

Tau Beyond Tangles: DNA Damage Response and Cytoskeletal Protein Crosstalk on Neurodegeneration


Abstract
:1. Introduction
2. Development and Repair of DNA Damage in Neurons
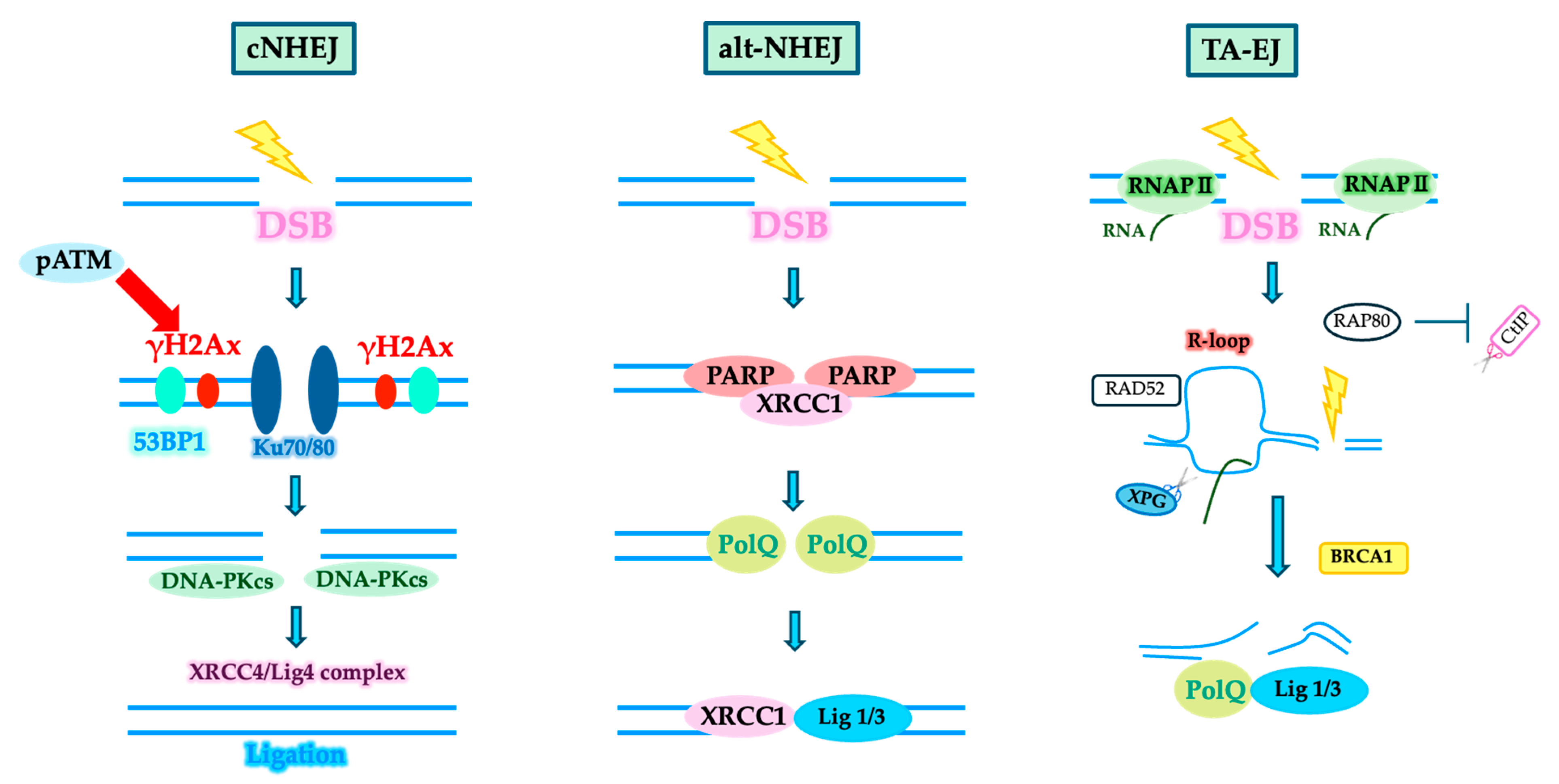
3. DSB Repair System
4. DDR and Cytoskeletal Proteins
4.1. DDR and Nuclear Membrane
4.2. DDR and Memory Formation
5. Disruption of DNA Damage Repair and Neurodegenerative Diseases
5.1. Neurodegenerative Diseases and Genomic Structural Changes
5.2. Presence of DSBs in the AD Brain
5.3. Relationship between DDR and Tau
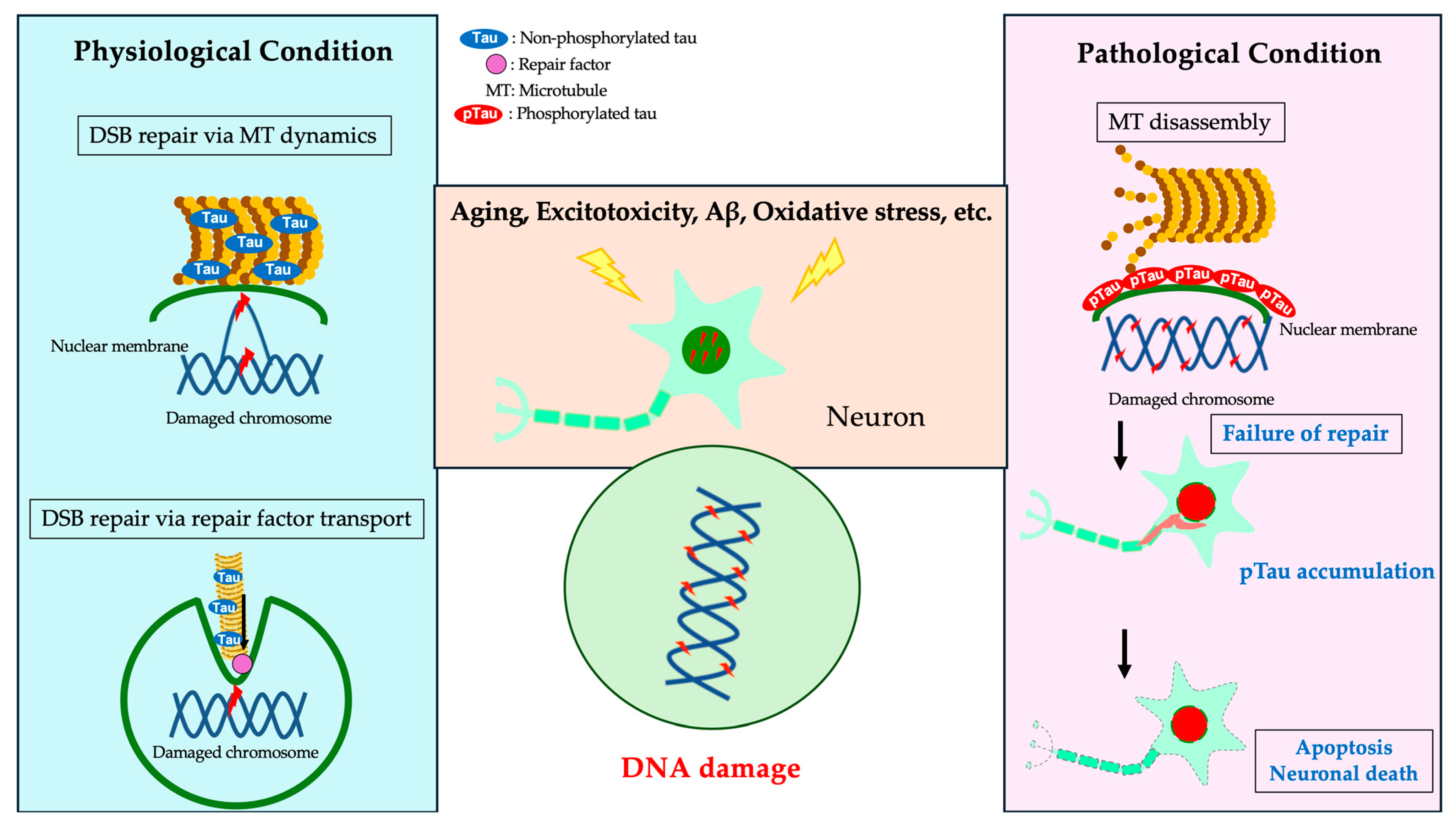
5.4. Microtubule Polymerization and DDR
5.5. DDR and Pathogenic Proteins in Neurodegenerative Diseases
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Crowe, S.L.; Movsesyan, V.A.; Jorgensen, T.J.; Kondratyev, A. Rapid phosphorylation of histone H2A.X following ionotropic glutamate receptor activation. Eur. J. Neurosci. 2006, 23, 2351–2361. [Google Scholar] [CrossRef] [PubMed]
- Shokrollahi, M.; Mekhail, K. Interphase microtubules in nuclear organization and genome maintenance. Trends Cell Biol. 2021, 31, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Suberbielle, E.; Sanchez, P.E.; Kravitz, A.V.; Wang, X.; Ho, K.; Eilertson, K.; Devidze, N.; Kreitzer, A.C.; Mucke, L. Physiologic brain activity causes DNA double-strand breaks in neurons, with exacerbation by amyloid-β. Nat. Neurosci. 2013, 16, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Madabhushi, R.; Gao, F.; Pfenning, A.R.; Pan, L.; Yamakawa, S.; Seo, J.; Rueda, R.; Phan, T.X.; Yamakawa, H.; Pao, P.C.; et al. Activity-Induced DNA Breaks Govern the Expression of Neuronal Early-Response Genes. Cell 2015, 161, 1592–1605. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, E.; Beach, T.; Shen, Y.; Li, R.; Chang, Y. Deficiency of the Mre11 DNA repair complex in Alzheimer’s disease brains. Brain Res. Mol. Brain Res. 2004, 128, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Lovell, M.A.; Markesbery, W.R. Oxidative DNA damage in mild cognitive impairment and late-stage Alzheimer’s disease. Nucleic Acids Res. 2007, 35, 7497–7504. [Google Scholar] [CrossRef]
- Lu, T.; Pan, Y.; Kao, S.Y.; Li, C.; Kohane, I.; Chan, J.; Yankner, B.A. Gene regulation and DNA damage in the ageing human brain. Nature 2004, 429, 883–891. [Google Scholar] [CrossRef]
- Jovasevic, V.; Zhang, H.; Sananbenesi, F.; Guedea, A.L.; Soman, K.V.; Wiktorowicz, J.E.; Fischer, A.; Radulovic, J. Primary cilia are required for the persistence of memory and stabilization of perineuronal nets. iScience 2021, 24, 102617. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.H.; Wang, H.; Soderling, S.H.; Yasuda, R. Loss of Cdc42 leads to defects in synaptic plasticity and remote memory recall. Elife 2014, 3, e02839. [Google Scholar] [CrossRef]
