
Expanding the Phenotypic Spectrum of Pathogenic KIAA0586 Variants: From Joubert Syndrome to Hydrolethalus Syndrome

 , ,
, , {kind=link}
Abstract
:1. Introduction
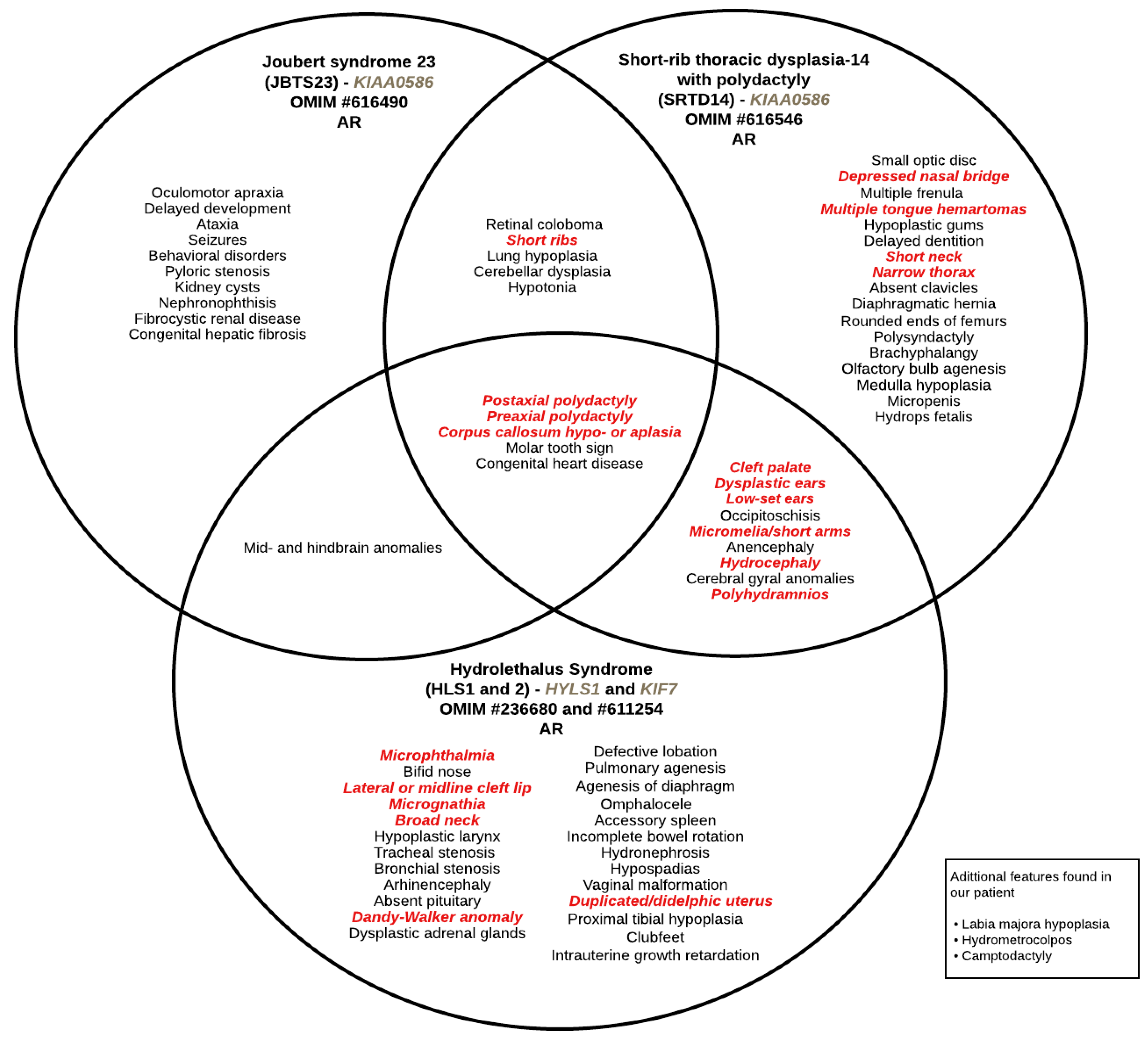
2. Results
2.1. Clinical Findings
2.2. Genetic Analysis Results
3. Discussion
4. Materials and Methods
Molecular Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Waters, A.M.; Beales, P.L. Ciliopathies: An expanding disease spectrum. Pediatr. Nephrol. 2011, 26, 1039–1056. [Google Scholar] [CrossRef] [PubMed]
- Braun, D.A.; Hildebrandt, F. Ciliopathies. Cold Spring Harb. Perspect. Biol. 2017, 9, a028191. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, F.; Benzing, T.; Katsanis, N. Ciliopathies. N. Engl. J. Med. 2011, 364, 1533–1543. [Google Scholar] [CrossRef] [PubMed]
- Alby, C.; Piquand, K.; Huber, C.; Megarbane, A.; Ichkou, A.; Legendre, M.; Pelluard, F.; Encha-Ravazi, F.; Abi-Tayeh, G.; Bessieres, B.; et al. Mutations in KIAA0586 cause lethal ciliopathies ranging from a hydrolethalus phenotype to short-rib polydactyly syndrome. Am. J. Hum. Genet. 2015, 97, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Malicdan, M.C.V.; Vilboux, T.; Stephen, J.; Maglic, D.; Mian, L.; Konzman, D.; Guo, J.; Yildirimli, D.; Bryant, J.; Fischer, R.; et al. Mutations in human homologue of chicken talpid3 gene (KIAA0586) cause a hybrid ciliopathy with overlapping features of Jeune and Joubert syndromes. J. Med. Genet. 2015, 52, 830–839. [Google Scholar] [CrossRef] [PubMed]
- Fraser, A.M.; Davey, M.G. TALPID3 in Joubert syndrome and related ciliopathy disorders. Curr. Opin. Genet. Dev. 2019, 56, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Lam, W.Y.; Tang, C.S.; So, M.T.; Yue, H.; Hsu, J.S.; Chung, P.H.; Nicholls, J.M.; Yeung, F.; Lee, C.D.; Ngo, D.N.; et al. Identification of a wide spectrum of ciliary gene mutations in nonsyndromic biliary atresia patients implicates ciliary dysfunction as a novel disease mechanism. EBioMedicine 2021, 71, 103530. [Google Scholar] [CrossRef] [PubMed]
- Bachmann-Gagescu, R.; Phelps, I.G.; Dempsey, J.C.; Sharma, V.A.; Ishak, G.E.; Boyle, E.A.; Wilson, M.; Marques, L.C.; Arslan, M.; University of Washington Center for Mendelian Genomics; et al. KIAA0586 is Mutated in Joubert Syndrome. Hum. Mutat. 2015, 36, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Ben, J.; Elworthy, S.; Ng, A.S.; van Eeden, F.; Ingham, P.W. Targeted mutation of the talpid3 gene in zebrafish reveals its conserved requirement for ciliogenesis and Hedgehog signaling across the vertebrates. Development 2011, 138, 4969–4978. [Google Scholar] [CrossRef]
