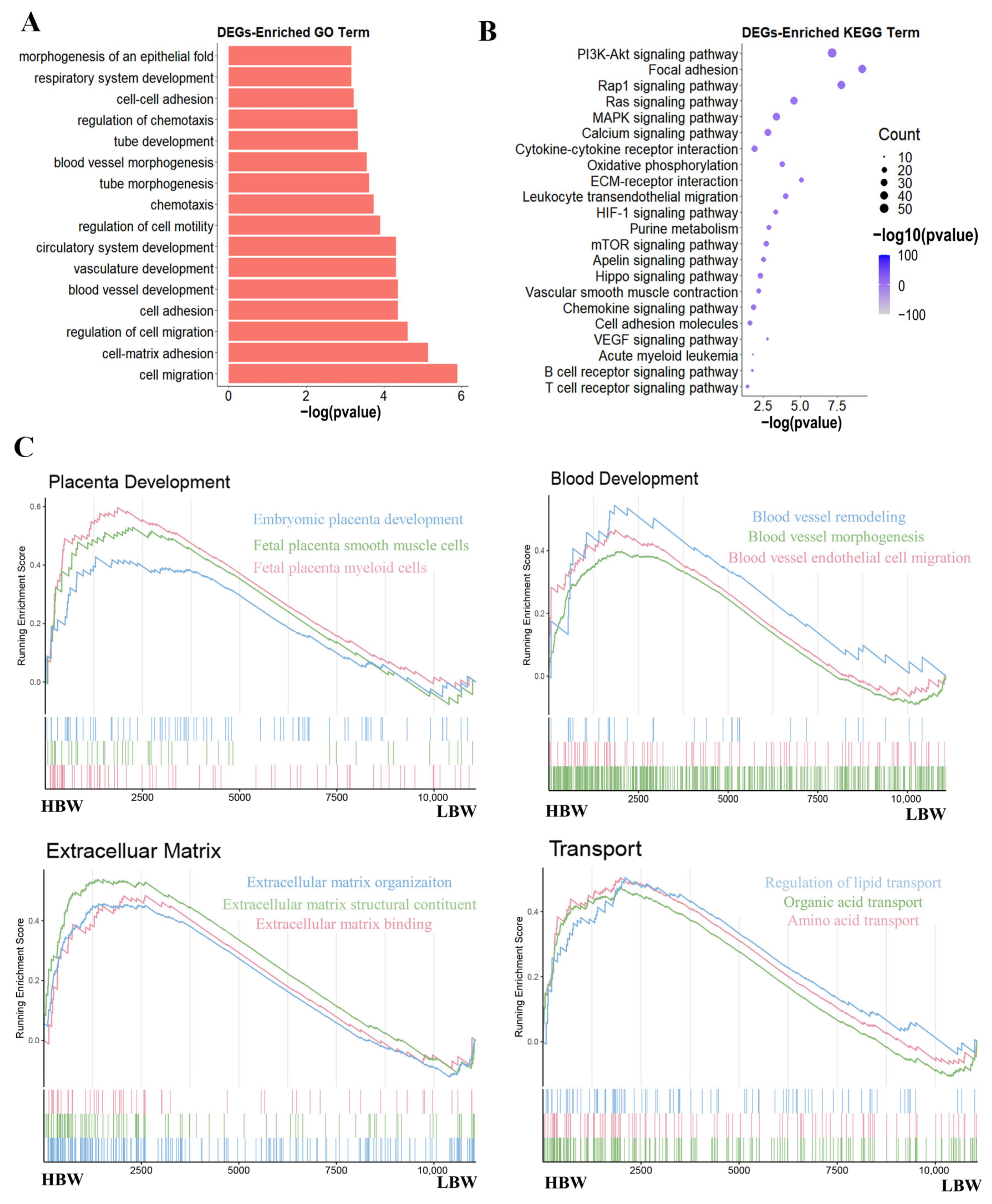
2.2. Functional Enrichment Analysis of Differentially Expressed Genes
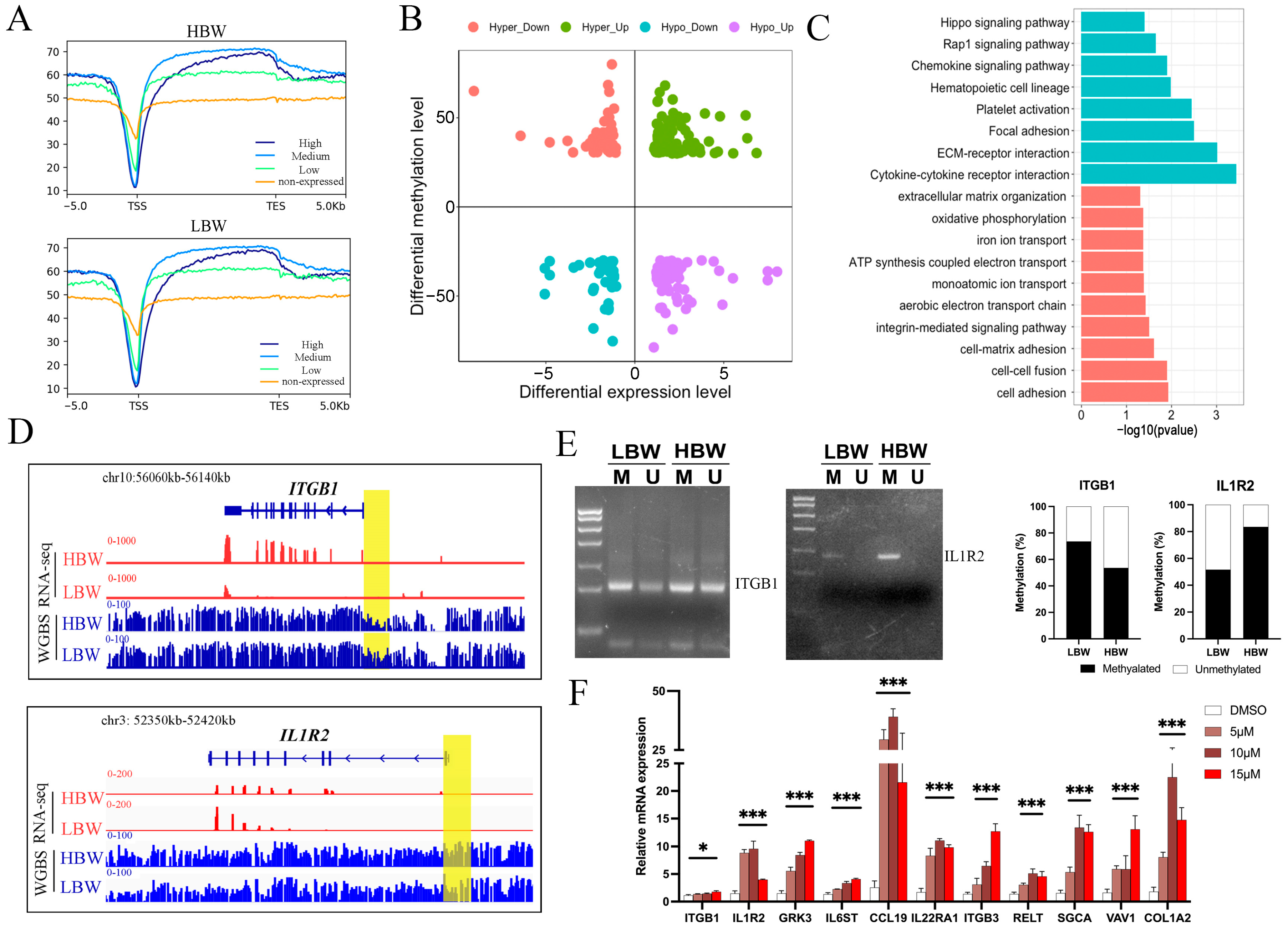
2.3. Characteristics of DNA Methylome in the HBW and LBW Placentas
2.4. Dynamic Changes in DNA Methylation Landscape in the HBW and LBW Placentas
2.5. Association between DNA Methylation and Gene Expression in the HBW and LBW Placentas
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Sample Collection
5.2. Cell Culture and 5-Aza Treatment
5.3. Whole Genome Bisulfite Sequencing and Data Analysis
5.4. RNA-Seq and Data Analysis
5.5. Real-Time Quantitative PCR (qPCR)
5.6. Methylation-Specific PCR (MSP)
5.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vilanova, C.S.; Hirakata, V.N.; de Souza Buriol, V.C.; Nunes, M.; Goldani, M.Z.; da Silva, C.H. The relationship between the different low birth weight strata of newborns with infant mortality and the influence of the main health determinants in the extreme south of Brazil. Popul. Health Metr. 2019, 17, 15. [Google Scholar] [CrossRef] [PubMed]
- Bateson, P.; Barker, D.; Clutton-Brock, T.; Deb, D.; D’Udine, B.; Foley, R.A.; Gluckman, P.; Godfrey, K.; Kirkwood, T.; Lahr, M.M.; et al. Developmental plasticity and human health. Nature 2004, 430, 419–421. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, J.R.; Hyde, M.J.; Gale, C.; Santhakumaran, S.; Modi, N. Preterm birth and the metabolic syndrome in adult life: A systematic review and meta-analysis. Pediatrics 2013, 131, e1240–e1263. [Google Scholar] [CrossRef] [PubMed]
- Huxley, R.; Owen, C.G.; Whincup, P.H.; Cook, D.G.; Rich-Edwards, J.; Smith, G.D.; Collins, R. Is birth weight a risk factor for ischemic heart disease in later life? Am. J. Clin. Nutr. 2007, 85, 1244–1250. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, D.A.; Ronalds, G.; Clark, H.; Smith, G.D.; Leon, D.A. Birth weight is inversely associated with incident coronary heart disease and stroke among individuals born in the 1950s: Findings from the Aberdeen Children of the 1950s prospective cohort study. Circulation 2005, 112, 1414–1418. [Google Scholar] [CrossRef] [PubMed]
- Law, C.M.; Shiell, A.W.; Newsome, C.A.; Syddall, H.E.; Shinebourne, E.A.; Fayers, P.M.; Martyn, C.N.; de Swiet, M. Fetal, infant, and childhood growth and adult blood pressure: A longitudinal study from birth to 22 years of age. Circulation 2002, 105, 1088–1092. [Google Scholar] [CrossRef] [PubMed]