- Tada, T.; Sheng, M. Molecular mechanisms of dendritic spine morphogenesis. Curr. Opin. Neurobiol. 2006, 16, 95–101. [Google Scholar] [CrossRef]
- Asada-Utsugi, M.; Uemura, K.; Kubota, M.; Noda, Y.; Tashiro, Y.; Uemura, T.M.; Yamakado, H.; Urushitani, M.; Takahashi, R.; Hattori, S.; et al. Mice with cleavage-resistant N-cadherin exhibit synapse anomaly in the hippocampus and outperformance in spatial learning tasks. Mol. Brain 2021, 14, 23. [Google Scholar] [CrossRef] [PubMed]
- Jovasevic, V.; Wood, E.M.; Cicvaric, A.; Zhang, H.; Petrovic, Z.; Carboncino, A.; Parker, K.K.; Bassett, T.E.; Moltesen, M.; Yamawaki, N.; et al. Formation of memory assemblies through the DNA-sensing TLR9 pathway. Nature 2024, 628, 145–153. [Google Scholar] [CrossRef]
- Rolls, E.T. The hippocampus, ventromedial prefrontal cortex, and episodic and semantic memory. Prog. Neurobiol. 2022, 217, 102334. [Google Scholar] [CrossRef]
- Xia, Y.; Prokop, S.; Giasson, B.I. “Don’t Phos Over Tau”: Recent developments in clinical biomarkers and therapies targeting tau phosphorylation in Alzheimer’s disease and other tauopathies. Mol. Neurodegener. 2021, 16, 37. [Google Scholar] [CrossRef] [PubMed]
- Asada-Utsugi, M.; Uemura, K.; Ayaki, T.; Uemura, M.T.; Minamiyama, S.; Hikiami, R.; Morimura, T.; Shodai, A.; Ueki, T.; Takahashi, R.; et al. Failure of DNA double-strand break repair by tau mediates Alzheimer’s disease pathology in vitro. Commun. Biol. 2022, 5, 358. [Google Scholar] [CrossRef] [PubMed]
- Madabhushi, R.; Pan, L.; Tsai, L.H. DNA damage and its links to neurodegeneration. Neuron 2014, 83, 266–282. [Google Scholar] [CrossRef]
- Welch, G.; Tsai, L.H. Mechanisms of DNA damage-mediated neurotoxicity in neurodegenerative disease. EMBO Rep. 2022, 23, e54217. [Google Scholar] [CrossRef]
- Li, Y.; Yuan, J. Role of deubiquitinating enzymes in DNA double-strand break repair. J. Zhejiang Univ. Sci. B 2021, 22, 63–72. [Google Scholar] [CrossRef]
- Mitrentsi, I.; Lou, J.; Kerjouan, A.; Verigos, J.; Reina-San-Martin, B.; Hinde, E.; Soutoglou, E. Heterochromatic repeat clustering imposes a physical barrier on homologous recombination to prevent chromosomal translocations. Mol. Cell 2022, 82, 2132–2147.e6. [Google Scholar] [CrossRef]
- Delint-Ramirez, I.; Konada, L.; Heady, L.; Rueda, R.; Jacome, A.S.V.; Marlin, E.; Marchioni, C.; Segev, A.; Kritskiy, O.; Yamakawa, S.; et al. Calcineurin dephosphorylates topoisomerase IIbeta and regulates the formation of neuronal-activity-induced DNA breaks. Mol. Cell 2022, 82, 3794–3809.e8. [Google Scholar] [CrossRef]