- Bangs, F.; Antonio, N.; Thongnuek, P.; Welten, M.; Davey, M.G.; Briscoe, J.; Tickle, C. Generation of mice with functional inactivation of talpid3, a gene first identified in chicken. Development 2011, 138, 3261–3272. [Google Scholar] [CrossRef]
- Davey, M.G.; Paton, I.R.; Yin, Y.; Schmidt, M.; Bangs, F.K.; Morrice, D.R.; Smith, T.G.; Buxton, P.; Stamataki, D.; Tanaka, M.; et al. The chicken talpid3 gene encodes a novel protein essential for Hedgehog signaling. Genes Dev. 2006, 20, 1365–1377. [Google Scholar] [CrossRef]
- Yin, Y.; Bangs, F.; Paton, I.R.; Prescott, A.; James, J.; Davey, M.G.; Whitley, P.; Genikhovich, G.; Technau, U.; Burt, D.W.; et al. The Talpid3 gene (KIAA0586) encodes a centrosomal protein that is essential for primary cilia formation. Development 2009, 136, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Cocciadiferro, D.; Agolini, E.; Digilio, M.C.; Sinibaldi, L.; Castori, M.; Silvestri, E.; Dotta, A.; Dallapiccola, B.; Novelli, A. The splice c.1815G>A variant in KIAA0586 results in a phenotype bridging short-rib-polydactyly and oral-facial-digital syndrome: A case report and literature review. Medicine 2020, 99, e19169. [Google Scholar] [CrossRef]
- Parisi, M.A. The molecular genetics of Joubert syndrome and related ciliopathies: The challenges of genetic and phenotypic heterogeneity. Transl. Sci. Rare Dis. 2019, 4, 25–49. [Google Scholar] [CrossRef]
- Salonen, R.; Herva, R.; Norio, R. The hydrolethalus syndrome: Delineation of a “new,” lethal malformation syndrome based on 28 patients. Clin. Genet. 1981, 19, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Paetau, A.; Honkala, H.; Salonen, R.; Ignatius, J.; Kestilä, M.; Herva, R. H7ydrolethalus syndrome: Neuropathology of 21 cases confirmed by HYLS1 gene mutation analysis. J. Neuropathol. Exp. Neurol. 2008, 67, 750–762. [Google Scholar] [CrossRef] [PubMed]
- Putoux, A.; Thomas, S.; Coene, K.L.; Davis, E.E.; Alanay, Y.; Ogur, G.; Uz, E.; Buzas, D.; Gomes, C.; Patrier, S.; et al. KIF7 mutations cause fetal hydrolethalus and acrocallosal syndromes. Nat. Genet. 2011, 43, 601–606. [Google Scholar] [CrossRef]
