Abstract
Mitochondrial stress, resulting from dysfunction and proteostasis disturbances, triggers the mitochondrial unfolded protein response (UPRMT), which activates gene encoding chaperones and proteases to restore mitochondrial function. Although ATFS-1 mediates mitochondrial stress UPRMT induction in C. elegans, the mechanisms relaying mitochondrial stress signals to the nucleus in mammals remain poorly defined. Here, we explored the role of protein kinase R (PKR), an eIF2α kinase activated by double-stranded RNAs (dsRNAs), in mitochondrial stress signaling. We found that UPRMT does not occur in cells lacking PKR, indicating its crucial role in this process. Mechanistically, we observed that dsRNAs accumulate within mitochondria under stress conditions, along with unprocessed mitochondrial transcripts. Furthermore, we demonstrated that accumulated mitochondrial dsRNAs in mouse embryonic fibroblasts (MEFs) deficient in the Bax/Bak channels are not released into the cytosol and do not induce the UPRMT upon mitochondrial stress, suggesting a potential role of the Bax/Bak channels in mediating the mitochondrial stress response. These discoveries enhance our understanding of how cells maintain mitochondrial integrity, respond to mitochondrial dysfunction, and communicate stress signals to the nucleus through retrograde signaling. This knowledge provides valuable insights into prospective therapeutic targets for diseases associated with mitochondrial stress.
1. Introduction
In the last few decades, mitochondria’s role in health and disease has gained interest exponentially. Aside from its well-known function as a cellular powerhouse, mitochondria possess other essential metabolic functions, such as calcium homeostasis and the synthesis of amino acids, fatty acids, and nucleotides [,,,]. Furthermore, mitochondria serve as signaling hubs to regulate programmed cell death and NLRP3 inflammasome activation [,]. To carry out these diverse set of functions, mitochondria contain over a thousand proteins, most of which are encoded by nuclear genes, imported from the cytosol, and properly folded in the mitochondria [,,]. Thus, the disruption of its proteostasis could result in mitochondrial dysfunction. To preserve the mitochondrial homeostasis upon the stressful condition, cells activate a transcriptional response known as the mitochondrial unfolded protein response (mitochondrial UPR, UPRMT) [].
UPRMT is well established in C. elegans, where the activating transcription factor associated with stress (ATFS-1) is the key mediator for this response. ATFS-1 has a nuclear localization signal (NLS) as well as N-terminal mitochondrial-targeting sequences (MTS) []. In a normal condition, ATFS-1 is imported into the mitochondria and degraded by the protease LONP-1 []. However, under mitochondrial dysfunction conditions, the mitochondrial import of ATFS-1 is reduced, resulting in the accumulation of ATFS-1 in the cytoplasm and then it traffics to the nucleus driven by its NLS to activate the transcription of its target genes [].
In contrast to the well-defined mechanism observed in C. elegans, the precise pathway by which the UPRMT is activated in mammalian cells remains elusive. However, several recent studies identified ATF5 as a functional ortholog of ATFS-1 []. In addition to ATF5, ATF4 and CHOP are also suggested to be involved in the UPRMT activation [,,,]. Beyond ATF5, ATF4, and CHOP, there has been considerable evolutionary divergence in UPRMT signaling between C. elegans and mammalian cells. This divergence includes the expansion of transcription factors and the incorporation of additional signaling mechanisms. It is of note that the expression of all three transcription factors requires the phosphorylation of the alpha subunit of the translation initiation factor 2 (eIF2α) and subsequent signaling pathway, collectively referred to as the integrated stress response (ISR) [].
The ISR is an evolutionary conserved signaling network for the cells to adapt to variable environmental and pathophysiological stresses, including a proteostasis defect, oxidative stress, nutrient deprivation, and viral infection []. These various stresses are sensed by four specific kinases responsible for eIF2α phosphorylation: PKR-like endoplasmic reticulum kinase (PERK), general control non-derepressible 2 (GCN2), protein kinase RNA-activated (PKR), and heme-regulated eIF2α kinase (HRI) []. The phosphorylated eIF2α inhibits the activity of eIF2B, a guanine nucleotide exchange factor for the eIF2-GDP complex, resulting in a global attenuation of protein synthesis [,]. Paradoxically, certain genes with upstream open reading frames (uORFs) in the 5′ UTR, such as ATF4 and CHOP, are preferentially translated []. These proteins function as transcription factors to induce various genes involved in regulating the stress response and fine-tuning the outcomes of these stresses [].
PKR, a well-established antiviral sensor, has the primary function to sense and respond to double-stranded RNA (dsRNA), a molecular pattern often associated with viral infections []. Upon binding to dsRNA, PKR undergoes autophosphorylation and activation, leading to eIF2α phosphorylation and global protein synthesis, thereby limiting viral replication and activating stress response pathways []. Interestingly, PKR has been demonstrated to be activated in uninfected cells by cellular endogenous dsRNAs during mitosis [], and further investigation revealed that mitochondrial RNAs constitute a major fraction of the PKR interactome []. On the other hand, mitochondrial dsRNAs have emerged as a potent damage-associated molecular pattern activating PKR and the ISR in several disease models [,,]. While an earlier study has implicated PKR’s role in UPRMT activation [], the interplay of mitochondrial dsRNAs and PKR activation in the UPRMT is yet to be addressed.
Recent studies have demonstrated increased ISR activation during mitochondrial stress [], suggesting a potential role for eIF2α phosphorylation and its kinases. While several studies have proposed that GCN2 [], PERK [], HRI [,], and PKR [] might sense mitochondrial dysfunction and transduce the signal to the ISR, the specific role of PKR in this process has not yet been elucidated. In this study, we revealed that PKR is involved in the activation of the UPRMT by sensing mitochondrial dsRNAs generated under mitochondrial stress conditions.
2. Results
2.1. Critical Role of ISR in the Activation of UPRMT
Although several studies have suggested that the ISR plays a pivotal role in triggering the UPRMT following mitochondrial stress, conclusive evidence directly linking the ISR to the UPRMT remains elusive. Given the critical role of eIF2α phosphorylation in initiating the ISR, we investigated whether it is essential for UPRMT activation under mitochondrial stress conditions. To this end, we utilized mouse embryonic fibroblasts (MEFs) that feature a homozygous alanine substitution at serine 51 of the eIF2α subunit (Eif2aA/A), effectively preventing its phosphorylation.
To induce mitochondrial stress, we treated the MEFs with gamitrinib-triphenylphosphonium (GTPP), which inhibits the mitochondrial chaperone HSP90 homolog TRAP1 [,]. As a positive control to induce the ISR, we used thapsigargin (TG), a SERCA inhibitor known to induce ER stress and subsequent eIF2α phosphorylation. As expected, the Eif2aA/A MEFs did not exhibit phosphorylated forms of eIF2α following treatment with GTPP and TG, in contrast to the increase in phosphorylated eIF2α observed in the wildtype Eif2aS/S MEFs treated with both chemicals (Figure 1A). In the Eif2aA/A MEFs, the expression of the UPRMT genes such as Hspd1 and Hspe1, as well as the ISR-related genes including Ddit3, Gdf15, and Fgf21, did not increase following treatment with either chemical, in contrast to the response observed in the Eif2aS/S MEFs (Figure 1C).
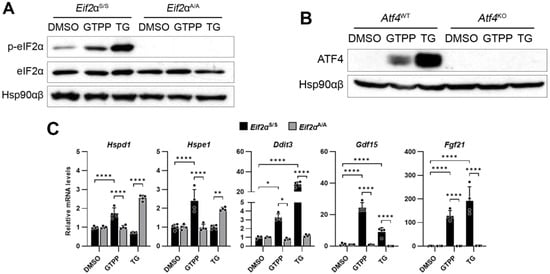
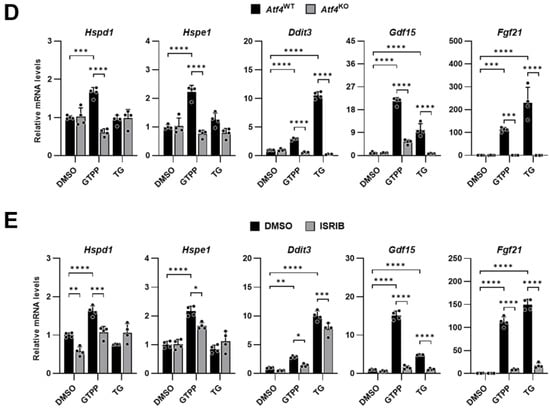
Figure 1.