- Zada, D.; Bronshtein, I.; Lerer-Goldshtein, T.; Garini, Y.; Appelbaum, L. Sleep increases chromosome dynamics to enable reduction of accumulating DNA damage in single neurons. Nat. Commun. 2019, 10, 895. [Google Scholar] [CrossRef] [PubMed]
- Shibata, A. Regulation of repair pathway choice at two-ended DNA double-strand breaks. Mutat. Res. 2017, 803–805, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Shibata, A.; Jeggo, P. A historical reflection on our understanding of radiation-induced DNA double strand break repair in somatic mammalian cells; interfacing the past with the present. Int. J. Radiat. Biol. 2019, 95, 945–956. [Google Scholar] [CrossRef]
- Yasuhara, T.; Kato, R.; Yamauchi, M.; Uchihara, Y.; Zou, L.; Miyagawa, K.; Shibata, A. RAP80 suppresses the vulnerability of R-loops during DNA double-strand break repair. Cell Rep. 2022, 38, 110335. [Google Scholar] [CrossRef] [PubMed]
- Caridi, C.P.; D’Agostino, C.; Ryu, T.; Zapotoczny, G.; Delabaere, L.; Li, X.; Khodaverdian, V.Y.; Amaral, N.; Lin, E.; Rau, A.R.; et al. Nuclear F-actin and myosins drive relocalization of heterochromatic breaks. Nature 2018, 559, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Paonessa, F.; Evans, L.D.; Solanki, R.; Larrieu, D.; Wray, S.; Hardy, J.; Jackson, S.P.; Livesey, F.J. Microtubules Deform the Nuclear Membrane and Disrupt Nucleocytoplasmic Transport in Tau-Mediated Frontotemporal Dementia. Cell Rep. 2019, 26, 582–593.e5. [Google Scholar] [CrossRef] [PubMed]
- Teng, J.; Rai, T.; Tanaka, Y.; Takei, Y.; Nakata, T.; Hirasawa, M.; Kulkarni, A.B.; Hirokawa, N. The KIF3 motor transports N-cadherin and organizes the developing neuroepithelium. Nat. Cell Biol. 2005, 7, 474–482. [Google Scholar] [CrossRef]
- Hirokawa, N.; Niwa, S.; Tanaka, Y. Molecular motors in neurons: Transport mechanisms and roles in brain function, development, and disease. Neuron 2010, 68, 610–638. [Google Scholar] [CrossRef]
- Shokrollahi, M.; Stanic, M.; Hundal, A.; Chan, J.N.Y.; Urman, D.; Jordan, C.A.; Hakem, A.; Espin, R.; Hao, J.; Krishnan, R.; et al. DNA double-strand break-capturing nuclear envelope tubules drive DNA repair. Nat. Struct. Mol. Biol. 2024. [Google Scholar] [CrossRef]
- Eftekharzadeh, B.; Daigle, J.G.; Kapinos, L.E.; Coyne, A.; Schiantarelli, J.; Carlomagno, Y.; Cook, C.; Miller, S.J.; Dujardin, S.; Amaral, A.S.; et al. Tau Protein Disrupts Nucleocytoplasmic Transport in Alzheimer’s Disease. Neuron 2018, 99, 925–940.e7. [Google Scholar] [CrossRef]
