
Neuroblastoma—A Review of Combination Immunotherapy
 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Treatment Methods in the Past and Now
3. Tumor Biomarkers
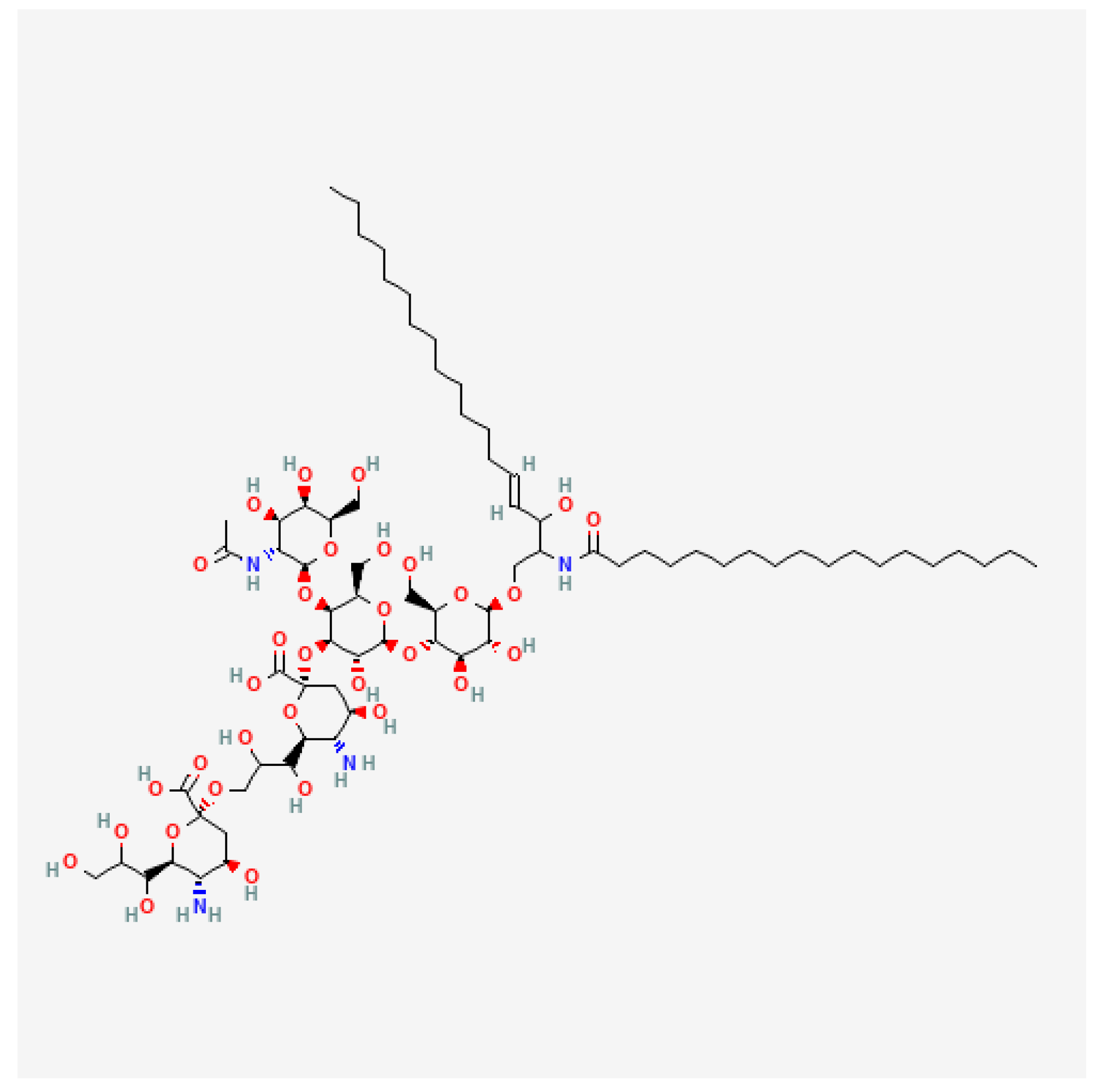
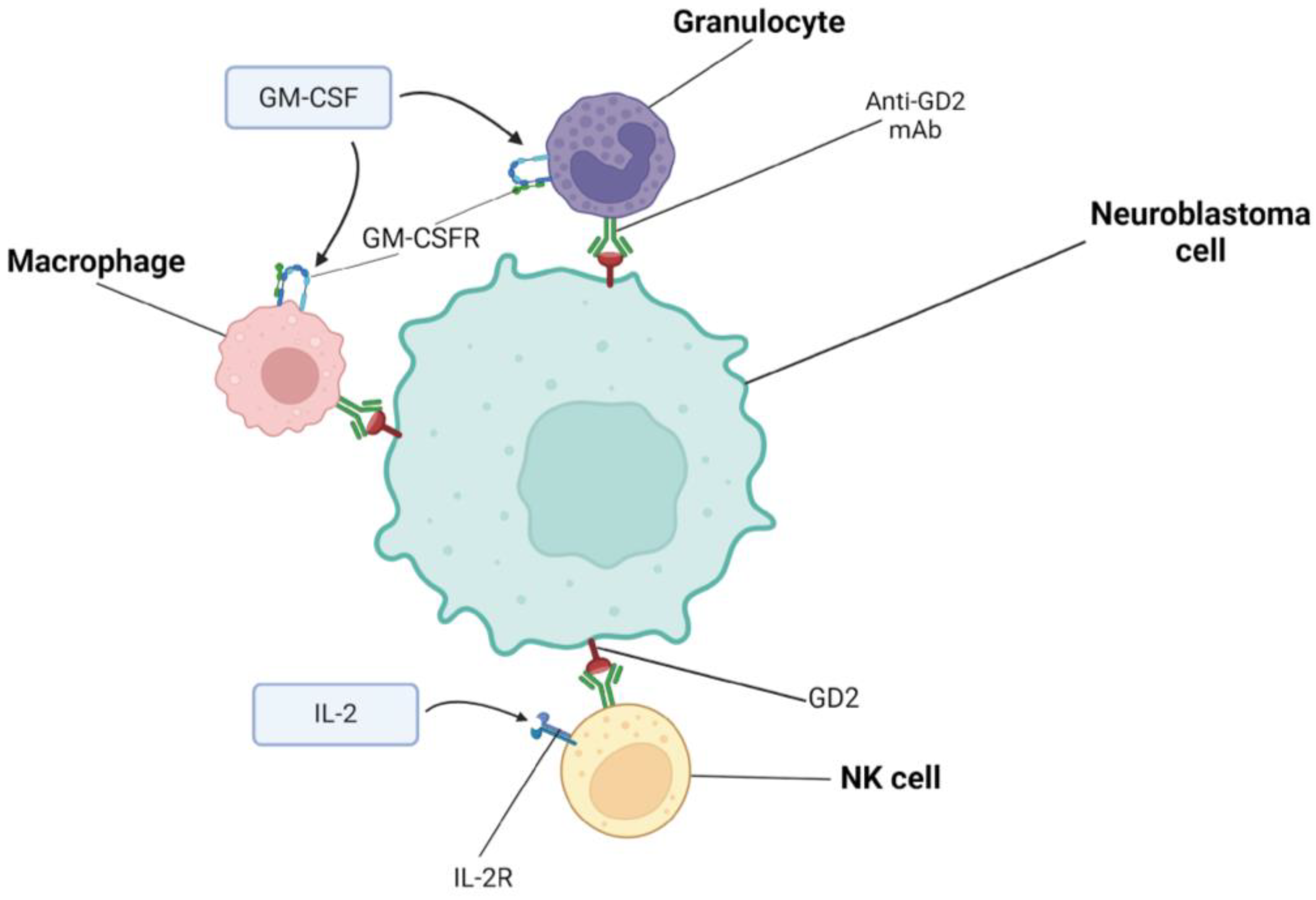
4. Combining Immunotherapies Based on Dinutuximab
4.1. Characteristics of Dinutuximab and Dinutuximab Beta
4.2. Dinutuximab and Irinotecan, Temozolomide, GM-CSF
4.3. Dinutuximab and Tipifarnib
4.4. Dinutuximab Beta and Irinotecan, Temozolomide

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Molecular Target | Combined Therapy Model | Common Toxicities | Uncommon Toxicities | ClinicalTrials.gov Identifier | References |
|---|---|---|---|---|---|---|
| Dinuxitimab | GD2 | Dinutuximab, irinotecan, temozolomide, GM-CSF | Fever Neutropenia Pain Diarrhea | Vomiting Thrombocytopenia | NCT01767194 | [38,48] |
| Dinuxitimab beta | GD2 | Dinutuximab beta, irinotecan, temozolomide | Leukopenia Neutropenia Fever Anemia Thrombocytopenia Hypertransaminemia Diarrhea | Tachycardia Vomiting Pain Anorexia Rash and itching Hypertension Capillary leak syndrome Allergy | NCT05272371 | [37,47,49] |
| Dinuxitimab beta | GD2 | Dinutuximab beta, haplo-SCT | Hypertransaminemia Anemia Proteinuria Fever Constipation Diarrhea Pain Skin toxicity | Leukopenia Neutropenia Thrombocytopenia Creatinine elevation Capillary leak syndrome Stomatitis Bilirubin elevation Allergy Nausea Vomiting Neurotoxicity Hypotension | NCT02258815 | [50,51,52] |
| Naxitamab | GD2 | Naxitamab, irinotecan, temozolomide, GM-CSF | Anemia Neutropenia Thrombocytopenia Hypotension Pain Hypertension Diarrhea | Bronchospasm Anorexia Urticaria Skin ulceration Vomiting Swelling of the larynx Postural disease Hypertransaminemia Neutropenic fever | - | [53] |
4.5. Dinutuximab Beta and Haplo-SCT
5. Combining Immunotherapies Based on Naxitamab
5.1. Characteristics of Naxitamab
5.2. Naxitamab and Irinotecan, Temozolomide, GM-CSF
5.3. Naxitamab and Nanofenretinide/Nanospermidine
6. Combining Immunotherapies Based on CAR-T
6.1. CAR-T and Immune Checkpoint Inhibitors
6.2. GD2-CAR-T and Bevacizumab
7. Future Directions
| Method | Treatment Elements | Results | Comments | References |
|---|---|---|---|---|
| Photothermal therapy + immune checkpoint inhibitor | Prussian blue nanoparticle + CpG oligodeoxynucleotides Anti-CTLA-4 | Complete tumor regression in mice | Mice developed immunity and memory response against cancer cells | [79] |
| YAP knockdown and dinutuximab | Genetic inhibition of YAP Dinutuximab, γδ T cells, and cyclophosphamide | Extended survival of mice | YAP inhibition increases sensitivity to dinutuximab | [94] |
| Oncolytic virus and immune checkpoint blockade | Seneca Valley Virus Anti-PD-1 mAb | Extended survival of mice | The combination increased the response rate compared to monotherapy | [100] |
| Spleen tyrosine kinase inhibitor and anti-PDL-1 monoclonal antibodies | Fostamatinib Anti-PDL1 mAb | Complete tumor regression in mice with small tumors (50 mm3) | Adding the radiation extended the survival of mice with large tumors | [122] |
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dalianis, T.; Lukoseviciute, M.; Holzhauser, S.; Kostopoulou, O.N. New Approaches Towards Targeted Therapy for Childhood sveenty. Anticancer Res. 2023, 43, 3829–3839. [Google Scholar] [CrossRef]
- Chung, C.; Boterberg, T.; Lucas, J.; Panoff, J.; Valteau-Couanet, D.; Hero, B.; Bagatell, R.; Hill-Kayser, C.E. Neuroblastoma. Pediatr. Blood Cancer 2021, 68, e28473. [Google Scholar] [CrossRef]
- Nguyen, R.; Thiele, C.J. Immunotherapy Approaches Targeting Neuroblastoma. Curr. Opin. Pediatr. 2021, 33, 19–25. [Google Scholar] [CrossRef]