- Whincup, P.H.; Kaye, S.J.; Owen, C.G.; Huxley, R.; Cook, D.G.; Anazawa, S.; Barrett-Connor, E.; Bhargava, S.K.; Birgisdottir, B.E.; Carlsson, S.; et al. Birth weight and risk of type 2 diabetes: A systematic review. JAMA 2008, 300, 2886–2897. [Google Scholar] [PubMed]
- Rehfeldt, C.; Kuhn, G. Consequences of birth weight for postnatal growth performance and carcass quality in pigs as related to myogenesis. J. Anim. Sci. 2006, 84 (Suppl. 13), E113–E123. [Google Scholar] [CrossRef] [PubMed]
- Andrés, S.; Valdés, C.; Santos, A.; Mateo, J.; Giráldez, F.J. Effects of Birth Weight on Animal Performance, Fattening Traits and Meat Quality of Lambs. Animals 2020, 10, 2364. [Google Scholar] [CrossRef]
- Vernunft, A.; Maass, M.; Brüssow, K.-P. Placental characteristics of German Landrace sows and their relationships to different fertility parameters. Czech J. Anim. Sci. 2018, 63, 339–346. [Google Scholar] [CrossRef]
- Trachsel, C.; Küker, S.; Nathues, H.; Grahofer, A. Influence of different sow traits on the expulsion and characteristics of the placenta in a free farrowing system. Theriogenology 2021, 161, 74–82. [Google Scholar] [CrossRef] [PubMed]
- van den Bosch, M.; Soede, N.; Kemp, B.; van den Brand, H. Sow Nutrition, Uterine Contractions, and Placental Blood Flow during the Peri-Partum Period and Short-Term Effects on Offspring: A Review. Animals 2023, 13, 910. [Google Scholar] [CrossRef] [PubMed]
- König, N.L.; Wähner, M.; Seeger, J.; Sigmarsson, H.L.; Kauffold, J. An investigation into uterine capacity based on litter and placental characteristics in two sow lines with different prolificacy (Danish Landrace x Danish Yorkshire versus German Saddleback). Reprod. Domest. Anim. 2021, 56, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Naeye, R.L. Do placental weights have clinical significance? Hum. Pathol. 1987, 18, 387–391. [Google Scholar] [CrossRef]
- Desforges, M.; Sibley, C.P. Placental nutrient supply and fetal growth. Int. J. Dev. Biol. 2009, 54, 377–390. [Google Scholar] [CrossRef]
- Dilworth, M.; Sibley, C. Transport across the placenta of mice and women. Placenta 2013, 34, S34–S39. [Google Scholar] [CrossRef]