Integrated stress response (ISR) is required for mitochondrial unfolded protein response (UPRMT) activation. Indicated mouse embryonic fibroblasts (MEFs) were treated with mitochondrial chaperone TRAP1 inhibitor gamitrinib-triphenylphosphonium (GTPP; 10 μM), ER stressor thapsigargin (TG; 300 nM), or DMSO control. (A,B) Cell lysates were obtained 2 h (A) and 4 h (B) after stress induction for Western blot analysis. Hsp90αβ was used as loading control. (C–E) Total RNAs were isolated from (C,D) indicated MEFs or (E) wildtype MEFs treated in the presence of ISR inhibitor (ISRIB; 500 nM) at 8 h after stress induction for RT-qPCR analysis to measure UPRMT- and ISR-related genes (n = 4). Also, 18S rRNA primers were used as internal control. Data are presented as mean ± SD. * p < 0.0332, ** p < 0.0021, *** p < 0.0002, and **** p < 0.0001.
Furthermore, we investigated the role of ATF4, which functions downstream of eIF2α phosphorylation in the ISR signaling pathway, in the mitochondrial stress response elicited by GTPP using ATF4 knock-out (Atf4KO) MEFs. In the absence of ATF4 protein expression in the Atf4KO cells (Figure 1B), the expression of the UPRMT-related genes was not induced following treatment with either GTPP or TG (Figure 1D). This lack of response underscores the critical role of ATF4 in UPRMT signaling.
The impact of the ISR on UPRMT induction was further confirmed through the application of ISRIB, an inhibitor of the ISR signaling pathway. Consistent with previous observations, treatment with ISRIB significantly attenuated the expression of genes related to the UPRMT and the ISR (Figure 1E). This corroborates the findings in Eif2aA/A MEFs, where the absence of eIF2α phosphorylation hindered the necessary signal transduction to activate these genes, emphasizing the indispensable role of the ISR, and particularly ATF4, in the regulation of the UPRMT.
2.2. PKR Mediation of ISR and UPRMT in Response to Mitochondrial Stress
The ISR is initiated by the phosphorylation of eIF2α, mediated by four kinases: GCN2, PERK, HRI, and PKR. While these kinases have been implicated in mitochondrial stress-mediated UPRMT activation, the specific role of PKR in this process remains unclear. Given the close relationship between the ISR and UPRMT induction following GTPP treatment, we focused on elucidating PKR’s contribution to ISR activation and the UPRMT under mitochondrial stress conditions.
We employed MEFs derived from PKR knock-out (PkrKO) mice to delineate the specific role of PKR in this pathway. To confirm the loss of PKR, we treated the MEFs with the viral dsRNA mimic poly(I:C) and assessed the expression of the Ifnb1 gene, which is specifically activated by double-stranded RNA and is a known downstream target of PKR. In the PkrKO MEFs, phosphorylation of eIF2α was absent, and there was a failure to induce Ifnb1 following transfection with the dsRNA analog poly(I:C), which is in marked contrast to the response observed in the PKR wildtype (PkrWT) MEFs (Figure 2A). We then treated both the PkrKO and PkrWT MEFs with GTPP and TG. The absence of PKR in the PkrKO MEFs resulted in reduced eIF2α phosphorylation in response to GTPP, whereas there was no significant difference in the eIF2α phosphorylation between the PkrKO and PkrWT MEFs when treated with TG (Figure 2B). In addition, Ifnb1 expression was induced in the PkrWT upon GTPP and TG treatment but not in the PkrKO MEFs (Figure 2C). This differential response suggests that GTPP-induced phosphorylation of eIF2α is contingent on the presence of PKR, highlighting its unique role in the cellular response to mitochondrial stress.
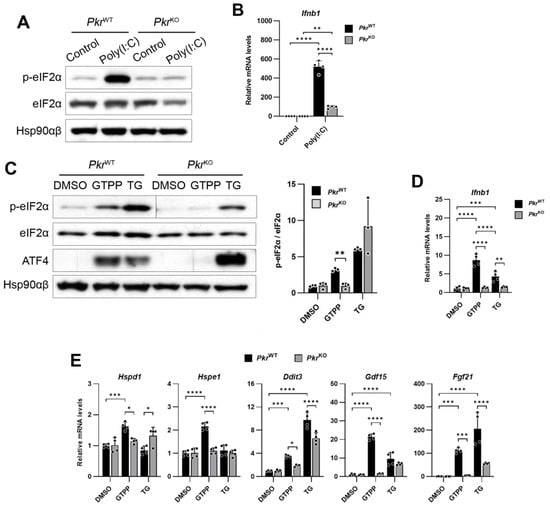
Figure 2.
Immortalized PkrWT and PkrKO MEFs were generated from PkrWT and PkrKO mice. (A,B) Indicated MEFs transfected with double-stranded RNA (dsRNA) mimic poly(I:C) (5 µg/mL) were analyzed for Western blot (A) at 6 h post-transfection or RT-qPCR (B) at 12 h after transfection. (C,D) Indicated MEFs were treated with 10 μM GTPP and 300 nM TG up to 8 h. (C) Cell lysates were obtained 2 h after treatment for Western blot analysis. (D,E) Total RNA was isolated at 8 h after treatment for RT-qPCR analysis to measure Ifnb1 and UPRMT- and ISR-related genes (n = 4). Hsp90αβ was used as loading control. Also, 18S rRNA primers were used as internal control. Data are presented as mean ± SD. * p < 0.0332, ** p < 0.0021, *** p < 0.0002, and **** p < 0.0001.
Furthermore, the expression of genes associated with the UPRMT and ISR was not induced by GTPP in the PkrKO MEFs, as compared to the PkrWT MEFs (Figure 2D). This absence of gene induction in PkrKO MEFs confirms PKR’s critical involvement in mediating the mitochondrial stress response via the ISR pathway, thereby underscoring the kinase’s essential role in cellular stress signaling mechanisms.
2.3. Double-Stranded RNA Formation and Localization in Response to Mitochondrial Stress
After confirming that PKR is essential for transferring the signal to activate the ISR and subsequently induce the UPRMT, we sought to understand how mitochondrial stress could trigger PKR activation. PKR is traditionally known to be activated by dsRNAs originating from either exogenous or endogenous origin [,,]. This led us to investigate potential sources of dsRNAs generated during mitochondrial stress.
To explore this, we first investigated the presence of dsRNAs in cells undergoing mitochondrial stress induced by GTPP treatment, employing the J2 antibody, which specifically recognizes dsRNAs within cells []. We observed the presence of dsRNAs recognized by the J2 antibody as early as one hour post-treatment (Figure 3A), with an increased puncta diameter of the J2 signal in a dose-dependent manner (Figure 3B).
Figure 3.
Wildtype MEFs were treated with mitochondrial chaperone TRAP1 inhibitor gamitrinib-triphenylphosphonium (GTPP) (A) at 10 µM concentration for indicated times or (B) at indicated concentrations for 1 h. Immunofluorescence staining was performed using monoclonal antibody J2 as marker for double-stranded RNAs (dsRNAs), MitoTracker Red CMXROS for mitochondria, and Hoecsht 33342 for nuclei. Yellow puncta diameter was measured across 3 images. (C) Orthogonal image of confocal microscopy on a single cell treated with GTPP was captured to observe complete localization, as represented by yellow puncta indicated by white arrows. Red and green lines indicate cross-section view on top and side panels. Scale bar, 10 µm. (D) Mitochondrial RNAs were isolated from wildtype MEFs treated with 20 µM GTPP or DMSO control for 1 h. The isolated RNAs were then digested using specific RNases. M, mock digestion. T1, single-stranded RNA-specific endonuclease RNase T1. III, dsRNA-specific endonuclease RNase III. Following digestion, samples were run on 1.5% agarose gel and visualized by EtBr staining (left). Densitometry was performed by measuring signal intensity normalized to the mock digestion (right). Data are presented as mean ± SD. * p < 0.0332, ** p < 0.0021, and **** p < 0.0001.