- Ash, S.; Askenasy, N. Immunotherapy for Neuroblastoma by Hematopoietic Cell Transplantation and Post-Transplant Immunomodulation. Crit. Rev. Oncol. Hematol. 2023, 185, 103956. [Google Scholar] [CrossRef]
- Qiu, B.; Matthay, K.K. Advancing Therapy for Neuroblastoma. Nat. Rev. Clin. Oncol. 2022, 19, 515–533. [Google Scholar] [CrossRef]
- DuBois, S.G.; Macy, M.E.; Henderson, T.O. High-Risk and Relapsed Neuroblastoma: Toward More Cures and Better Outcomes. Am. Soc. Clin. Oncol. Educ. Book 2022, 42, 768–780. [Google Scholar] [CrossRef]
- Whittle, S.B.; Smith, V.; Doherty, E.; Zhao, S.; McCarty, S.; Zage, P.E. Overview and Recent Advances in the Treatment of Neuroblastoma. Expert Rev. Anticancer Ther. 2017, 17, 369–386. [Google Scholar] [CrossRef]
- Croteau, N.; Nuchtern, J.; LaQuaglia, M.P. Management of Neuroblastoma in Pediatric Patients. Surg. Oncol. Clin. N. Am. 2021, 30, 291–304. [Google Scholar] [CrossRef]
- Pastor, E.R.; Mousa, S.A. Current Management of Neuroblastoma and Future Direction. Crit. Rev. Oncol. Hematol. 2019, 138, 38–43. [Google Scholar] [CrossRef]
- Newman, E.A.; Abdessalam, S.; Aldrink, J.H.; Austin, M.; Heaton, T.E.; Bruny, J.; Ehrlich, P.; Dasgupta, R.; Baertschiger, R.M.; Lautz, T.B.; et al. Update on Neuroblastoma. J. Pediatr. Surg. 2019, 54, 383–389. [Google Scholar] [CrossRef]
- Hallett, A.; Traunecker, H. A Review and Update on Neuroblastoma. Paediatr. Child Health 2012, 22, 103–107. [Google Scholar] [CrossRef]
- Anderson, J.; Majzner, R.G.; Sondel, P.M. Immunotherapy of Neuroblastoma: Facts and Hopes. Clin. Cancer Res. 2022, 28, 3196–3206. [Google Scholar] [CrossRef]
- Bartolucci, D.; Montemurro, L.; Raieli, S.; Lampis, S.; Pession, A.; Hrelia, P.; Tonelli, R. MYCN Impact on High-Risk Neuroblastoma: From Diagnosis and Prognosis to Targeted Treatment. Cancers 2022, 14, 4421. [Google Scholar] [CrossRef]
- Morandi, F.; Sabatini, F.; Podestà, M.; Airoldi, I. Immunotherapeutic Strategies for Neuroblastoma: Present, Past and Future. Vaccines 2021, 9, 43. [Google Scholar] [CrossRef]
- Bhoopathi, P.; Mannangatti, P.; Emdad, L.; Das, S.K.; Fisher, P.B. The Quest to Develop an Effective Therapy for Neuroblastoma. J. Cell Physiol. 2021, 236, 7775–7791. [Google Scholar] [CrossRef]
- Smith, V.; Foster, J. High-Risk Neuroblastoma Treatment Review. Children 2018, 5, 114. [Google Scholar] [CrossRef]
- Weinstein, J.L.; Katzenstein, H.M.; Cohn, S.L. Advances in the Diagnosis and Treatment of Neuroblastoma. Oncologist 2003, 8, 278–292. [Google Scholar] [CrossRef]
- Yeung, V.; Gabriel, M.; Padhye, B.D. Late Effects and Treatment Related Morbidity Associated with Treatment of Neuroblastoma Patients in a Tertiary Paediatric Centre. Cancer Rep. 2023, 6, e1738. [Google Scholar] [CrossRef]
- Elzembely, M.M.; Dahlberg, A.E.; Pinto, N.; Leger, K.J.; Chow, E.J.; Park, J.R.; Carpenter, P.A.; Baker, K.S. Late Effects in High-risk Neuroblastoma Survivors Treated with High-dose Chemotherapy and Stem Cell Rescue. Pediatr. Blood Cancer 2019, 66, e27421. [Google Scholar] [CrossRef]
- Friedman, D.; Henderson, T. Late Effects and Survivorship Issues in Patients with Neuroblastoma. Children 2018, 5, 107. [Google Scholar] [CrossRef]
- Shawraba, F.; Hammoud, H.; Mrad, Y.; Saker, Z.; Fares, Y.; Harati, H.; Bahmad, H.F.; Nabha, S. Biomarkers in Neuroblastoma: An Insight into Their Potential Diagnostic and Prognostic Utilities. Curr. Treat. Options Oncol. 2021, 22, 102. [Google Scholar] [CrossRef]
- Trigg, R.M.; Shaw, J.A.; Turner, S.D. Opportunities and Challenges of Circulating Biomarkers in Neuroblastoma. Open Biol. 2019, 9, 190056. [Google Scholar] [CrossRef]