- Wong, X.; Stewart, C.L. The Laminopathies and the Insights They Provide into the Structural and Functional Organization of the Nucleus. Annu. Rev. Genom. Hum. Genet. 2020, 21, 263–288. [Google Scholar] [CrossRef]
- Earle, A.J.; Kirby, T.J.; Fedorchak, G.R.; Isermann, P.; Patel, J.; Iruvanti, S.; Moore, S.A.; Bonne, G.; Wallrath, L.L.; Lammerding, J. Mutant lamins cause nuclear envelope rupture and DNA damage in skeletal muscle cells. Nat. Mater. 2020, 19, 464–473. [Google Scholar] [CrossRef]
- Cho, S.; Vashisth, M.; Abbas, A.; Majkut, S.; Vogel, K.; Xia, Y.; Ivanovska, I.L.; Irianto, J.; Tewari, M.; Zhu, K.; et al. Mechanosensing by the Lamina Protects against Nuclear Rupture, DNA Damage, and Cell-Cycle Arrest. Dev. Cell 2019, 49, 920–935.e5. [Google Scholar] [CrossRef]
- Kirby, T.J.; Zahr, H.C.; Fong, E.H.H.; Lammerding, J. Eliminating elevated p53 signaling fails to rescue skeletal muscle defects or extend survival in lamin A/C-deficient mice. Cell Death Discov. 2024, 10, 245. [Google Scholar] [CrossRef]
- Yap, E.L.; Greenberg, M.E. Activity-Regulated Transcription: Bridging the Gap between Neural Activity and Behavior. Neuron 2018, 100, 330–348. [Google Scholar] [CrossRef]
- Pollina, E.A.; Gilliam, D.T.; Landau, A.T.; Lin, C.; Pajarillo, N.; Davis, C.P.; Harmin, D.A.; Yap, E.L.; Vogel, I.R.; Griffith, E.C.; et al. A NPAS4-NuA4 complex couples synaptic activity to DNA repair. Nature 2023, 614, 732–741. [Google Scholar] [CrossRef]
- Nonaka, M.; Kim, R.; Sharry, S.; Matsushima, A.; Takemoto-Kimura, S.; Bito, H. Towards a better understanding of cognitive behaviors regulated by gene expression downstream of activity-dependent transcription factors. Neurobiol. Learn. Mem. 2014, 115, 21–29. [Google Scholar] [CrossRef]
- Lin, Y.; Bloodgood, B.L.; Hauser, J.L.; Lapan, A.D.; Koon, A.C.; Kim, T.K.; Hu, L.S.; Malik, A.N.; Greenberg, M.E. Activity-dependent regulation of inhibitory synapse development by Npas4. Nature 2008, 455, 1198–1204. [Google Scholar] [CrossRef]
- Welch, G.M.; Boix, C.A.; Schmauch, E.; Davila-Velderrain, J.; Victor, M.B.; Dileep, V.; Bozzelli, P.L.; Su, Q.; Cheng, J.D.; Lee, A.; et al. Neurons burdened by DNA double-strand breaks incite microglia activation through antiviral-like signaling in neurodegeneration. Sci. Adv. 2022, 8, eabo4662. [Google Scholar] [CrossRef]
- Shanbhag, N.M.; Evans, M.D.; Mao, W.; Nana, A.L.; Seeley, W.W.; Adame, A.; Rissman, R.A.; Masliah, E.; Mucke, L. Early neuronal accumulation of DNA double strand breaks in Alzheimer’s disease. Acta Neuropathol. Commun. 2019, 7, 77. [Google Scholar] [CrossRef]