- Mee, L.; Honkala, H.; Kopra, O.; Vesa, J.; Finnilä, S.; Visapää, I.; Sang, T.K.; Jackson, G.R.; Salonen, R.; Kestilä, M.; et al. Hydrolethalus syndrome is caused by a missense mutation in a novel gene HYLS1. Hum. Mol. Genet. 2005, 14, 1475–1488. [Google Scholar] [CrossRef] [PubMed]
- Curinha, A.; Huang, Z.; Anglen, T.; Strong, M.A.; Gliech, C.R.; Jewett, C.E.; Friskes, A.; Holland, A.J. Centriole structural integrity defects are a crucial feature of Hydrolethalus Syndrome. bioRxiv 2024. [Google Scholar] [CrossRef]
- Dafinger, C.; Liebau, M.C.; Elsayed, S.M.; Hellenbroich, Y.; Boltshauser, E.; Korenke, G.C.; Fabretti, F.; Janecke, A.R.; Ebermann, I.; Nürnberg, G.; et al. Mutations in KIF7 link Joubert syndrome with Sonic Hedgehog signaling and microtubule dynamics. J. Clin. Investig. 2011, 121, 2662–2667. [Google Scholar] [CrossRef]
- Aksu Uzunhan, T.; Ertürk, B.; Aydın, K.; Ayaz, A.; Altunoğlu, U.; Yarar, M.H.; Gezdirici, A.; İçağasıoğlu, D.F.; Gökpınar İli, E.; Uyanık, B.; et al. Clinical and genetic spectrum from a prototype of ciliopathy: Joubert syndrome. Clin. Neurol. Neurosurg. 2023, 224, 107560. [Google Scholar] [CrossRef]
- Parisi, M.; Glass, I. Joubert syndrome and related disorders. In GeneReviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Joubert, M.; Eisenring, J.J.; Andermann, F. Familial dysgenesis of the vermis: A syndrome of hyperventilation, abnormal eye movements and retardation. Neurology 1968, 18, 302–303. [Google Scholar] [PubMed]
- Vilboux, T.; Doherty, D.A.; Glass, I.A.; Parisi, M.A.; Phelps, I.G.; Cullinane, A.R.; Zein, W.; Brooks, B.P.; Heller, T.; Soldatos, A.; et al. Molecular genetic findings and clinical correlations in 100 patients with Joubert syndrome and related disorders prospectively evaluated at a single center. Genet. Med. 2017, 19, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Stephen, L.A.; Tawamie, H.; Davis, G.M.; Tebbe, L.; Nürnberg, P.; Nürnberg, G.; Thiele, H.; Thoenes, M.; Boltshauser, E.; Uebe, S.; et al. TALPID3 controls centrosome and cell polarity and the human ortholog KIAA0586 is mutated in Joubert syndrome (JBTS23). eLife 2015, 4, e08077. [Google Scholar] [CrossRef]
- Huber, C.; Cormier-Daire, V. Ciliary disorder of the skeleton. Am. J. Med. Genet. 2012, 160C, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Schmidts, M.; Vodopiutz, J.; Christou-Savina, S.; Cortes, C.R.; McInerney-Leo, A.M.; Emes, R.D.; Arts, H.H.; Tuysuz, B.; D’Silva, J.; Leo, P.J.; et al. Mutations in the gene encoding IFT dynein complex component WDR34 cause Jeune asphyxiating thoracic dystrophy. Am. J. Hum. Genet. 2013, 93, 932–944. [Google Scholar] [CrossRef]