- Machy, P.; Mortier, E.; Birklé, S. Biology of GD2 Ganglioside: Implications for Cancer Immunotherapy. Front. Pharmacol. 2023, 14, 1249929. [Google Scholar] [CrossRef]
- Chan, G.C.-F.; Chan, C.M. Anti-GD2 Directed Immunotherapy for High-Risk and Metastatic Neuroblastoma. Biomolecules 2022, 12, 358. [Google Scholar] [CrossRef]
- Shao, C.; Anand, V.; Andreeff, M.; Battula, V.L. Ganglioside GD2: A Novel Therapeutic Target in Triple-negative Breast Cancer. Ann. N. Y. Acad. Sci. 2022, 1508, 35–53. [Google Scholar] [CrossRef]
- Balis, F.M.; Busch, C.M.; Desai, A.V.; Hibbitts, E.; Naranjo, A.; Bagatell, R.; Irwin, M.; Fox, E. The Ganglioside G D2 as a Circulating Tumor Biomarker for Neuroblastoma. Pediatr. Blood Cancer 2020, 67, e28031. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 6450346, GD2 Ganglioside. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/GD2-Ganglioside (accessed on 25 June 2024).
- Sait, S.; Modak, S. Anti-GD2 Immunotherapy for Neuroblastoma. Expert Rev. Anticancer Ther. 2017, 17, 889–904. [Google Scholar] [CrossRef]
- Dhillon, S. Dinutuximab: First Global Approval. Drugs 2015, 75, 923–927. [Google Scholar] [CrossRef]
- Wang, W. NK Cell-Mediated Antibody-Dependent Cellular Cytotoxicity in Cancer Immunotherapy. Front. Immunol. 2015, 6, 368. [Google Scholar] [CrossRef]
- Iannello, A.; Ahmad, A. Role of Antibody-Dependent Cell-Mediated Cytotoxicity in the Efficacy of Therapeutic Anti-Cancer Monoclonal Antibodies. Cancer Metastasis Rev. 2005, 24, 487–499. [Google Scholar] [CrossRef]
- Golay, J.; Taylor, R.P. The Role of Complement in the Mechanism of Action of Therapeutic Anti-Cancer MAbs. Antibodies 2020, 9, 58. [Google Scholar] [CrossRef]
- Philippova, J.; Shevchenko, J.; Sennikov, S. GD2-Targeting Therapy: A Comparative Analysis of Approaches and Promising Directions. Front. Immunol. 2024, 15, 1371345. [Google Scholar] [CrossRef]
- Blom, T.; Lurvink, R.; Aleven, L.; Mensink, M.; Wolfs, T.; Dierselhuis, M.; van Eijkelenburg, N.; Kraal, K.; van Noesel, M.; van Grotel, M.; et al. Treatment-Related Toxicities During Anti-GD2 Immunotherapy in High-Risk Neuroblastoma Patients. Front. Oncol. 2021, 10, 601076. [Google Scholar] [CrossRef]
- Yu, A.L.; Gilman, A.L.; Ozkaynak, M.F.; Naranjo, A.; Diccianni, M.B.; Gan, J.; Hank, J.A.; Batova, A.; London, W.B.; Tenney, S.C.; et al. Long-Term Follow-up of a Phase III Study of Ch14.18 (Dinutuximab) + Cytokine Immunotherapy in Children with High-Risk Neuroblastoma: COG Study ANBL0032. Clin. Cancer Res. 2021, 27, 2179–2189. [Google Scholar] [CrossRef]
- Szanto, C.L.; Cornel, A.M.; Tamminga, S.M.; Delemarre, E.M.; de Koning, C.C.H.; van den Beemt, D.A.M.H.; Dunnebach, E.; Tas, M.L.; Dierselhuis, M.P.; Tytgat, L.G.A.M.; et al. Immune Monitoring during Therapy Reveals Activitory and Regulatory Immune Responses in High-Risk Neuroblastoma. Cancers 2021, 13, 2096. [Google Scholar] [CrossRef]
- Olgun, N.; Cecen, E.; Ince, D.; Kizmazoglu, D.; Baysal, B.; Onal, A.; Ozdogan, O.; Guleryuz, H.; Cetingoz, R.; Demiral, A.; et al. Dinutuximab Beta plus Conventional Chemotherapy for Relapsed/Refractory High-Risk Neuroblastoma: A Single-Center Experience. Front. Oncol. 2022, 12, 1041443. [Google Scholar] [CrossRef]
- Mody, R.; Yu, A.L.; Naranjo, A.; Zhang, F.F.; London, W.B.; Shulkin, B.L.; Parisi, M.T.; Servaes, S.-E.-N.; Diccianni, M.B.; Hank, J.A.; et al. Irinotecan, Temozolomide, and Dinutuximab with GM-CSF in Children with Refractory or Relapsed Neuroblastoma: A Report from the Children’s Oncology Group. J. Clin. Oncol. 2020, 38, 2160–2169. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 60838, Irinotecan. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Irinotecan (accessed on 25 June 2024).