A further analysis on the localization of these dsRNAs showed that they overlapped with MitoTracker, a fluorescent dye that labels mitochondria in live cells (Figure 3A,B). An orthogonal view of a single cell treated with GTPP revealed overlapping signals between the J2 antibody (dsRNAs) and MitoTracker (mitochondria), indicating colocalization (Figure 3C). This finding suggests that the dsRNAs generated by GTPP treatment are located within the mitochondria.
To further explore the origin of dsRNAs upon GTPP treatment, we isolated mitochondrial RNAs from MEFs treated with either DMSO or GTPP for 2 h. These mitochondrial RNAs were then treated with strand-specific endoribonucleases to assess their structural characteristics. The treatment of mitochondrial RNAs from the GTPP-treated cells with RNase T1, which specifically targets single-stranded RNA, resulted in fewer degradation signals compared to those from the DMSO-treated controls (Figure 3D). Conversely, treatment with RNase III, which specifically targets dsRNAs, yielded more degradation signals in mitochondrial RNAs from the GTPP-treated cells compared to the controls (Figure 3D). This pattern indicates a higher abundance of dsRNA structures in the mitochondrial RNAs from cells exposed to GTPP, suggesting stress-induced alterations in RNA conformation.
2.4. Impaired Pre-Processing of Primary Transcripts and Mitochondrial dsRNA Accumulation
Mitochondrial RNA transcription, driven by mitochondrial RNA polymerase, produces primary transcripts composed of polycistronic units with multiple genes. These undergo detailed processing, including endonucleolytic cleavage, to separate individual tRNA, rRNA, and mRNA molecules. Disruptions in the assembly or function of mitochondrial ribonucleoprotein complexes responsible for RNA processing can lead to the accumulation of unprocessed or partially processed RNAs. Furthermore, due to bidirectional transcription from mitochondrial DNA’s light and heavy strands, such disturbances in mitochondrial proteostasis may lead to the generation of complementary RNA molecules, increasing the likelihood of dsRNA formation.
To validate this hypothesis, we initially assessed the impact of RNA transcription on the formation of dsRNAs during mitochondrial stress. Consistent with previous observations, pronounced dsRNA detection was evident through J2 antibody staining following treatment with GTPP (Figure 4A). The inhibition of transcription with Actinomycin D significantly reduced this staining, highlighting the dependence of dsRNA formation on active transcription under conditions of mitochondrial stress (Figure 4A). Importantly, ER stress induced by TG did not provoke J2 antibody staining, indicating that dsRNA formation is uniquely associated with disturbances in mitochondrial proteostasis (Figure 4A).
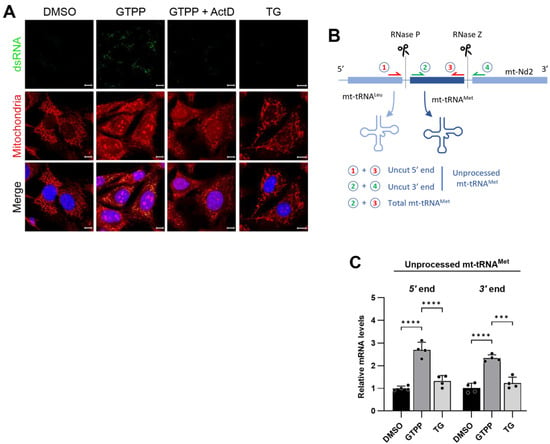
Figure 4.
(A) Wildtype MEFs were treated with 10 µM GTPP alone or in the presence of transcription inhibitor actinomycin D (Act D; 15 µM) or 300 nM TG. Immunofluorescence staining was performed as described in Figure 3. (B) Schematic of mitochondrial polycistronic RNA processing represented by tRNAMet. RNase P and RNase Z cleave tRNAMet at its 5′ and 3′ ends, respectively. (C) Total RNAs were isolated from wildtype MEFs at 8 h after treated with 10 µM GTPP, TG 300 nM, or DMSO control for RT-qPCR analysis to measure mitochondrial RNA processing. Unprocessed mitochondrial tRNAMet fragments were measured by using primer pairs 1 and 3, or 2 and 4, as shown in (B) [for details, please refer to Materials and Methods Section 4.4]. The resulting Ct values were normalized to the Ct values of total mitochondrial tRNAMet as obtained from primer pair 2 and 3 (n = 4). Data values presented as mean ± SD. *** p < 0.0002, and **** p < 0.0001.
Further investigations concentrated on the status of pre-RNA processing, under the hypothesis that mitochondrial stress disrupts this essential function, leading to the accumulation of unprocessed RNA segments that may subsequently form dsRNA. To examine this, we conducted a quantitative analysis of the levels of unprocessed tRNAMet using qRT-PCR. We specifically designed primers targeting the 5′ region of tRNAMet adjacent to the tRNALeu region and the 3′ region adjacent to the Nd1 region of the primary transcript (Figure 4B). These segments are typically processed by RNase P and RNase Z, respectively []. Following treatment with GTPP, there was a significant increase in the levels of these amplicons, indicating an increase in the amount of unprocessed tRNAMet (Figure 4C). Conversely, treatment with TG did not result in a similar increase in unprocessed tRNAMet levels (Figure 4C). This pattern of RNA processing, or the lack thereof, directly correlates with dsRNA formation, as detected by J2 antibody staining. This finding confirms the specificity of dsRNA formation to disruptions in mitochondrial proteostasis as opposed to ER proteostasis.
2.5. Cytosolic Translocation of Mitochondrial dsRNA and Its Role in PKR Activation
Next, we investigated whether dsRNAs accumulated in the mitochondria upon GTPP-induced stress could activate PKR and subsequently induce the UPRMT in a manner similar to direct GTPP treatment. To this end, we isolated the total mitochondrial RNAs from the GTPP-treated and control DMSO-treated MEFs and transfected them into PkrWT and PkrKO MEFs to evaluate the induction of the UPRMT.
The mitochondrial RNAs isolated from the GTPP-treated MEFs significantly phosphorylated eIF2α, to a similar extent as observed with poly(I:C) treatment, in the PkrWT MEFs but not in the PkrKO MEFs, indicating a PKR-dependent mechanism (Figure 5A). Conversely, the mitochondrial RNAs from the DMSO-treated MEFs did not induce phosphorylation of eIF2α in either the PkrWT or PkrKO MEFs. Furthermore, the mitochondrial RNAs from the GTPP-treated MEFs triggered the expression of both UPRMT-related and ISR-related genes in the PkrWT MEFs but not in the PkrKO MEFs. Intriguingly, both the poly(I:C) and mitochondrial RNAs from the GTPP-treated MEFs induced the expression of Ifnb1 to a much greater extent than the UPR-related genes exclusively in the PkrWT MEFs and not in the PkrKO MEFs (Figure 5B,C). These findings strongly suggest that the dsRNAs accumulated in mitochondria under GTPP-induced mitochondrial stress can activate the UPRMT signaling pathway through PKR-dependent mechanisms.
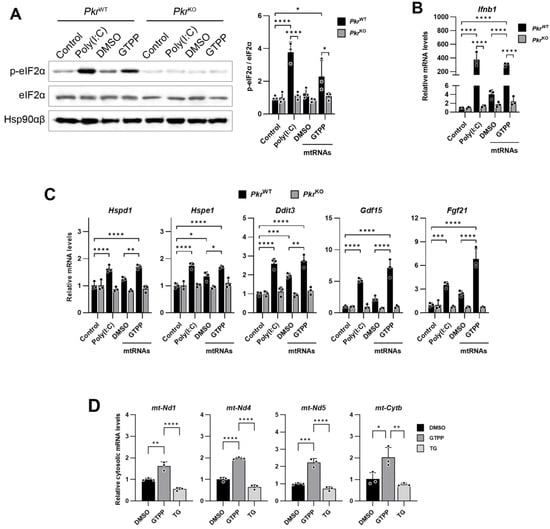
Figure 5.