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Rahnama, F.; Shafiei, F.; Gluckman, P.D.; Mitchell, M.D.; Lobie, P.E. Epigenetic regulation of human trophoblastic cell migration and invasion. Endocrinology 2006, 147, 5275–5283. [Google Scholar] [CrossRef]
- Serman, L.; Vlahović, M.; Sijan, M.; Bulić-Jakus, F.; Serman, A.; Sincić, N.; Matijević, R.; Jurić-Lekić, G.; Katusić, A. The impact of 5-azacytidine on placental weight, glycoprotein pattern and proliferating cell nuclear antigen expression in rat placenta. Placenta 2007, 28, 803–811. [Google Scholar] [CrossRef]
- Tan, B.; Zhou, C.; Zang, X.; Zhao, X.; Xiao, L.; Zeng, J.; Hong, L.; Wu, Z.; Gu, T. Integrated Analysis of DNA Methylation and Gene Expression in Porcine Placental Development. Int. J. Mol. Sci. 2023, 24, 5169. [Google Scholar] [CrossRef] [PubMed]
- Valente, M.; Turoczi, S.; Kantane, P.; Marazzi, G.; Hulot, J.S.; Sassoon, D. Elucidating a role for Pw1/Peg3 in placenta vascular formation. Arch. Cardiovasc. Dis. Suppl. 2020, 12, 206–207. [Google Scholar]
- Argente, J.; Mehls, O.; Barrios, V. Growth and body composition in very young SGA children. Pediatr. Nephrol. 2010, 25, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Maccani, M.A.; Marsit, C.J. Epigenetics in the placenta. Am. J. Reprod. Immunol. 2009, 62, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, J.A.; Winn, V.D. Vasculogenesis and Angiogenesis in the IUGR Placenta. Semin. Perinatol. 2008, 32, 172–177. [Google Scholar] [CrossRef]
- Meegdes, B.H.; Ingenhoes, R.; Peeters, L.L.; Exalto, N. Early pregnancy wastage: Relationship between chorionic vascularization and embryonic development. Fertil. Steril. 1988, 49, 216–220. [Google Scholar] [CrossRef]
- Shibuya, M. Vascular Endothelial Growth Factor (VEGF) and Its Receptor (VEGFR) Signaling in Angiogenesis: A Crucial Target for Anti- and Pro-Angiogenic Therapies. Genes Cancer 2011, 2, 1097–1105. [Google Scholar] [CrossRef]
- Kaiser, S.; Koch, Y.; Kühnel, E.; Sharma, N.; Gellhaus, A.; Kuckenberg, P.; Schorle, H.; Winterhager, E. Reduced Gene Dosage of Tfap2c Impairs Trophoblast Lineage Differentiation and Alters Maternal Blood Spaces in the Mouse Placenta. Biol. Reprod. 2015, 93, 31. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wu, D.; Zhang, S.; Zhang, M.; Zhou, Y.; An, X.; Li, Q.; Li, Z. Transcription Factor TFAP2C Affects Porcine Early Embryo Development via Regulating Epigenetic Modification. Reprod. BioMed. Online 2023, 103772. [Google Scholar] [CrossRef]