- National Center for Biotechnology Information. PubChem Compound Summary for CID 5394, Temozolomide. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Temozolomide (accessed on 25 June 2024).
- Bergaggio, E.; Tai, W.-T.; Aroldi, A.; Mecca, C.; Landoni, E.; Nüesch, M.; Mota, I.; Metovic, J.; Molinaro, L.; Ma, L.; et al. ALK Inhibitors Increase ALK Expression and Sensitize Neuroblastoma Cells to ALK.CAR-T Cells. Cancer Cell 2023, 41, 2100–2116.e10. [Google Scholar] [CrossRef]
- Lerman, B.J.; Li, Y.; Carlowicz, C.; Granger, M.; Cash, T.; Sadanand, A.; Somers, K.; Ranavaya, A.; Weiss, B.D.; Choe, M.; et al. Progression-Free Survival and Patterns of Response in Patients with Relapsed High-Risk Neuroblastoma Treated with Irinotecan/Temozolomide/Dinutuximab/Granulocyte-Macrophage Colony-Stimulating Factor. J. Clin. Oncol. 2023, 41, 508–516. [Google Scholar] [CrossRef]
- Gilardi, M.; Wang, Z.; Proietto, M.; Chillà, A.; Calleja-Valera, J.L.; Goto, Y.; Vanoni, M.; Janes, M.R.; Mikulski, Z.; Gualberto, A.; et al. Tipifarnib as a Precision Therapy for HRAS-Mutant Head and Neck Squamous Cell Carcinomas. Mol. Cancer Ther. 2020, 19, 1784–1796. [Google Scholar] [CrossRef]
- Usha, L.; Dehghan-Paz, I.; van Golen, K.L.; Il’yasova, D. Tipifarnib and Farnesyltransferase Inhibitors in the Treatment of Inflammatory Breast Cancer: Is the Story over? A Review. Orphan Drugs Res. Rev. 2013, 2013, 11–21. [Google Scholar] [CrossRef]
- Liu, X.; Wills, C.A.; Chen, L.; Zhang, J.; Zhao, Y.; Zhou, M.; Sundstrom, J.M.; Schell, T.; Spiegelman, V.S.; Young, M.M.; et al. Small Extracellular Vesicles Induce Resistance to Anti-GD2 Immunotherapy Unveiling Tipifarnib as an Adjunct to Neuroblastoma Immunotherapy. J. Immunother. Cancer 2022, 10, e004399. [Google Scholar] [CrossRef]
- Capuano, C.; Pighi, C.; Battella, S.; De Federicis, D.; Galandrini, R.; Palmieri, G. Harnessing CD16-Mediated NK Cell Functions to Enhance Therapeutic Efficacy of Tumor-Targeting MAbs. Cancers 2021, 13, 2500. [Google Scholar] [CrossRef]
- Wieczorek, A.; Zaniewska-Tekieli, A.; Ehlert, K.; Pawinska-Wasikowska, K.; Balwierz, W.; Lode, H. Dinutuximab Beta Combined with Chemotherapy in Patients with Relapsed or Refractory Neuroblastoma. Front. Oncol. 2023, 13, 1082771. [Google Scholar] [CrossRef]
- U.S. National Library of Medicine. ClinicalTrials.gov. Irinotecan Hydrochloride and Temozolomide with Temsirolimus or Dinutuximab in Treating Younger Patients with Refractory or Relapsed Neuroblastoma. Available online: https://clinicaltrials.gov/study/NCT01767194 (accessed on 25 June 2024).
- U.S. National Library of Medicine. ClinicalTrials.gov. Immunotherapy with Dinutuximab Beta in Combination with Chemotherapy for the Treatment of Patients with Primary Neuroblastoma Refractory to Standard Therapy and with Relapsed or Progressive Disease (ChIm-NB-PL). Available online: https://clinicaltrials.gov/study/NCT05272371 (accessed on 25 June 2024).
- Flaadt, T.; Ebinger, M.; Schreiber, M.; Ladenstein, R.L.; Simon, T.; Lode, H.N.; Hero, B.; Schuhmann, M.U.; Schäfer, J.; Paulsen, F.; et al. Multimodal Therapy with Consolidating Haploidentical Stem Cell Transplantation and Dinutuximab Beta for Patients with High-Risk Neuroblastoma and Central Nervous System Relapse. J. Clin. Med. 2023, 12, 6196. [Google Scholar] [CrossRef]
- Flaadt, T.; Ladenstein, R.L.; Ebinger, M.; Lode, H.N.; Arnardóttir, H.B.; Poetschger, U.; Schwinger, W.; Meisel, R.; Schuster, F.R.; Döring, M.; et al. Anti-GD2 Antibody Dinutuximab Beta and Low-Dose Interleukin 2 After Haploidentical Stem-Cell Transplantation in Patients with Relapsed Neuroblastoma: A Multicenter, Phase I/II Trial. J. Clin. Oncol. 2023, 41, 3135–3148. [Google Scholar] [CrossRef]
- U.S. National Library of Medicine. ClinicalTrials.gov. CH14.18 1021 Antibody and IL2 After Haplo SCT in Children with Relapsed Neuroblastoma. Available online: https://clinicaltrials.gov/study/NCT02258815 (accessed on 25 June 2024).
- Muñoz, J.; Larrosa, C.; Chamorro, S.; Perez-Jaume, S.; Simao, M.; Sanchez-Sierra, N.; Varo, A.; Gorostegui, M.; Castañeda, A.; Garraus, M.; et al. Early Salvage Chemo-Immunotherapy with Irinotecan, Temozolomide and Naxitamab Plus GM-CSF (HITS) for Patients with Primary Refractory High-Risk Neuroblastoma Provide the Best Chance for Long-Term Outcomes. Cancers 2023, 15, 4837. [Google Scholar] [CrossRef]
- Markham, A. Naxitamab: First Approval. Drugs 2021, 81, 291–296. [Google Scholar] [CrossRef]
- Mora, J. Dinutuximab for the Treatment of Pediatric Patients with High-Risk Neuroblastoma. Expert Rev. Clin. Pharmacol. 2016, 9, 647–653. [Google Scholar] [CrossRef]
- Balaguer, J.; García Hidalgo, L.; Hladun, R.; Márquez Vega, C.; Pérez Alonso, V. Recent Evidence-Based Clinical Guide for the Use of Dinutuximab Beta in Pediatric Patients with Neuroblastoma. Target. Oncol. 2023, 18, 77–93. [Google Scholar] [CrossRef]
- Qarziba (dinutuximab beta) Summary of Product Characteristics (28/01/2020). Available online: https://www.ema.europa.eu/en/documents/product-information/qarziba-epar-product-information_en.pdf (accessed on 24 June 2024).