Mitochondrial RNAs were isolated from wildtype MEFs treated with 20 µM GTPP or DMSO control for 1 h and were then used to transfect into indicated MEFs. Poly(I:C) was used as positive control. (A) Cell lysates were obtained 6 h after transfection for Western blot analysis. Hsp90αβ was used as loading control. (B,C) Total RNAs were isolated 12 h after transfection for RT-qPCR analysis to measure (B) dsRNA-responsive Ifnb1 and (C) UPRMT- and ISR-related genes. (n = 4). (D) Wildtype MEFs were treated with 20 µM GTPP, 300 nM TG, or DMSO control. Total RNAs were isolated from cytosolic fraction at 1 h after stress induction for RT-qPCR analysis to measure mitochondrial transcript levels (n = 3). Data values presented as mean ± SD. * p < 0.0332, ** p < 0.0021, *** p < 0.0002, and **** p < 0.0001.
Given the cytosolic localization of PKR and the mitochondrial accumulation of dsRNAs, we next explored the potential mechanism by which mitochondrial dsRNAs might activate PKR. We hypothesized that the accumulated mitochondrial dsRNAs could be released into the cytosol, thereby activating cytosolic PKR. To investigate this possibility, we examined the presence of mitochondria-originated dsRNAs in cytoplasmic RNAs isolated from cells treated with either DMSO or GTPP. Our findings revealed a significant increase in the cytosolic presence of mitochondrial transcripts, including Nd1, Nd4, Nd5, and Cytb, following GTPP treatment (Figure 5D). In contrast, TG treatment, which induces ER stress, did not result in an increase in these mitochondrial transcripts in the cytosol, suggesting that mitochondrial RNAs are specifically exported to the cytoplasm under conditions of mitochondrial stress induced by GTPP. This indicates a possible pathway for PKR activation through the release of mitochondrial dsRNAs into the cytosol during mitochondrial stress.
2.6. Bax/Bak Channels Export Mitochondrial dsRNAs upon Mitochondrial Stress
Next, we explored the mechanisms through which mitochondrial dsRNAs are translocated into the cytosol to activate PKR and the subsequent signaling pathways. Given that mitochondria are bounded by a double membrane, specific pathways must exist for the export of contents, including RNAs, into the cytosol. Previously, it was demonstrated that the Bax/Bak channels mediate the translocation of cytochrome c, which triggers apoptosis []. Subsequently, it was discovered that mitochondrial DNAs also utilize the Bax/Bak channels to reach the cytosol and induce an innate immune response by activating pathways, such as cGAS-STING [,]. More recently, several reports have suggested that mitochondrial RNAs can similarly exploit the Bax/Bak channels for their translocation into the cytosol [,,].
Based on our understanding of mitochondrial dsRNA dynamics, we hypothesized that accumulated mitochondrial dsRNAs might be released into the cytosol via the Bax/Bak channel. To test this hypothesis, we employed Bax/Bak double-knock-out (Bax/BakdKO) MEFs, treating them with GTPP to induce mitochondrial stress and subsequently monitoring the presence of mitochondrial RNAs in the cytosol. In the wildtype control (Bax/BakWT) MEFs, a significant increase in cytosolic mitochondrial RNAs was observed (Figure 6A). However, this increase was absent in the cytosol of the Bax/BakdKO MEFs (Figure 6A), indicating a potential role of the Bax/Bak channels in mitochondrial RNA export. Furthermore, the phosphorylation of eIF2α, typically induced by GTPP treatment in the Bax/BakWT MEFs, was not observed in the Bax/BakdKO MEFs (Figure 6B). Similarly, the expression levels of the genes related to the UPRMT and ISR were significantly reduced in the Bax/BakdKO MEFs compared to the Bax/BakWT MEFs (Figure 6C).
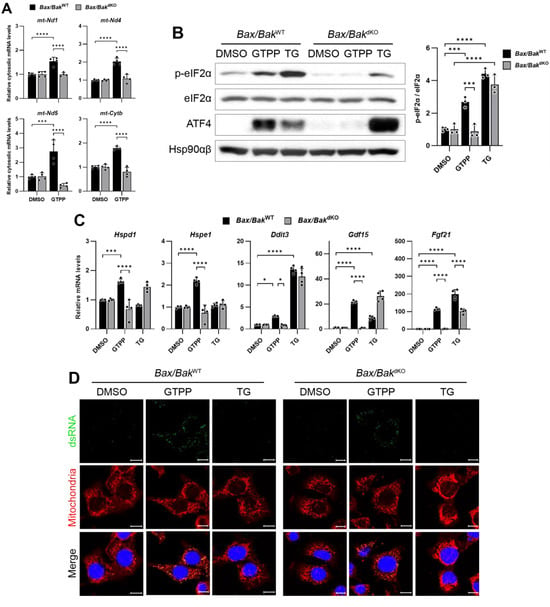
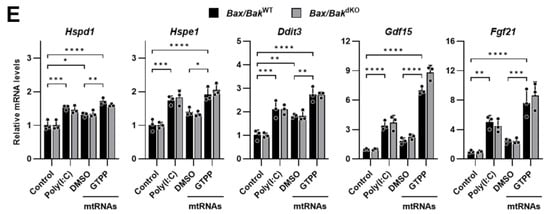
Figure 6.
(A) Indicated MEFs were treated with mitochondrial chaperone TRAP1 inhibitor gamitrinib-triphenylphosphonium (GTPP; 10 µM) or DMSO control. Cytosolic RNAs were isolated at 8 h after stress induction for RT-qPCR analysis to measure mitochondrial transcript levels. (B,C) Indicated MEFs were treated with 10 µM GTPP, thapsigargin (TG; 300 nM), or DMSO control. (B) Cell lysates were obtained 2 h after stress induction for Western blot analysis. Densitometry was performed by measuring p-eIF2α signal normalized to total eIF2α. Hsp90αβ was used as loading control. (C) Total RNAs were isolated at 8 h after stress induction for RT-qPCR to measure UPRMT- and ISR-related genes (n = 4). (D) Immunofluorescence staining was performed 1 h after stress induction to observe dsRNA accumulation. (E) MEFs were transfected with mitochondrial RNAs isolated from GTPP- or DMSO-treated cells, and total RNAs were isolated 12 h after transfection for RT-qPCR analysis to measure UPRMT- and ISR-related genes (n = 3). Data values presented as mean ± SD. * p < 0.0332, ** p < 0.0021, *** p < 0.0002, and **** p < 0.0001.
Importantly, there was no significant difference in the J2 staining, which indicates dsRNA presence, between the GTPP-treated Bax/BakWT and Bax/BakdKO MEFs (Figure 6D). This suggests that the diminished induction of the UPRMT in Bax/BakdKO MEFs was not due to a reduced accumulation of mitochondrial dsRNAs. Moreover, when mitochondrial RNAs isolated from the GTPP-treated MEFs were used to induce the UPRMT and ISR-related genes, no differences were observed between the Bax/BakWT and Bax/BakdKO MEFs (Figure 6E).
These results collectively suggest that the translocation of dsRNAs from stressed mitochondria through the Bax/Bak channels plays a crucial role in triggering PKR activation and the subsequent induction of the UPRMT. This underscores the importance of mitochondrial dsRNA export mechanisms in cellular stress signaling pathways.
3. Discussion
While the phosphorylation of eIF2α and the subsequent induction of ATF4 are established as critical mediators of mitochondrial stress signaling leading to the activation of the UPRMT in mammalian systems, the specific pathways translating mitochondrial stress signals into cytosolic eIF2α phosphorylation remain unclear. In this study, we explored the role of PKR within this signaling cascade. Our findings indicate that in the absence of PKR, the UPRMT is not initiated under conditions of mitochondrial stress, underscoring PKR’s essential role in detecting mitochondrial stress signals and mediating the phosphorylation of eIF2α. Furthermore, we observed that dsRNAs, which accumulate within mitochondria during stress, are transported into the cytosol where they activate PKR. Importantly, our data reveals that in the absence of the Bax/Bak channel, dsRNAs do not reach the cytosol nor trigger the UPRMT, identifying this channel as the crucial conduit for dsRNA translocation from the mitochondria to the cytosol. These findings collectively demonstrate that PKR is an indispensable kinase for phosphorylating eIF2α and initiating the UPRMT. This activation process is facilitated by the accumulation of dsRNAs within mitochondria and its subsequent release into the cytosol via the Bax/Bak channels under mitochondrial stress conditions (Figure 7).