- Girirajan, S.; Eichler, E.E. Phenotypic variability and genetic susceptibility to genomic disorders. Hum. Mol. Genet. 2010, 19, R176–R187. [Google Scholar] [CrossRef]
- Reiter, J.F.; Leroux, M.R. Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell Biol. 2017, 18, 533–547. [Google Scholar] [CrossRef]
- Kurolap, A.; Mory, A.; Simchoni, S.; Krajden Haratz, K.; Malinger, G.; Birnbaum, R.; Baris Feldman, H.; Yaron, Y. Upgrading an intronic TMEM67 variant of unknown significance to likely pathogenic through RNA studies and community data sharing. Prenat. Diagn. 2022, 42, 1484–1487. [Google Scholar] [CrossRef]
- Hiraide, T.; Shimizu, K.; Okumura, Y.; Miyamoto, S.; Nakashima, M.; Ogata, T.; Saitsu, H. A deep intronic TCTN2 variant activating a cryptic exon predicted by SpliceRover in a patient with Joubert syndrome. J. Hum. Genet. 2023, 68, 499–505. [Google Scholar] [CrossRef]
- Marshall, A.E.; Lemire, G.; Liang, Y.; Davila, J.; Couse, M.; Care4Rare Canada Consortium; Boycott, K.M.; Kernohan, K.D. RNA sequencing reveals deep intronic CEP120 variant: A report of the diagnostic odyssey for two siblings with Joubert syndrome type 31. Am. J. Med. Genet. A 2024, 194, e63485. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deconte, D.; Diniz, B.L.; Hartmann, J.K.; de Souza, M.A.; Zottis, L.F.F.; Zen, P.R.G.; Rosa, R.F.M.; Fiegenbaum, M. Expanding the Phenotypic Spectrum of Pathogenic KIAA0586 Variants: From Joubert Syndrome to Hydrolethalus Syndrome. Int. J. Mol. Sci. 2024, 25, 7900. https://doi.org/10.3390/ijms25147900
Deconte D, Diniz BL, Hartmann JK, de Souza MA, Zottis LFF, Zen PRG, Rosa RFM, Fiegenbaum M. Expanding the Phenotypic Spectrum of Pathogenic KIAA0586 Variants: From Joubert Syndrome to Hydrolethalus Syndrome. International Journal of Molecular Sciences. 2024; 25(14):7900. https://doi.org/10.3390/ijms25147900
Chicago/Turabian StyleDeconte, Desirée, Bruna Lixinski Diniz, Jéssica K. Hartmann, Mateus A. de Souza, Laira F. F. Zottis, Paulo Ricardo Gazzola Zen, Rafael F. M. Rosa, and Marilu Fiegenbaum. 2024. "Expanding the Phenotypic Spectrum of Pathogenic KIAA0586 Variants: From Joubert Syndrome to Hydrolethalus Syndrome" International Journal of Molecular Sciences 25, no. 14: 7900. https://doi.org/10.3390/ijms25147900