- Castañeda, A.; Gorostegui, M.; Miralles, S.L.; Chamizo, A.; Patiño, S.C.; Flores, M.A.; Garraus, M.; Lazaro, J.J.; Santa-Maria, V.; Varo, A.; et al. How We Approach the Treatment of Patients with High-Risk Neuroblastoma with Naxitamab: Experience from the Hospital Sant Joan de Déu in Barcelona, Spain. ESMO Open 2022, 7, 100462. [Google Scholar] [CrossRef]
- Mora, J.; Castañeda, A.; Gorostegui, M.; Varo, A.; Perez-Jaume, S.; Simao, M.; Muñoz, J.; Garraus, M.; Larrosa, C.; Salvador, N.; et al. Naxitamab Combined with Granulocyte-Macrophage Colony-Stimulating Factor as Consolidation for High-Risk Neuroblastoma Patients in First Complete Remission under Compassionate Use—Updated Outcome Report. Cancers 2023, 15, 2535. [Google Scholar] [CrossRef]
- Mody, R.; Naranjo, A.; Van Ryn, C.; Yu, A.L.; London, W.B.; Shulkin, B.L.; Parisi, M.T.; Servaes, S.-E.-N.; Diccianni, M.B.; Sondel, P.M.; et al. Irinotecan–Temozolomide with Temsirolimus or Dinutuximab in Children with Refractory or Relapsed Neuroblastoma (COG ANBL1221): An Open-Label, Randomised, Phase 2 Trial. Lancet Oncol. 2017, 18, 946–957. [Google Scholar] [CrossRef]
- Galassi, L.; Rossi, M.; Lodeserto, P.; Lenzi, M.; Borsetti, F.; Voltattorni, M.; Farruggia, G.; Blasi, P.; Orienti, I. Naxitamab Activity in Neuroblastoma Cells Is Enhanced by Nanofenretinide and Nanospermidine. Pharmaceutics 2023, 15, 648. [Google Scholar] [CrossRef]
- Orienti, I.; Francescangeli, F.; De Angelis, M.L.; Fecchi, K.; Bongiorno-Borbone, L.; Signore, M.; Peschiaroli, A.; Boe, A.; Bruselles, A.; Costantino, A.; et al. A New Bioavailable Fenretinide Formulation with Antiproliferative, Antimetabolic, and Cytotoxic Effects on Solid Tumors. Cell Death Dis. 2019, 10, 529. [Google Scholar] [CrossRef]
- Lodeserto, P.; Rossi, M.; Blasi, P.; Farruggia, G.; Orienti, I. Nanospermidine in Combination with Nanofenretinide Induces Cell Death in Neuroblastoma Cell Lines. Pharmaceutics 2022, 14, 1215. [Google Scholar] [CrossRef]
- Kaczanowska, S.; Murty, T.; Alimadadi, A.; Contreras, C.F.; Duault, C.; Subrahmanyam, P.B.; Reynolds, W.; Gutierrez, N.A.; Baskar, R.; Wu, C.J.; et al. Immune Determinants of CAR-T Cell Expansion in Solid Tumor Patients Receiving GD2 CAR-T Cell Therapy. Cancer Cell 2024, 42, 35–51.e8. [Google Scholar] [CrossRef]
- Yeku, O.O.; Longo, D.L. CAR T Cells for Neuroblastoma. N. Engl. J. Med. 2023, 388, 1328–1331. [Google Scholar] [CrossRef]
- Lutskovich, D.; Meleshko, A.; Katsin, M. State of the Art and Perspectives of CAR-T Cell Therapy for Neuroblastoma. Cytotherapy 2024, in press. [Google Scholar] [CrossRef] [PubMed]
- García-García, L.; Sánchez, E.G.; Ivanova, M.; Pastora, K.; Alcántara-Sánchez, C.; García-Martínez, J.; Martín-Antonio, B.; Ramírez, M.; González-Murillo, Á. Choosing T-Cell Sources Determines CAR-T Cell Activity in Neuroblastoma. Front. Immunol. 2024, 15, 1375833. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef]
- Del Bufalo, F.; De Angelis, B.; Caruana, I.; Del Baldo, G.; De Ioris, M.A.; Serra, A.; Mastronuzzi, A.; Cefalo, M.G.; Pagliara, D.; Amicucci, M.; et al. GD2-CART01 for Relapsed or Refractory High-Risk Neuroblastoma. N. Engl. J. Med. 2023, 388, 1284–1295. [Google Scholar] [CrossRef]
- Toews, K.; Grunewald, L.; Schwiebert, S.; Klaus, A.; Winkler, A.; Ali, S.; Zirngibl, F.; Astrahantseff, K.; Wagner, D.L.; Henssen, A.G.; et al. Central Memory Phenotype Drives Success of Checkpoint Inhibition in Combination with CAR T Cells. Mol. Carcinog. 2020, 59, 724–735. [Google Scholar] [CrossRef] [PubMed]
- Bocca, P.; Di Carlo, E.; Caruana, I.; Emionite, L.; Cilli, M.; De Angelis, B.; Quintarelli, C.; Pezzolo, A.; Raffaghello, L.; Morandi, F.; et al. Bevacizumab-Mediated Tumor Vasculature Remodelling Improves Tumor Infiltration and Antitumor Efficacy of GD2-CAR T Cells in a Human Neuroblastoma Preclinical Model. Oncoimmunology 2018, 7, e1378843. [Google Scholar] [CrossRef] [PubMed]
- Zuo, S.; Sho, M.; Sawai, T.; Kanehiro, H.; Maeda, K.; Yoshida, M.; Tsukada, R.; Nomura, M.; Okuyama, H. Potential Role of the PD-L1 Expression and Tumor-Infiltrating Lymphocytes on Neuroblastoma. Pediatr. Surg. Int. 2020, 36, 137–143. [Google Scholar] [CrossRef]
- Heczey, A.; Louis, C.U.; Savoldo, B.; Dakhova, O.; Durett, A.; Grilley, B.; Liu, H.; Wu, M.F.; Mei, Z.; Gee, A.; et al. CAR T Cells Administered in Combination with Lymphodepletion and PD-1 Inhibition to Patients with Neuroblastoma. Mol. Ther. 2017, 25, 2214–2224. [Google Scholar] [CrossRef] [PubMed]
- Vanneman, M.; Dranoff, G. Combining Immunotherapy and Targeted Therapies in Cancer Treatment. Nat. Rev. Cancer 2012, 12, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Xu, L.; Liang, C.; Wang, C.; Peng, R.; Liu, Z. Photothermal Therapy with Immune-Adjuvant Nanoparticles Together with Checkpoint Blockade for Effective Cancer Immunotherapy. Nat. Commun. 2016, 7, 13193. [Google Scholar] [CrossRef]
- Tao, Y.; Ju, E.; Ren, J.; Qu, X. Immunostimulatory Oligonucleotides-Loaded Cationic Graphene Oxide with Photothermally Enhanced Immunogenicity for Photothermal/Immune Cancer Therapy. Biomaterials 2014, 35, 9963–9971. [Google Scholar] [CrossRef]