- Kim, J.; Huang, A.Y.; Johnson, S.L.; Lai, J.; Isacco, L.; Jeffries, A.M.; Miller, M.B.; Lodato, M.A.; Walsh, C.A.; Lee, E.A. Prevalence and mechanisms of somatic deletions in single human neurons during normal aging and in DNA repair disorders. Nat. Commun. 2022, 13, 5918. [Google Scholar] [CrossRef]
- Okhovat, M.; VanCampen, J.; Nevonen, K.A.; Harshman, L.; Li, W.; Layman, C.E.; Ward, S.; Herrera, J.; Wells, J.; Sheng, R.R.; et al. TAD evolutionary and functional characterization reveals diversity in mammalian TAD boundary properties and function. Nat. Commun. 2023, 14, 8111. [Google Scholar] [CrossRef]
- Dileep, V.; Boix, C.A.; Mathys, H.; Marco, A.; Welch, G.M.; Meharena, H.S.; Loon, A.; Jeloka, R.; Peng, Z.; Bennett, D.A.; et al. Neuronal DNA double-strand breaks lead to genome structural variations and 3D genome disruption in neurodegeneration. Cell 2023, 186, 4404–4421.e20. [Google Scholar] [CrossRef]
- Meharena, H.S.; Marco, A.; Dileep, V.; Lockshin, E.R.; Akatsu, G.Y.; Mullahoo, J.; Watson, L.A.; Ko, T.; Guerin, L.N.; Abdurrob, F.; et al. Down-syndrome-induced senescence disrupts the nuclear architecture of neural progenitors. Cell Stem Cell 2022, 29, 116–130.e7. [Google Scholar] [CrossRef]
- Sun, J.H.; Zhou, L.; Emerson, D.J.; Phyo, S.A.; Titus, K.R.; Gong, W.; Gilgenast, T.G.; Beagan, J.A.; Davidson, B.L.; Tassone, F.; et al. Disease-Associated Short Tandem Repeats Co-localize with Chromatin Domain Boundaries. Cell 2018, 175, 224–238.e15. [Google Scholar] [CrossRef]
- Thadathil, N.; Delotterie, D.F.; Xiao, J.; Hori, R.; McDonald, M.P.; Khan, M.M. DNA Double-Strand Break Accumulation in Alzheimer’s Disease: Evidence from Experimental Models and Postmortem Human Brains. Mol. Neurobiol. 2021, 58, 118–131. [Google Scholar] [CrossRef]
- Mano, T.; Nagata, K.; Nonaka, T.; Tarutani, A.; Imamura, T.; Hashimoto, T.; Bannai, T.; Koshi-Mano, K.; Tsuchida, T.; Ohtomo, R.; et al. Neuron-specific methylome analysis reveals epigenetic regulation and tau-related dysfunction of BRCA1 in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, E9645–E9654. [Google Scholar] [CrossRef]
- Sultan, A.; Nesslany, F.; Violet, M.; Begard, S.; Loyens, A.; Talahari, S.; Mansuroglu, Z.; Marzin, D.; Sergeant, N.; Humez, S.; et al. Nuclear tau, a key player in neuronal DNA protection. J. Biol. Chem. 2011, 286, 4566–4575. [Google Scholar] [CrossRef]
- Violet, M.; Delattre, L.; Tardivel, M.; Sultan, A.; Chauderlier, A.; Caillierez, R.; Talahari, S.; Nesslany, F.; Lefebvre, B.; Bonnefoy, E.; et al. A major role for Tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditions. Front. Cell Neurosci. 2014, 8, 84. [Google Scholar] [CrossRef]