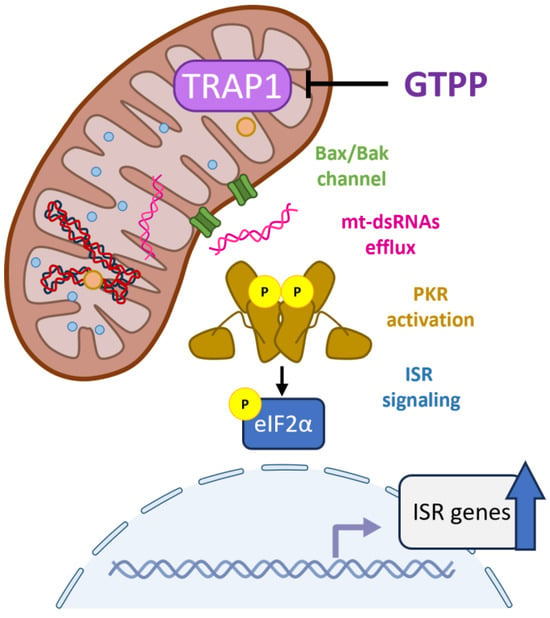
Figure 7.
Role of mitochondrial dsRNAs and PKR in mitochondrial stress signal relay for UPRMT. When mitochondrial proteostasis is disturbed, mitochondrial RNA processing is impaired, leading to the accumulation of double-stranded mitochondrial RNAs. These dsRNAs are then exported to the cytosol via the Bax/Bak channel. In the cytosol, PKR binds to these mitochondrial dsRNAs and activates the integrated stress response (ISR) signaling cascade by phosphorylating eIF2α leading to the upregulation of ISR target genes.
Following the identification of ATFS-1 as a mediator of the UPRMT in C. elegans, considerable research has been directed toward identifying a similar system to induce the UPRMT in mammalian cells. It was found that the bZIP transcription factors ATF5, ATF4, and CHOP are associated with transcriptional activation of UPRMT-related genes [,,,,]. Interestingly, these transcription factors are the downstream target of eIF2α phosphorylation, and studies have demonstrated that eIF2α phosphorylation and ISR induction are crucial for the initiation of the UPRMT in response to mitochondrial stress. This raised questions about the mechanisms by which mitochondrial stress signals are relayed to the cytosol to trigger eIF2α phosphorylation. While it has been suggested that all four eIF2α kinases are implicated in UPRMT activation upon mitochondrial stress, only HRI has been directly linked to activation by mitochondrial stress through its interaction with DELE1. Under mitochondrial stress, protease OMA1 is activated, and it cleaves DELE1 leading to its accumulation in the cytosol where it interacts with HRI, activating its eIF2α kinase activity [,]. Additionally, the crosstalk between mitochondria and the endoplasmic reticulum (ER) may activate PERK [], while reactive oxygen species (ROS) generated during mitochondrial stress could potentially activate other kinases, including HRI, GCN2, and PKR [,,]. Specifically regarding PKR, although several studies have implicated it in the mitochondrial stress-initiated UPRMT, the precise mechanisms remained unclear. Initially, PKR was discovered to mediate the UPRMT induction in a DSS-induced colitis model [], but exactly how PKR was activated was not explored. On the other hand, PKR was revealed to bind to endogenous dsRNAs, particularly of mitochondrial origin, and control eIF2α phosphorylation during mitosis and stress [,]. Subsequent studies demonstrated that mitochondrial dsRNAs in various disease models activate the ISR activation through PKR [,,]. These findings suggest that mitochondrial dsRNAs might be the signal from stressed mitochondria to induce the UPRMT, although this correlation has not been definitively established. In this study, we provide evidence that PKR is crucial for activating the UPRMT in response to mitochondrial stress induced by GTPP treatment. Furthermore, we observed that dsRNAs generated during GTPP treatment accumulate within the mitochondria, adding a new layer to our understanding of how mitochondrial distress signals are communicated to the cellular stress response machinery.
The presence of dsRNAs has traditionally been associated with viral infections, whether from RNA or DNA viruses. However, accumulating evidence suggests that dsRNAs can also be produced endogenously in various pathophysiological states []. In this study, we observed that GTPP-induced mitochondrial stress leads to the accumulation of dsRNAs within the mitochondria. Due to the circular form and bidirectional transcription of mitochondrial DNA, mitochondrial transcripts can form dsRNAs, which are typically recycled by a degradasome complex composed of the exoribonuclease polynucleotide phosphorylase (PNPase) and the ATP-dependent RNA helicase SUPV3L1 [,]. When the function of this degradasome is compromised, there is an increased formation of dsRNAs within the mitochondria [,]. Additionally, improper RNA processing of mitochondrial transcripts can also lead to the generation of mitochondrial dsRNAs []. GTPP, by inhibiting TRAP1, a mitochondrial chaperone, likely disturbs proteostasis, impairing the processing of mitochondrial transcripts and the degradation of unwanted dsRNAs by the degradasome []. In our study, GTPP treatment resulted in the accumulation of unprocessed mitochondrial tRNAMet, suggesting that disturbances in mitochondrial proteostasis may impair RNA processing mechanisms, thereby contributing to dsRNA generation under stress conditions. Furthermore, we found that halting transcription with actinomycin D eliminated dsRNA formation in mitochondria, indicating that newly synthesized primary transcripts are the main sources of dsRNAs.
In this study, we demonstrated that dsRNAs accumulate upon GTPP-induced mitochondrial stress and that these mitochondrial dsRNAs can activate PKR and subsequently induce the UPRMT signaling pathway. Given that mitochondria are enclosed by a double membrane, the translocation of accumulated dsRNAs from the mitochondria to the cytosol requires specific gateways. Among the various channels in the mitochondrial outer membrane, the Bax/Bak channel protein is well known for facilitating the release of cytochrome c release to initiate apoptosis. In addition, recent publications suggest that mitochondria utilize this channel to translocate the macromolecules, including mitochondrial DNA []. Interestingly, some studies have demonstrated that mitochondrial dsRNAs are also released into the cytosol via the Bax/Bak channels []. Consistent with these findings, we observed that GTPP-induced mitochondrial stress could not induce PKR activation or subsequent UPRMT signaling in the absence of Bax/Bak, even though dsRNAs clearly accumulated in Bax/BakdKO MEFs. In Bax/BakdKO MEFs, dsRNAs were not released into the cytosol. However, the accumulated dsRNAs in Bax/BakdKO MEFs were able to induce UPRMT signaling when introduced into the cells by transfection, suggesting that the lack of UPRMT induction in Bax/BakdKO MEFs is due to the blockade of dsRNA translocation.
The findings in this study will enhance our understanding of how mitochondrial integrity is maintained and how stress signals are communicated to the nucleus via retrograde signaling. They provide a foundation for further research into therapeutic strategies targeting mitochondrial stress responses, which could be beneficial in treating diseases characterized by mitochondrial dysfunction. The identification of PKR and the Bax/Bak channels as key components in this pathway opens new avenues for developing interventions aimed at mitigating the effects of mitochondrial stress and enhancing cellular resilience. Furthermore, the role of dsRNA formation under mitochondrial stress conditions and its subsequent involvement in PKR activation and UPRMT induction elucidates the intricate mechanisms of cellular stress responses and identifies potential molecular targets for therapeutic intervention.