- Guo, L.; Yan, D.D.; Yang, D.; Li, Y.; Wang, X.; Zalewski, O.; Yan, B.; Lu, W. Combinatorial Photothermal and Immuno Cancer Therapy Using Chitosan-Coated Hollow Copper Sulfide Nanoparticles. ACS Nano 2014, 8, 5670–5681. [Google Scholar] [CrossRef]
- Zhou, F.; Wu, S.; Song, S.; Chen, W.R.; Resasco, D.E.; Xing, D. Antitumor Immunologically Modified Carbon Nanotubes for Photothermal Therapy. Biomaterials 2012, 33, 3235–3242. [Google Scholar] [CrossRef] [PubMed]
- Cano-Mejia, J.; Shukla, A.; Ledezma, D.K.; Palmer, E.; Villagra, A.; Fernandes, R. CpG-Coated Prussian Blue Nanoparticles-Based Photothermal Therapy Combined with Anti-CTLA-4 Immune Checkpoint Blockade Triggers a Robust Abscopal Effect against Neuroblastoma. Transl. Oncol. 2020, 13, 100823. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, P.B.; Sweeney, E.E.; Ramanujam, A.S.; Fernandes, R. Photothermal Therapies to Improve Immune Checkpoint Blockade for Cancer. Int. J. Hyperth. 2020, 37, 34–49. [Google Scholar] [CrossRef]
- Sekhri, P.; Ledezma, D.K.; Shukla, A.; Sweeney, E.E.; Fernandes, R. The Thermal Dose of Photothermal Therapy Generates Differential Immunogenicity in Human Neuroblastoma Cells. Cancers 2022, 14, 1447. [Google Scholar] [CrossRef]
- Zhou, W.; Lim, A.; Edderkaoui, M.; Osipov, A.; Wu, H.; Wang, Q.; Pandol, S. Role of YAP Signaling in Regulation of Programmed Cell Death and Drug Resistance in Cancer. Int. J. Biol. Sci. 2024, 20, 15–28. [Google Scholar] [CrossRef]
- Ricco, N.; Kron, S.J. Statins in Cancer Prevention and Therapy. Cancers 2023, 15, 3948. [Google Scholar] [CrossRef] [PubMed]
- Mokhtari, R.B.; Ashayeri, N.; Baghaie, L.; Sambi, M.; Satari, K.; Baluch, N.; Bosykh, D.A.; Szewczuk, M.R.; Chakraborty, S. The Hippo Pathway Effectors YAP/TAZ-TEAD Oncoproteins as Emerging Therapeutic Targets in the Tumor Microenvironment. Cancers 2023, 15, 3468. [Google Scholar] [CrossRef]
- Shim, J.; Lee, Y.J.; Janssen, K.; Fritz, A.; Goldsmith, K.C. Abstract 2889: The Yes-Associated Protein Plays a Role in Tumor Formation, Stemness, and Therapy Response in High Risk Neuroblastoma. Cancer Res. 2019, 79, 2889. [Google Scholar] [CrossRef]
- Shim, J.; Goldsmith, K.C. A New Player in Neuroblastoma: YAP and Its Role in the Neuroblastoma Microenvironment. Cancers 2021, 13, 4650. [Google Scholar] [CrossRef]
- Coggins, G.E.; Farrel, A.; Rathi, K.S.; Hayes, C.M.; Scolaro, L.; Rokita, J.L.; Maris, J.M. YAP1 Mediates Resistance to MEK1/2 Inhibition in Neuroblastomas with Hyperactivated RAS Signaling. Cancer Res. 2019, 79, 6204–6214. [Google Scholar] [CrossRef]
- Shen, X.; Xu, X.; Xie, C.; Liu, H.; Yang, D.; Zhang, J.; Wu, Q.; Feng, W.; Wang, L.; Du, L.; et al. YAP Promotes the Proliferation of Neuroblastoma Cells through Decreasing the Nuclear Location of P27 Kip1 Mediated by Akt. Cell Prolif. 2020, 53, e12734. [Google Scholar] [CrossRef]
- Yang, C.; Tan, J.; Zhu, J.; Wang, S.; Wei, G. YAP Promotes Tumorigenesis and Cisplatin Resistance in Neuroblastoma. Oncotarget 2017, 8, 37154–37163. [Google Scholar] [CrossRef]
- Özşengezer, S.K.; Altun, Z.; Olgun, N. Neuroblastoma and Hippo Signaling Pathway. J. Behcet Uz Child. Hosp. 2021, 11, 1–8. [Google Scholar] [CrossRef]
- Takemoto, M.; Tanaka, T.; Tsuji, R.; Togashi, Y.; Higashi, M.; Fumino, S.; Tajiri, T. The Synergistic Antitumor Effect of Combination Therapy with a MEK Inhibitor and YAP Inhibitor on PERK-Positive Neuroblastoma. Biochem. Biophys. Res. Commun. 2021, 570, 41–46. [Google Scholar] [CrossRef]
- Shim, J.; Lee, J.Y.; Jonus, H.C.; Arnold, A.; Schnepp, R.W.; Janssen, K.M.; Maximov, V.; Goldsmith, K.C. YAP-Mediated Repression of HRK Regulates Tumor Growth, Therapy Response, and Survival Under Tumor Environmental Stress in Neuroblastoma. Cancer Res. 2020, 80, 4741–4753. [Google Scholar] [CrossRef]
- Belgiovine, C.; Mebelli, K.; Raffaele, A.; De Cicco, M.; Rotella, J.; Pedrazzoli, P.; Zecca, M.; Riccipetitoni, G.; Comoli, P. Pediatric Solid Cancers: Dissecting the Tumor Microenvironment to Improve the Results of Clinical Immunotherapy. Int. J. Mol. Sci. 2024, 25, 3225. [Google Scholar] [CrossRef]
- Pilgrim, A.A.; Jonus, H.C.; Ho, A.; Cole, A.; Shim, J.; Goldsmith, K.C. Abstract 3545: The Yes-Associated Protein (YAP) Regulates GD2 Immunotherapy Response in High-Risk Neuroblastoma. Cancer Res. 2023, 83, 3545. [Google Scholar] [CrossRef]
- Pilgrim, A.A.; Jonus, H.C.; Ho, A.; Cole, A.C.; Shim, J.; Goldsmith, K.C. The Yes-Associated Protein (YAP) Is Associated with Resistance to Anti-GD2 Immunotherapy in Neuroblastoma through Downregulation of ST8SIA1. Oncoimmunology 2023, 12, 2240678. [Google Scholar] [CrossRef]
- Lovatt, C.; Parker, A.L. Oncolytic Viruses and Immune Checkpoint Inhibitors: The “Hot” New Power Couple. Cancers 2023, 15, 4178. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-T.; Dai, S.-Y.; Zhan, Y.; Yang, R.; Chen, D.-Q.; Li, Y.; Zhou, E.-Q.; Dong, R. Progress of Oncolytic Virotherapy for Neuroblastoma. Front. Pediatr. 2022, 10, 1055729. [Google Scholar] [CrossRef] [PubMed]