- Yang, Y.; Lei, T.; Du, S.; Tong, R.; Wang, H.; Yang, J.; Huang, J.; Sun, M.; Wang, Y.; Dong, Z. Nuclear GSK3beta induces DNA double-strand break repair by phosphorylating 53BP1 in glioblastoma. Int. J. Oncol. 2018, 52, 709–720. [Google Scholar] [CrossRef]
- Guo, T.; Noble, W.; Hanger, D.P. Roles of tau protein in health and disease. Acta Neuropathol. 2017, 133, 665–704. [Google Scholar] [CrossRef]
- Ittner, A.; Ittner, L.M. Dendritic Tau in Alzheimer’s Disease. Neuron 2018, 99, 13–27. [Google Scholar] [CrossRef]
- Tuerde, D.; Kimura, T.; Miyasaka, T.; Furusawa, K.; Shimozawa, A.; Hasegawa, M.; Ando, K.; Hisanaga, S.I. Isoform-independent and -dependent phosphorylation of microtubule-associated protein tau in mouse brain during postnatal development. J. Biol. Chem. 2018, 293, 1781–1793. [Google Scholar] [CrossRef]
- Rico, T.; Gilles, M.; Chauderlier, A.; Comptdaer, T.; Magnez, R.; Chwastyniak, M.; Drobecq, H.; Pinet, F.; Thuru, X.; Buee, L.; et al. Tau Stabilizes Chromatin Compaction. Front. Cell Dev. Biol. 2021, 9, 740550. [Google Scholar] [CrossRef]
- Rico, T.; Denechaud, M.; Caillierez, R.; Comptdaer, T.; Adriaenssens, E.; Buee, L.; Lefebvre, B. Cancer Cells Upregulate Tau to Gain Resistance to DNA Damaging Agents. Cancers 2022, 15, 116. [Google Scholar] [CrossRef]
- Frost, B.; Hemberg, M.; Lewis, J.; Feany, M.B. Tau promotes neurodegeneration through global chromatin relaxation. Nat. Neurosci. 2014, 17, 357–366. [Google Scholar] [CrossRef]
- Fernandez-Nogales, M.; Santos-Galindo, M.; Merchan-Rubira, J.; Hoozemans, J.J.M.; Rabano, A.; Ferrer, I.; Avila, J.; Hernandez, F.; Lucas, J.J. Tau-positive nuclear indentations in P301S tauopathy mice. Brain Pathol. 2017, 27, 314–322. [Google Scholar] [CrossRef]
- Gil, L.; Federico, C.; Pinedo, F.; Bruno, F.; Rebolledo, A.B.; Montoya, J.J.; Olazabal, I.M.; Ferrer, I.; Saccone, S. Aging dependent effect of nuclear tau. Brain Res. 2017, 1677, 129–137. [Google Scholar] [CrossRef]
- Ulrich, G.; Salvade, A.; Boersema, P.; Cali, T.; Foglieni, C.; Sola, M.; Picotti, P.; Papin, S.; Paganetti, P. Phosphorylation of nuclear Tau is modulated by distinct cellular pathways. Sci. Rep. 2018, 8, 17702. [Google Scholar] [CrossRef]
- Schaser, A.J.; Osterberg, V.R.; Dent, S.E.; Stackhouse, T.L.; Wakeham, C.M.; Boutros, S.W.; Weston, L.J.; Owen, N.; Weissman, T.A.; Luna, E.; et al. Alpha-synuclein is a DNA binding protein that modulates DNA repair with implications for Lewy body disorders. Sci. Rep. 2019, 9, 10919. [Google Scholar] [CrossRef]