4. Materials and Methods
4.1. Animal, Cell Culture, and Chemical Treatment
All the cells (listed in Table 1) were maintained in DMEM (Corning, NY, USA, 10-013-CV) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (Corning, NY, USA, 30-002-CI) and 1% (v/v) penicillin/streptomycin (Corning, NY, USA, 35-015-CV) as described in a previous study []. The Bax/BakdKO MEFs cell line and its respective wildtype counterpart (Bax/BakWT) were obtained from the American Type Culture Collections (ATCC, Manassas, VA, USA; CRL-2913, CRL-2907). The PkrKO mice were purchased from Australian Phenomics Facility (APF, APB631) at the Australian National University and were backcrossed to the C57BL/6 strain for at least six generations. The PkrWT and PkrKO mice were maintained in a 12 h dark/light cycle. All the animal care and experiment procedures were conducted according to the protocols and guidelines approved by Soonchunhyang University Animal Care and Use Committee (SCH23-0020). The primary PkrWT and PkrKO MEFs were isolated from the PkrWT and PkrKO mice, respectively, and immortalized by SV40 T antigen as previously described []. The MEFs were treated with DMSO, gamitrinib-triphenylphosphonium (GTPP; LegoChem Biosciences, Daejeon, Republic of Korea), thapsigargin (TG; Sigma, Livonia, MI, USA, T9033), ISRIB (Sigma, MI, USA, SML0843), and actinomycin D (Sigma, MI, USA, A9415) at the indicated concentrations and time points.

Table 1.
MEFs cell lines used in this study.
4.2. Subcellular RNAs Isolation and Transfection
Subcellular fractionation was performed using the Mitochondrial Isolation Kit for Cultured Cells (ThermoFisher, Carlsbad, CA, USA, 89874) according to the manufacturer’s instruction. The RNA was extracted from the cytoplasmic fraction and mitochondrial pellet using RiboEx™ LS (GeneAll, Seoul, Republic of Korea, 301-001) and RiboEx™ (GeneAll, Seoul, Republic of Korea, 302-001), respectively. The extracted RNA was then treated with RNase-free Turbo™ DNase (Invitrogen, Carlsbad, CA, USA, AM2238). For transfection of the mitochondrial RNAs, the MEFs were transfected in 12-well plates at 80% confluency with either low-molecular-weight poly(I:C) (Invivogen, CA, USA, TLRL-PICW) or mitochondrial RNAs at a concentration of 5 µg/mL using Lipofectamine™ RNAiMAX (Invitrogen, CA, USA, 13778075) with 1 µg:1 µL ratio. For the enzymatic treatment, 1 µg of nucleic acids was digested with 1 U of RNase T1 (Ambion, Naugatuck, CT, USA, AM2283) or RNase III (Ambion, CT, USA, AM2290) according to the manufacturer’s instructions. The isolated RNAs were run on 1.5% agarose gel and visualized by EtBr staining.
4.3. Reverse Transcription and qPCR Analysis
The total RNA was isolated from the indicated MEFs using RiboEx™ (GeneAll, Seoul, Republic of Korea, 302-001) and reverse transcribed using ReverTra Ace™ qPCR RT Master Mix (Toyobo, Tokyo, Japan, TOFSQ-201). A real-time qRT-PCR was performed using TOPreal™ SYBR Green qPCR PreMIX (Enzynomics, Daejeon, Republic of Korea, RT501M) on QuantStudio™ 1 (Applied Biosystems, Waltham, MA, USA). The primers used in this study are listed in Table 2.
Table 2.
Primers used in this study.
4.4. Mitochondrial RNA Processing Analysis
The MEFs were seeded at 80% confluency on a 12-well plate and treated with GTPP (10 µM) or TG (300 nM) for 8 h. Total RNA isolation, cDNA synthesis, and qRT-PCR were performed as described above. Unprocessed mitochondrial tRNAMet fragments were detected using primer pairs annealing at specific regions: (1) the 5′ end of tRNAMet 5′ (forward) and an internal region of tRNAMet (reverse), and (2) an internal region of tRNAMet (forward) and 3′ end downstream of tRNAMet (reverse). These primers specifically amplify the cleavage site of the RNase P at the 5′ end and RNase Z at 3′ end of tRNAMet, respectively. The Ct values from each target were normalized to the Ct values of total tRNAMet obtained using internal primers for tRNAMet (forward and reverse). The primers used for this analysis are listed in Table 2.
4.5. Immunoblotting
Protein lysates were prepared in RIPA lysis buffer (25 mM Tris-HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, and 0.1% SDS) supplemented with the Xpert protease inhibitor cocktail (Rockland, PA, USA, P3100) and Xpert phosphatase inhibitor cocktail (Rockland, USA, P3200). The protein concentrations were measured using the Pierce™ BCA Protein Assay Kit (ThermoFisher, CA, USA, 23225). A total of 10–20 µg of protein was mixed with Laemmli sample buffer (Bio-Rad, Hercules, CA, USA, BR1610747) containing 10% (v/v) 2-mercaptoethanol (Sigma Aldrich, Ann Arbor, MI, USA, M6250) and boiled at 100 °C for 5 min before loading onto an SDS-PAGE gel. The membranes were immunoblotted overnight at 4 °C with the following primary antibodies: ATF4 (Cell Signaling Technology, Danvers, MA, USA, #11815), p-eIF2α (Abcam, Fremont, CA, USA, ab32157), eIF2α (Cell Signaling Technology, MA, USA#9722), and Hsp90αβ (Santa Cruz, Dallas, TX, USA, sc-13119) using dilution according to the manufacturer’s recommendations. The antibody signals were visualized on medical X-ray films using the Agfa CP1000 automatic film processor. The band intensity was quantified by ImageJ software version 1.52.
4.6. Immunofluorescence Staining
The MEFs were seeded at 80% confluency on a 24-well cell imaging plate (Eppendorf, Framingham, MA, USA, 0030741021) one day before treatment. Prior to fixation, the MEFs were incubated with 5 µM MitoTracker™ Red CMXRos (ThermoFisher, CA, USA, M5710) for 15 min in the absence of serum. The MEFs were then washed twice with PBS and fixed with 4% (w/v) paraformaldehyde for 20 min at room temperature. Following fixation, the MEFs were washed three times with PBS and incubated with 0.3% Triton X-100 and 1% BSA in PBS for 45 min. The MEFs were then probed with J2 antibody (1:400, Scicons, 10010200) overnight at 4 °C. After washing three times with PBS, the MEFs were incubated with Alexa Fluor 488-conjugated secondary antibody (1:400; Jackson ImmunoResearch, West Grove, PA, USA, JAC-115-545-003) for 2 h at room temperature. Hoechst 33342 (ThermoFisher, CA, USA, H3570) was added during the final PBS wash to stain the nuclei. Confocal microscopy imaging was performed using Zeiss LSM 710. The yellow dsRNA puncta number and size were quantified using CellProfiler, software version 4.2.6.
4.7. Statistical Analysis
All the data values are presented as the means ± SD. The statistical significance of difference between the groups was assessed using the unpaired Student’s t test for single comparison and one-way or two-way ANOVA followed by Bonferroni’s post hoc test for multiple comparison. All the statistical analyses were performed using GraphPad Prism v9.5.0.