- Muscolini, M.; Tassone, E.; Hiscott, J. Oncolytic Immunotherapy: Can’t Start a Fire without a Spark. Cytokine Growth Factor Rev. 2020, 56, 94–101. [Google Scholar] [CrossRef]
- LaRocca, C.J.; Warner, S.G. Oncolytic Viruses and Checkpoint Inhibitors: Combination Therapy in Clinical Trials. Clin. Transl. Med. 2018, 7, 35. [Google Scholar] [CrossRef]
- Hallenbeck, P.L.; Chada, S.; Sankar, N.; Chauhan, A. Oncolytic Seneca Valley Virus (SVV-001) Overcomes Checkpoint Inhibitor Resistance and Demonstrates a Systemic Anti-Tumor Response in a Syngeneic Tumor Model. Endocr. Abstr. 2023, 89, B5. [Google Scholar] [CrossRef]
- Chiu, M.; Armstrong, E.J.L.; Jennings, V.; Foo, S.; Crespo-Rodriguez, E.; Bozhanova, G.; Patin, E.C.; McLaughlin, M.; Mansfield, D.; Baker, G.; et al. Combination Therapy with Oncolytic Viruses and Immune Checkpoint Inhibitors. Expert Opin. Biol. Ther. 2020, 20, 635–652. [Google Scholar] [CrossRef]
- Sivanandam, V.; LaRocca, C.J.; Chen, N.G.; Fong, Y.; Warner, S.G. Oncolytic Viruses and Immune Checkpoint Inhibition: The Best of Both Worlds. Mol. Ther. Oncolytics 2019, 13, 93–106. [Google Scholar] [CrossRef]
- Franco-Luzón, L.; González-Murillo, Á.; Alcántara-Sánchez, C.; García-García, L.; Tabasi, M.; Huertas, A.L.; Chesler, L.; Ramírez, M. Systemic Oncolytic Adenovirus Delivered in Mesenchymal Carrier Cells Modulate Tumor Infiltrating Immune Cells and Tumor Microenvironment in Mice with Neuroblastoma. Oncotarget 2020, 11, 347–361. [Google Scholar] [CrossRef]
- Mazar, J.; Brooks, J.K.; Peloquin, M.; Rosario, R.; Sutton, E.; Longo, M.; Drehner, D.; Westmoreland, T.J. The Oncolytic Activity of Zika Viral Therapy in Human Neuroblastoma In Vivo Models Confers a Major Survival Advantage in a CD24-Dependent Manner. Cancer Res. Commun. 2024, 4, 65–80. [Google Scholar] [CrossRef]
- Tanimoto, T.; Tazawa, H.; Ieda, T.; Nouso, H.; Tani, M.; Oyama, T.; Urata, Y.; Kagawa, S.; Noda, T.; Fujiwara, T. Elimination of MYCN-Amplified Neuroblastoma Cells by Telomerase-Targeted Oncolytic Virus via MYCN Suppression. Mol. Ther. Oncolytics 2020, 18, 14–23. [Google Scholar] [CrossRef]
- Gillory, L.A.; Megison, M.L.; Stewart, J.E.; Mroczek-Musulman, E.; Nabers, H.C.; Waters, A.M.; Kelly, V.; Coleman, J.M.; Markert, J.M.; Gillespie, G.Y.; et al. Preclinical Evaluation of Engineered Oncolytic Herpes Simplex Virus for the Treatment of Neuroblastoma. PLoS ONE 2013, 8, e77753. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Dutuor, A.; Tao, L.; Fu, X.; Zhang, X. Virotherapy with a Type 2 Herpes Simplex Virus–Derived Oncolytic Virus Induces Potent Antitumor Immunity against Neuroblastoma. Clin. Cancer Res. 2007, 13, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Bauer, D.F.; Pereboeva, L.; Gillespie, G.Y.; Cloud, G.A.; Elzafarany, O.; Langford, C.; Markert, J.M.; Lamb, L.S., Jr. Effect of HSV-IL12 Loaded Tumor Cell-Based Vaccination in a Mouse Model of High-Grade Neuroblastoma. J. Immunol. Res. 2016, 2016, 2568125. [Google Scholar] [CrossRef] [PubMed]
- Melen, G.J.; Franco-Luzón, L.; Ruano, D.; González-Murillo, Á.; Alfranca, A.; Casco, F.; Lassaletta, Á.; Alonso, M.; Madero, L.; Alemany, R.; et al. Influence of Carrier Cells on the Clinical Outcome of Children with Neuroblastoma Treated with High Dose of Oncolytic Adenovirus Delivered in Mesenchymal Stem Cells. Cancer Lett. 2016, 371, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Moreno, L.; Teira, P.; Croop, J.M.; Gerber, N.U.; André, N.; Aerts, I.; Gros Subias, L.; De Wilde, B.; Bautista, F.; Turpin, B.; et al. A Phase 1, First-in-Child, Multicenter Study to Evaluate the Safety and Efficacy of the Oncolytic Herpes Virus Talimogene Laherparepvec in Pediatric Patients with Advanced Solid Tumors. Front. Pediatr. 2023, 11, 1183295. [Google Scholar] [CrossRef] [PubMed]
- Burke, M.J.; Ahern, C.; Weigel, B.J.; Poirier, J.T.; Rudin, C.M.; Chen, Y.; Cripe, T.P.; Bernhardt, M.B.; Blaney, S.M. Phase I Trial of Seneca Valley Virus (NTX-010) in Children with Relapsed/Refractory Solid Tumors: A Report of the Children’s Oncology Group. Pediatr. Blood Cancer 2015, 62, 743–750. [Google Scholar] [CrossRef]
- Streby, K.A.; Currier, M.A.; Triplet, M.; Ott, K.; Dishman, D.J.; Vaughan, M.R.; Ranalli, M.A.; Setty, B.; Skeens, M.A.; Whiteside, S.; et al. First-in-Human Intravenous Seprehvir in Young Cancer Patients: A Phase 1 Clinical Trial. Mol. Ther. 2019, 27, 1930–1938. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.I.; Curti, B.; Daniels, G.A.; Hallmeyer, S.; Whitman, E.D.; Lutzky, J.; Spitler, L.E.; Zhou, K.; Bommareddy, P.K.; Grose, M.; et al. Clinical Responses of Oncolytic Coxsackievirus A21 (V937) in Patients with Unresectable Melanoma. J. Clin. Oncol. 2021, 39, 3829–3838. [Google Scholar] [CrossRef] [PubMed]
- Chesney, J.A.; Puzanov, I.; Collichio, F.A.; Singh, P.; Milhem, M.M.; Glaspy, J.; Hamid, O.; Ross, M.; Friedlander, P.; Garbe, C.; et al. Talimogene Laherparepvec in Combination with Ipilimumab versus Ipilimumab Alone for Advanced Melanoma: 5-Year Final Analysis of a Multicenter, Randomized, Open-Label, Phase II Trial. J. Immunother. Cancer 2023, 11, e006270. [Google Scholar] [CrossRef] [PubMed]