- Yoon, Y.S.; You, J.S.; Kim, T.K.; Ahn, W.J.; Kim, M.J.; Son, K.H.; Ricarte, D.; Ortiz, D.; Lee, S.J.; Lee, H.J. Senescence and impaired DNA damage responses in alpha-synucleinopathy models. Exp. Mol. Med. 2022, 54, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Weng, S.M.; Arzberger, T.; May, S.; Rentzsch, K.; Kremmer, E.; Schmid, B.; Kretzschmar, H.A.; Cruts, M.; Van Broeckhoven, C.; et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 2013, 339, 1335–1338. [Google Scholar] [CrossRef]
- Imamura, T.; Fujita, K.; Tagawa, K.; Ikura, T.; Chen, X.; Homma, H.; Tamura, T.; Mao, Y.; Taniguchi, J.B.; Motoki, K.; et al. Identification of hepta-histidine as a candidate drug for Huntington’s disease by in silico-in vitro- in vivo-integrated screens of chemical libraries. Sci. Rep. 2016, 6, 33861. [Google Scholar] [CrossRef]
- Enokido, Y.; Tamura, T.; Ito, H.; Arumughan, A.; Komuro, A.; Shiwaku, H.; Sone, M.; Foulle, R.; Sawada, H.; Ishiguro, H.; et al. Mutant huntingtin impairs Ku70-mediated DNA repair. J. Cell Biol. 2010, 189, 425–443. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.; Li, X.; Zhu, X.; Liu, X.; Guo, C.; Jia, D.; Tang, T.S. Determining the Fate of Neurons in SCA3: ATX3, a Rising Decision Maker in Response to DNA Stresses and Beyond. Front. Cell Dev. Biol. 2020, 8, 619911. [Google Scholar] [CrossRef] [PubMed]
- Gall-Duncan, T.; Luo, J.; Jurkovic, C.M.; Fischer, L.A.; Fujita, K.; Deshmukh, A.L.; Harding, R.J.; Tran, S.; Mehkary, M.; Li, V.; et al. Antagonistic roles of canonical and Alternative-RPA in disease-associated tandem CAG repeat instability. Cell 2023, 186, 4898–4919.e25. [Google Scholar] [CrossRef]
- Tamaki, Y.; Shodai, A.; Morimura, T.; Hikiami, R.; Minamiyama, S.; Ayaki, T.; Tooyama, I.; Furukawa, Y.; Takahashi, R.; Urushitani, M. Elimination of TDP-43 inclusions linked to amyotrophic lateral sclerosis by a misfolding-specific intrabody with dual proteolytic signals. Sci. Rep. 2018, 8, 6030. [Google Scholar] [CrossRef]
- Tamaki, Y.; Ross, J.P.; Alipour, P.; Castonguay, C.E.; Li, B.; Catoire, H.; Rochefort, D.; Urushitani, M.; Takahashi, R.; Sonnen, J.A.; et al. Spinal cord extracts of amyotrophic lateral sclerosis spread TDP-43 pathology in cerebral organoids. PLoS Genet. 2023, 19, e1010606. [Google Scholar] [CrossRef]
- Mitra, J.; Guerrero, E.N.; Hegde, P.M.; Liachko, N.F.; Wang, H.; Vasquez, V.; Gao, J.; Pandey, A.; Taylor, J.P.; Kraemer, B.C.; et al. Motor neuron disease-associated loss of nuclear TDP-43 is linked to DNA double-strand break repair defects. Proc. Natl. Acad. Sci. USA 2019, 116, 4696–4705. [Google Scholar] [CrossRef]
- Konopka, A.; Atkin, J.D. DNA Damage, Defective DNA Repair, and Neurodegeneration in Amyotrophic Lateral Sclerosis. Front. Aging Neurosci. 2022, 14, 786420. [Google Scholar] [CrossRef]