- Franczyk, M.; Lopucki, M.; Stachowicz, N.; Morawska, D.; Kankofer, M. Extracellular matrix proteins in healthy and re-tained placentas, comparing hemochorial and synepitheliochorial placentas. Placenta 2017, 50, 19–24. [Google Scholar] [CrossRef]
- Chrzanowska-Wodnicka, M. Distinct functions for Rap1 signaling in vascular morphogenesis and dysfunction. Exp. Cell Res. 2013, 319, 2350–2359. [Google Scholar] [CrossRef] [PubMed]
- Robertson, S.A.; Chin, P.-Y.; Femia, J.G.; Brown, H.M. Embryotoxic cytokines—Potential roles in embryo loss and fetal programming. J. Reprod. Immunol. 2018, 125, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Lan, M.; Li, J.; Dong, Q.; Li, X.; Liu, B.; Li, G.; Wang, H.; Zhang, Z.; Zhu, B. The proinflammatory cytokine TNFα induces DNA demethylation–dependent and –independent activation of interleukin-32 expression. J. Biol. Chem. 2019, 294, 6785–6795. [Google Scholar] [CrossRef] [PubMed]
- Crespi, B.J. Why and How Imprinted Genes Drive Fetal Programming. Front. Endocrinol. 2020, 10, 940. [Google Scholar] [CrossRef] [PubMed]
- Moore, G.E.; Ishida, M.; Demetriou, C.; Al-Olabi, L.; Leon, L.J.; Thomas, A.C.; Abu-Amero, S.; Frost, J.M.; Stafford, J.L.; Chaoqun, Y.; et al. The role and interaction of imprinted genes in human fetal growth. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20140074. [Google Scholar] [CrossRef] [PubMed]
- Gondret, F.; Lefaucheur, L.; Juin, H.; Louveau, I.; Lebret, B. Low birth weight is associated with enlarged muscle fiber area and impaired meat tenderness of the longissimus muscle in pigs. J. Anim. Sci. 2006, 84, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Mallmann, A.L.; Camilotti, E.; Fagundes, D.P.; Vier, C.E.; Mellagi, A.P.G.; Ulguim, R.R.; Bernardi, M.L.; Orlando, U.A.D.; Gonçalves, M.A.D.; Kummer, R.; et al. Impact of feed intake during late gestation on piglet birth weight and reproductive performance: A dose-response study performed in gilts. J. Anim. Sci. 2019, 97, 1262–1272. [Google Scholar] [CrossRef]
- Wallace, J.M.; Bourke, D.A.; Aitken, R.P.; Milne, J.S.; Hay, W.W., Jr. Placental glucose transport in growth-restricted pregnancies induced by overnourishing adolescent sheep. J. Physiol. 2003, 547 Pt 1, 85–94. [Google Scholar] [CrossRef]