- Puzanov, I.; Milhem, M.M.; Minor, D.; Hamid, O.; Li, A.; Chen, L.; Chastain, M.; Gorski, K.S.; Anderson, A.; Chou, J.; et al. Talimogene Laherparepvec in Combination with Ipilimumab in Previously Untreated, Unresectable Stage IIIB-IV Melanoma. J. Clin. Oncol. 2016, 34, 2619–2626. [Google Scholar] [CrossRef] [PubMed]
- Nassiri, F.; Patil, V.; Yefet, L.S.; Singh, O.; Liu, J.; Dang, R.M.A.; Yamaguchi, T.N.; Daras, M.; Cloughesy, T.F.; Colman, H.; et al. Oncolytic DNX-2401 Virotherapy plus Pembrolizumab in Recurrent Glioblastoma: A Phase 1/2 Trial. Nat. Med. 2023, 29, 1370–1378. [Google Scholar] [CrossRef]
- Monge, C.; Xie, C.; Myojin, Y.; Coffman, K.; Hrones, D.M.; Wang, S.; Hernandez, J.M.; Wood, B.J.; Levy, E.B.; Juburi, I.; et al. Phase I/II Study of PexaVec in Combination with Immune Checkpoint Inhibition in Refractory Metastatic Colorectal Cancer. J. Immunother. Cancer 2023, 11, e005640. [Google Scholar] [CrossRef]
- Guan, J.; Sun, K.; Guerrero, C.A.; Zheng, J.; Xu, Y.; Mathur, S.; Teh, B.S.; Farach, A.; Zhang, J.; Butler, E.; et al. A Phase 2 Study of In Situ Oncolytic Virus Therapy and Stereotactic Body Radiation Therapy Followed by Pembrolizumab in Metastatic Non-Small Cell Lung Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2024, 118, 1531–1540. [Google Scholar] [CrossRef] [PubMed]
- Hallenbeck, P.L.; Chada, S.; Morris, Z.S.; Bates, A.M.; Erbe, A.K. Abstract LB039: Oncolytic Seneca Valley Virus (SVV) Overcomes Resistance to Checkpoint Inhibitor Therapies in Neuroendocrine and Melanoma Murine Models Expressing the Receptor for SVV. Cancer Res. 2021, 81, LB039. [Google Scholar] [CrossRef]
- Cooper, N.; Ghanima, W.; Hill, Q.A.; Nicolson, P.L.; Markovtsov, V.; Kessler, C. Recent Advances in Understanding Spleen Tyrosine Kinase (SYK) in Human Biology and Disease, with a Focus on Fostamatinib. Platelets 2023, 34, 2131751. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Fostamatinib: First Global Approval. Drugs 2018, 78, 959–963. [Google Scholar] [CrossRef] [PubMed]
- Rohila, D.; Park, I.H.; Pham, T.V.; Jones, R.; Tapia, E.; Liu, K.X.; Tamayo, P.; Yu, A.; Sharabi, A.B.; Joshi, S. Targeting Macrophage Syk Enhances Responses to Immune Checkpoint Blockade and Radiotherapy in High-Risk Neuroblastoma. Front. Immunol. 2023, 14, 1148317. [Google Scholar] [CrossRef]
- Beaudry, A.; Jacques-Ricard, S.; Darracq, A.; Sgarioto, N.; Garcia, A.; García, T.R.; Lemieux, W.; Béland, K.; Haddad, E.; Cordeiro, P.; et al. Repurposing Disulfiram, an Alcohol-Abuse Drug, in Neuroblastoma Causes KAT2A Downregulation and in Vivo Activity with a Water/Oil Emulsion. Sci. Rep. 2023, 13, 16443. [Google Scholar] [CrossRef] [PubMed]
- Dufour, P.; Lang, J.-M.; Giron, C.; Duclos, B.; Haehnel, P.; Jaeck, D.; Jung, J.-M.; Oberling, F. Sodium Ditiocarb as Adjuvant Immunotherapy for High Risk Breast Cancer: A Randomized Study. Biotherapy 1993, 6, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Nechushtan, H.; Hamamreh, Y.; Nidal, S.; Gotfried, M.; Baron, A.; Shalev, Y.I.; Nisman, B.; Peretz, T.; Peylan-Ramu, N. A Phase IIb Trial Assessing the Addition of Disulfiram to Chemotherapy for the Treatment of Metastatic Non-Small Cell Lung Cancer. Oncologist 2015, 20, 366–367. [Google Scholar] [CrossRef]
- Skrott, Z.; Mistrik, M.; Andersen, K.K.; Friis, S.; Majera, D.; Gursky, J.; Ozdian, T.; Bartkova, J.; Turi, Z.; Moudry, P.; et al. Alcohol-Abuse Drug Disulfiram Targets Cancer via P97 Segregase Adaptor NPL4. Nature 2017, 552, 194–199. [Google Scholar] [CrossRef]

| Pretreatment Risk Group | Proportion of Patients | Current Treatment |
|---|---|---|
| Very low and low | 55% [2] | Observation alone or surgical resection |
| Intermediate | 9% [2] | Neoadjuvant chemotherapy, surgical resection |
| High | 36% [2] | Intensive multimodality treatment including: (I) Induction: surgical resection, multiagent chemotherapy (II) Consolidation: autologous stem cell transplant, high-dose chemotherapy, radiotherapy (III) Maintenance: anti-GD2 immunotherapy, isotretinoin |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pieniążek, B.; Cencelewicz, K.; Bździuch, P.; Młynarczyk, Ł.; Lejman, M.; Zawitkowska, J.; Derwich, K. Neuroblastoma—A Review of Combination Immunotherapy. Int. J. Mol. Sci. 2024, 25, 7730. https://doi.org/10.3390/ijms25147730
Pieniążek B, Cencelewicz K, Bździuch P, Młynarczyk Ł, Lejman M, Zawitkowska J, Derwich K. Neuroblastoma—A Review of Combination Immunotherapy. International Journal of Molecular Sciences. 2024; 25(14):7730. https://doi.org/10.3390/ijms25147730
Chicago/Turabian StylePieniążek, Barbara, Katarzyna Cencelewicz, Patrycja Bździuch, Łukasz Młynarczyk, Monika Lejman, Joanna Zawitkowska, and Katarzyna Derwich. 2024. "Neuroblastoma—A Review of Combination Immunotherapy" International Journal of Molecular Sciences 25, no. 14: 7730. https://doi.org/10.3390/ijms25147730