- Kodavati, M.; Wang, H.; Guo, W.; Mitra, J.; Hegde, P.M.; Provasek, V.; Rao, V.H.M.; Vedula, I.; Zhang, A.; Mitra, S.; et al. FUS unveiled in mitochondrial DNA repair and targeted ligase-1 expression rescues repair-defects in FUS-linked motor neuron disease. Nat. Commun. 2024, 15, 2156. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Liang, J.; Chen, C.; Chen, J.; Shen, Y.; Sun, S.; Li, L. C9orf72 functions in the nucleus to regulate DNA damage repair. Cell Death Differ. 2023, 30, 716–730. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Pathogenic Proteins | Neurodegenerative Diseases | Subcellular Localization | DDR-Related Functions |
|---|---|---|---|
| MAPT (Tau) | AD, ALS, FTLD, PSP, CBD, PiD, AGD, CTE | Cytoplasm, Nucleus, Axon, Dendrite, Cell membrane | 53BP1 transport (Breast cancer cell) [53] Chromatin remodeling (Breast cancer cell) [52,54] DSB repair on KD and KO [15,47] |
| Amyloid-β | AD | Cell surface | Enhancing of DSB [17,37,45] |
| TDP-43 | ALS, FTLD, LATE, Perry disease, FOSMN | Nucleus, Cytoplasm, Mitochondria | XRCC4/Lig4 complex transport [65] Interaction with Ku70 (HEK293 cell) [66] KD enhances the R loops and DSB (HeLa cell) [66] |
| FUS | ALS, FTLD | Nucleus, Cytoplasm | mtDNA Ligase IIIα (mtLig3) transport BER [67] |
| C9orf72 repeat peptide | ALS, FTLD | Nucleus, Cytoplasm, Endsome, Lysosome, Axon | Interaction with DNA-PKcs complex [68] |
| SOD1 | ALS, STAHP | Cytoplasm, Nucleus | DNA protection from oxdative damage [66] |
| HTT | Huntington disease | Nucleus, Cytoplasm, Early endosome | Interaction with Ku70 [61,62] |
| α-Synuclein | PD | Cytoplasm membrane, Nucleus, Synapse | Overexpression decreases NHEJ and MRE11 expression [58,59] |
| ATX3 | SCA3 | Nucleus, Nucleus matrix, Lysosome membrane | Interaction with PNKP, MDC1, Ku70, Chk1, HTT, DNA-PKcs, 53BP1 and p97 [63] |
| ATX1 | SCA1 | Nucleus, Cytoplasm | Increased expression of RPA1, RPA2, RPA3 and Alternative-RPA [64] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asada-Utsugi, M.; Urushitani, M. Tau Beyond Tangles: DNA Damage Response and Cytoskeletal Protein Crosstalk on Neurodegeneration. Int. J. Mol. Sci. 2024, 25, 7906. https://doi.org/10.3390/ijms25147906
Asada-Utsugi M, Urushitani M. Tau Beyond Tangles: DNA Damage Response and Cytoskeletal Protein Crosstalk on Neurodegeneration. International Journal of Molecular Sciences. 2024; 25(14):7906. https://doi.org/10.3390/ijms25147906
Chicago/Turabian StyleAsada-Utsugi, Megumi, and Makoto Urushitani. 2024. "Tau Beyond Tangles: DNA Damage Response and Cytoskeletal Protein Crosstalk on Neurodegeneration" International Journal of Molecular Sciences 25, no. 14: 7906. https://doi.org/10.3390/ijms25147906