- Matheson, S.M.; Bünger, L.; Dwyer, C.M. Genetic Parameters for Fitness and Neonatal Behavior Traits in Sheep. Behav. Genet. 2012, 42, 899–911. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, L.A.; Spiegel, A.K.; Ing, N.H.; Johnson, G.A.; Bazer, F.W.; Burghardt, R.C. Functional effects of transforming growth factor β on adhesive properties of porcine trophectoderm. Endocrinology 2005, 146, 3933–3942. [Google Scholar] [CrossRef]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Akalin, A.; Kormaksson, M.; Li, S.; Garrett-Bakelman, F.E.; Figueroa, M.E.; Melnick, A.; Mason, C.E. methylKit: A comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012, 13, R87. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.-G.; He, Q.-Y. ChIPseeker: An R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015, 31, 2382–2383. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2012, 14, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2013, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Mi, H.; Huang, X.; Muruganujan, A.; Tang, H.; Mills, C.; Kang, D.; Thomas, P.D. PANTHER version 11: Expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017, 45, D183–D189. [Google Scholar] [CrossRef]
- Bu, D.; Luo, H.; Huo, P.; Wang, Z.; Zhang, S.; He, Z.; Wu, Y.; Zhao, L.; Liu, J.; Guo, J. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Li, L.C.; Dahiya, R. MethPrimer: Designing primers for methylation PCRs. Bioinformatics 2002, 18, 1427–1431. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, B.; Xiao, L.; Wang, Y.; Zhou, C.; Huang, H.; Li, Z.; Hong, L.; Cai, G.; Wu, Z.; Gu, T. Comprehensive Analysis of Placental DNA Methylation Changes and Fetal Birth Weight in Pigs. Int. J. Mol. Sci. 2024, 25, 7702. https://doi.org/10.3390/ijms25147702
Tan B, Xiao L, Wang Y, Zhou C, Huang H, Li Z, Hong L, Cai G, Wu Z, Gu T. Comprehensive Analysis of Placental DNA Methylation Changes and Fetal Birth Weight in Pigs. International Journal of Molecular Sciences. 2024; 25(14):7702. https://doi.org/10.3390/ijms25147702
Chicago/Turabian StyleTan, Baohua, Liyao Xiao, Yongzhong Wang, Chen Zhou, Huijun Huang, Zicong Li, Linjun Hong, Gengyuan Cai, Zhenfang Wu, and Ting Gu. 2024. "Comprehensive Analysis of Placental DNA Methylation Changes and Fetal Birth Weight in Pigs" International Journal of Molecular Sciences 25, no. 14: 7702. https://doi.org/10.3390/ijms25147702