Author Contributions
Conceptualization, J.H. and F.K.; Methodology, F.K., S.P., K.A.N., R.E. and D.L.; software, F.K.; formal analysis, F.K. and J.H.; writing—original draft preparation, F.K. and J.H.; supervision, J.H.; funding acquisition, J.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Research Foundation of Korea (NRF-2022R1A2C1010676, NRF-2019R1A2C1085284, and NRF-2019R1A5A8083404).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data available on request from the authors.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef] [PubMed]
- King, N. Amino Acids and the Mitochondria. In Advances in Biochemistry in Health and Disease, Mitochondria; Schaffer, S., Suleiman, M., Eds.; Springer: New York, NY, USA, 2007; Volume 2. [Google Scholar]
- Nowinski, S.M.; Solmonson, A.; Rusin, S.F.; Maschek, J.A.; Bensard, C.L.; Fogarty, S.; Jeong, M.Y.; Lettlova, S.; Berg, J.A.; Morgan, J.T.; et al. Mitochondrial fatty acid synthesis coordinates oxidative metabolism in mammalian mitochondria. Elife 2020, 9, e58041. [Google Scholar] [CrossRef] [PubMed]
- Wang, L. Mitochondrial purine and pyrimidine metabolism and beyond. Nucleosides Nucleotides Nucleic Acids 2016, 35, 578–594. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Kelly, B.; O’Neill, L.A.J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 2017, 18, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Haastrup, M.O.; Vikramdeo, K.S.; Singh, S.; Singh, A.P.; Dasgupta, S. The Journey of Mitochondrial Protein Import and the Roadmap to Follow. Int. J. Mol. Sci. 2023, 24, 2479. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, O.; Pfanner, N.; Meisinger, C. Mitochondrial protein import: From proteomics to functional mechanisms. Nat. Rev. Mol. Cell Biol. 2010, 11, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Ingman, M.; Gyllensten, U. mtDB: Human Mitochondrial Genome Database, a resource for population genetics and medical sciences. Nucleic Acids Res. 2006, 34, D749–D751. [Google Scholar] [CrossRef] [PubMed]
- Haynes, C.M.; Ron, D. The mitochondrial UPR—Protecting organelle protein homeostasis. J. Cell Sci. 2010, 123, 3849–3855. [Google Scholar] [CrossRef] [PubMed]
- Haynes, C.M.; Yang, Y.; Blais, S.P.; Neubert, T.A.; Ron, D. The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol. Cell 2010, 37, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Nargund, A.M.; Pellegrino, M.W.; Fiorese, C.J.; Baker, B.M.; Haynes, C.M. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 2012, 337, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.F.; Schulz, A.M.; Pellegrino, M.W.; Lu, Y.; Shaham, S.; Haynes, C.M. Maintenance and propagation of a deleterious mitochondrial genome by the mitochondrial unfolded protein response. Nature 2016, 533, 416–419. [Google Scholar] [CrossRef] [PubMed]
- Fiorese, C.J.; Schulz, A.M.; Lin, Y.F.; Rosin, N.; Pellegrino, M.W.; Haynes, C.M. The Transcription Factor ATF5 Mediates a Mammalian Mitochondrial UPR. Curr. Biol. 2016, 26, 2037–2043. [Google Scholar] [CrossRef] [PubMed]
- Quiros, P.M.; Prado, M.A.; Zamboni, N.; D’Amico, D.; Williams, R.W.; Finley, D.; Gygi, S.P.; Auwerx, J. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol. 2017, 216, 2027–2045. [Google Scholar] [CrossRef] [PubMed]
- Kaspar, S.; Oertlin, C.; Szczepanowska, K.; Kukat, A.; Senft, K.; Lucas, C.; Brodesser, S.; Hatzoglou, M.; Larsson, O.; Topisirovic, I.; et al. Adaptation to mitochondrial stress requires CHOP-directed tuning of ISR. Sci. Adv. 2021, 7, eabf0971. [Google Scholar] [CrossRef] [PubMed]
- Tyynismaa, H.; Carroll, C.J.; Raimundo, N.; Ahola-Erkkila, S.; Wenz, T.; Ruhanen, H.; Guse, K.; Hemminki, A.; Peltola-Mjosund, K.E.; Tulkki, V.; et al. Mitochondrial myopathy induces a starvation-like response. Hum. Mol. Genet. 2010, 19, 3948–3958. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.A.; Nikkanen, J.; Yatsuga, S.; Jackson, C.; Wang, L.; Pradhan, S.; Kivela, R.; Pessia, A.; Velagapudi, V.; Suomalainen, A. mTORC1 Regulates Mitochondrial Integrated Stress Response and Mitochondrial Myopathy Progression. Cell Metab. 2017, 26, 419–428.e415. [Google Scholar] [CrossRef] [PubMed]
- Young, S.K.; Wek, R.C. Upstream Open Reading Frames Differentially Regulate Gene-specific Translation in the Integrated Stress Response. J. Biol. Chem. 2016, 291, 16927–16935. [Google Scholar] [CrossRef] [PubMed]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [PubMed]
- Hershey, J.W.; Sonenberg, N.; Mathews, M.B. Principles of translational control: An overview. Cold Spring Harb. Perspect. Biol. 2012, 4, a011528. [Google Scholar] [CrossRef] [PubMed]
- Pavitt, G.D.; Ron, D. New insights into translational regulation in the endoplasmic reticulum unfolded protein response. Cold Spring Harb. Perspect. Biol. 2012, 4, a012278. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.G.; Hur, S. Cellular origins of dsRNA, their recognition and consequences. Nat. Rev. Mol. Cell Biol. 2021, 23, 286–301. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.A.; Gil, J.; Ventoso, I.; Guerra, S.; Domingo, E.; Rivas, C.; Esteban, M. Impact of protein kinase PKR in cell biology: From antiviral to antiproliferative action. Microbiol. Mol. Biol. Rev. 2006, 70, 1032–1060. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Lee, J.H.; Park, J.E.; Cho, J.; Yi, H.; Kim, V.N. PKR is activated by cellular dsRNAs during mitosis and acts as a mitotic regulator. Genes Dev. 2014, 28, 1310–1322. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Park, J.; Kim, S.; Kim, M.; Kang, M.G.; Kwak, C.; Kang, M.; Kim, B.; Rhee, H.W.; Kim, V.N. PKR Senses Nuclear and Mitochondrial Signals by Interacting with Endogenous Double-Stranded RNAs. Mol. Cell 2018, 71, 1051–1063.e6. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhang, M.; Wang, W.; Qu, S.; Liu, M.; Rong, W.; Yang, W.; Liang, H.; Zeng, C.; Zhu, X.; et al. Polynucleotide phosphorylase protects against renal tubular injury via blocking mt-dsRNA-PKR-eIF2alpha axis. Nat. Commun. 2023, 14, 1223. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, K.; Choi, Y.S.; Ku, J.; Kim, H.; Kharbash, R.; Yoon, J.; Lee, Y.S.; Kim, J.H.; Lee, Y.J.; et al. Mitochondrial double-stranded RNAs govern the stress response in chondrocytes to promote osteoarthritis development. Cell Rep. 2022, 40, 111178. [Google Scholar] [CrossRef]
- Yoon, J.; Lee, M.; Ali, A.A.; Oh, Y.R.; Choi, Y.S.; Kim, S.; Lee, N.; Jang, S.G.; Park, S.; Chung, J.H.; et al. Mitochondrial double-stranded RNAs as a pivotal mediator in the pathogenesis of Sjögren’s syndrome. Mol. Ther. Nucleic Acids 2022, 30, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Rath, E.; Berger, E.; Messlik, A.; Nunes, T.; Liu, B.; Kim, S.C.; Hoogenraad, N.; Sans, M.; Sartor, R.B.; Haller, D. Induction of dsRNA-activated protein kinase links mitochondrial unfolded protein response to the pathogenesis of intestinal inflammation. Gut 2012, 61, 1269–1278. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.M.; Nargund, A.M.; Sun, T.; Haynes, C.M. Protective coupling of mitochondrial function and protein synthesis via the eIF2alpha kinase GCN-2. PLoS Genet. 2012, 8, e1002760. [Google Scholar] [CrossRef] [PubMed]
- Rainbolt, T.K.; Atanassova, N.; Genereux, J.C.; Wiseman, R.L. Stress-regulated translational attenuation adapts mitochondrial protein import through Tim17A degradation. Cell Metab. 2013, 18, 908–919. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Aviles, G.; Liu, Y.; Tian, R.; Unger, B.A.; Lin, Y.T.; Wiita, A.P.; Xu, K.; Correia, M.A.; Kampmann, M. Mitochondrial stress is relayed to the cytosol by an OMA1-DELE1-HRI pathway. Nature 2020, 579, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Fessler, E.; Eckl, E.M.; Schmitt, S.; Mancilla, I.A.; Meyer-Bender, M.F.; Hanf, M.; Philippou-Massier, J.; Krebs, S.; Zischka, H.; Jae, L.T. A pathway coordinated by DELE1 relays mitochondrial stress to the cytosol. Nature 2020, 579, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.H.; Plescia, J.; Song, H.Y.; Meli, M.; Colombo, G.; Beebe, K.; Scroggins, B.; Neckers, L.; Altieri, D.C. Combinatorial drug design targeting multiple cancer signaling networks controlled by mitochondrial Hsp90. J. Clin. Investig. 2009, 119, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.H.; Tavecchio, M.; Goel, H.L.; Hsieh, C.C.; Garlick, D.S.; Raskett, C.M.; Lian, J.B.; Stein, G.S.; Languino, L.R.; Altieri, D.C. Targeted inhibition of mitochondrial Hsp90 suppresses localised and metastatic prostate cancer growth in a genetic mouse model of disease. Br. J. Cancer 2011, 104, 629–634. [Google Scholar] [CrossRef]
- Corbet, G.A.; Burke, J.M.; Bublitz, G.R.; Tay, J.W.; Parker, R. dsRNA-induced condensation of antiviral proteins modulates PKR activity. Proc. Natl. Acad. Sci. USA 2022, 119, e2204235119. [Google Scholar] [CrossRef] [PubMed]
- Youssef, O.A.; Safran, S.A.; Nakamura, T.; Nix, D.A.; Hotamisligil, G.S.; Bass, B.L. Potential role for snoRNAs in PKR activation during metabolic stress. Proc. Natl. Acad. Sci. USA 2015, 112, 5023–5028. [Google Scholar] [CrossRef] [PubMed]
- Schonborn, J.; Oberstraβ, J.; Breyel, E.; Tittgen, J.; Schumacher, J.; Lukacs, N. Monoclonal antibodies to double-stranded RNA as probes of RNA structure in crude nucleic acid extracts. Nucleic Acids Res. 1991, 19, 2993–3000. [Google Scholar] [CrossRef] [PubMed]
- Holzmann, J.; Frank, P.; Loffler, E.; Bennett, K.L.; Gerner, C.; Rossmanith, W. RNase P without RNA: Identification and functional reconstitution of the human mitochondrial tRNA processing enzyme. Cell 2008, 135, 462–474. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zheng, J.; Nussinov, R.; Ma, B. Release of Cytochrome C from Bax Pores at the Mitochondrial Membrane. Sci. Rep. 2017, 7, 2635. [Google Scholar] [CrossRef] [PubMed]
- White, M.J.; McArthur, K.; Metcalf, D.; Lane, R.M.; Cambier, J.C.; Herold, M.J.; van Delft, M.F.; Bedoui, S.; Lessene, G.; Ritchie, M.E.; et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell 2014, 159, 1549–1562. [Google Scholar] [CrossRef] [PubMed]
- McArthur, K.; Whitehead, L.W.; Heddleston, J.M.; Li, L.; Padman, B.S.; Oorschot, V.; Geoghegan, N.D.; Chappaz, S.; Davidson, S.; San Chin, H.; et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 2018, 359, eaao6047. [Google Scholar] [CrossRef] [PubMed]
- Dhir, A.; Dhir, S.; Borowski, L.S.; Jimenez, L.; Teitell, M.; Rotig, A.; Crow, Y.J.; Rice, G.I.; Duffy, D.; Tamby, C.; et al. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature 2018, 560, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Tigano, M.; Vargas, D.C.; Tremblay-Belzile, S.; Fu, Y.; Sfeir, A. Nuclear sensing of breaks in mitochondrial DNA enhances immune surveillance. Nature 2021, 591, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Krieger, M.R.; Abrahamian, M.; He, K.L.; Atamdede, S.; Hakimjavadi, H.; Momcilovic, M.; Ostrow, D.; Maggo, S.D.; Tsang, Y.P.; Gai, X.; et al. Trafficking of mitochondrial double-stranded RNA from mitochondria to the cytosol. Life Sci. Alliance 2024, 7, e202302396. [Google Scholar] [CrossRef]
- Celardo, I.; Costa, A.C.; Lehmann, S.; Jones, C.; Wood, N.; Mencacci, N.E.; Mallucci, G.R.; Loh, S.H.; Martins, L.M. Mitofusin-mediated ER stress triggers neurodegeneration in pink1/parkin models of Parkinson’s disease. Cell Death Dis. 2016, 7, e2271. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Reyes, I.; Sanchez-Arago, M.; Cuezva, J.M. AMPK and GCN2-ATF4 signal the repression of mitochondria in colon cancer cells. Biochem. J. 2012, 444, 249–259. [Google Scholar] [CrossRef] [PubMed]
- McEwen, E.; Kedersha, N.; Song, B.; Scheuner, D.; Gilks, N.; Han, A.; Chen, J.J.; Anderson, P.; Kaufman, R.J. Heme-regulated inhibitor kinase-mediated phosphorylation of eukaryotic translation initiation factor 2 inhibits translation, induces stress granule formation, and mediates survival upon arsenite exposure. J. Biol. Chem. 2005, 280, 16925–16933. [Google Scholar] [CrossRef]
- Borowski, L.S.; Dziembowski, A.; Hejnowicz, M.S.; Stepien, P.P.; Szczesny, R.J. Human mitochondrial RNA decay mediated by PNPase-hSuv3 complex takes place in distinct foci. Nucleic Acids Res. 2013, 41, 1223–1240. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.D.; Shu, Z.; Lieser, S.A.; Chen, P.L.; Lee, W.H. Human mitochondrial SUV3 and polynucleotide phosphorylase form a 330-kDa heteropentamer to cooperatively degrade double-stranded RNA with a 3′-to-5′ directionality. J. Biol. Chem. 2009, 284, 20812–20821. [Google Scholar] [CrossRef] [PubMed]
- Pajak, A.; Laine, I.; Clemente, P.; El-Fissi, N.; Schober, F.A.; Maffezzini, C.; Calvo-Garrido, J.; Wibom, R.; Filograna, R.; Dhir, A.; et al. Defects of mitochondrial RNA turnover lead to the accumulation of double-stranded RNA in vivo. PLoS Genet. 2019, 15, e1008240. [Google Scholar] [CrossRef]
- Wiatrek, D.M.; Candela, M.E.; Sedmik, J.; Oppelt, J.; Keegan, L.P.; O’Connell, M.A. Activation of innate immunity by mitochondrial dsRNA in mouse cells lacking p53 protein. RNA 2019, 25, 713–726. [Google Scholar] [CrossRef] [PubMed]
- Munch, C.; Harper, J.W. Mitochondrial unfolded protein response controls matrix pre-RNA processing and translation. Nature 2016, 534, 710–713. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Lim, Y.; Lee, D.; Elvira, R.; Lee, J.M.; Lee, M.R.; Han, J. Modulation of Protein Synthesis by eIF2alpha Phosphorylation Protects Cell from Heat Stress-Mediated Apoptosis. Cells 2018, 7, 254. [Google Scholar] [CrossRef] [PubMed]
- Scheuner, D.; Song, B.; McEwen, E.; Liu, C.; Laybutt, R.; Gillespie, P.; Saunders, T.; Bonner-Weir, S.; Kaufman, R.J. Translational Control Is Required for the Unfolded Protein Response and In Vivo Glucose Homeostasis. Mol. Cell 2001, 7, 1165–1176. [Google Scholar] [CrossRef] [PubMed]
- Hettmann, T.; Barton, K.; Leiden, J.M. Microphthalmia due to p53-mediated apoptosis of anterior lens epithelial cells in mice lacking the CREB-2 transcription factor. Dev. Biol. 2000, 222, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-L.; Reis, L.F.L.; Pavlovic, J.P.; Aguzzi, A.; Schafer, R.; Kumar, A.; Williams, B.R.G.; Aguet, M.; Weissmann, C. Deficient signaling in mice devoid of doublestranded RNA-dependent protein kinase. EMBO J. 1995, 14, 6095–6106. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.C.; Zong, W.X.; Cheng, E.H.; Lindsten, T.; Panoutsakopoulou, V.; Ross, A.J.; Roth, K.A.; MacGregor, G.R.; Thompson, C.B.; Korsmeyer, S.J. Proapoptotic BAX and BAK: A requisite gateway to mitochondrial dysfunction and death. Science 2001, 292, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Knudson, C.M.; Tung, K.S.K.; Tourtellotte, W.G.; Brown, G.A.J.; Korsmeyer, S.J. Bax-Deficient Mice with Lymphoid Hyperplasia and Male Germ Cell Death. Sci. Adv. 1995, 270, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Lindsten, T.; Ross, A.J.; Zong, W.-X.; Shiels, H.A.; Mahar, P.; Wei, M.; Eng, V.M.; Simon, M.C.; Ma, A.; King, A.; et al. The Combined Functions of Proapoptotic Bcl-2 Family Members Bak and Bax Are Essential for Normal Development of Multiple Tissues. Mol. Cell 2000, 6, 1389–1399. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).