

Molecular Mechanisms Underlying the Anticancer Properties of Pitavastatin against Cervical Cancer Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
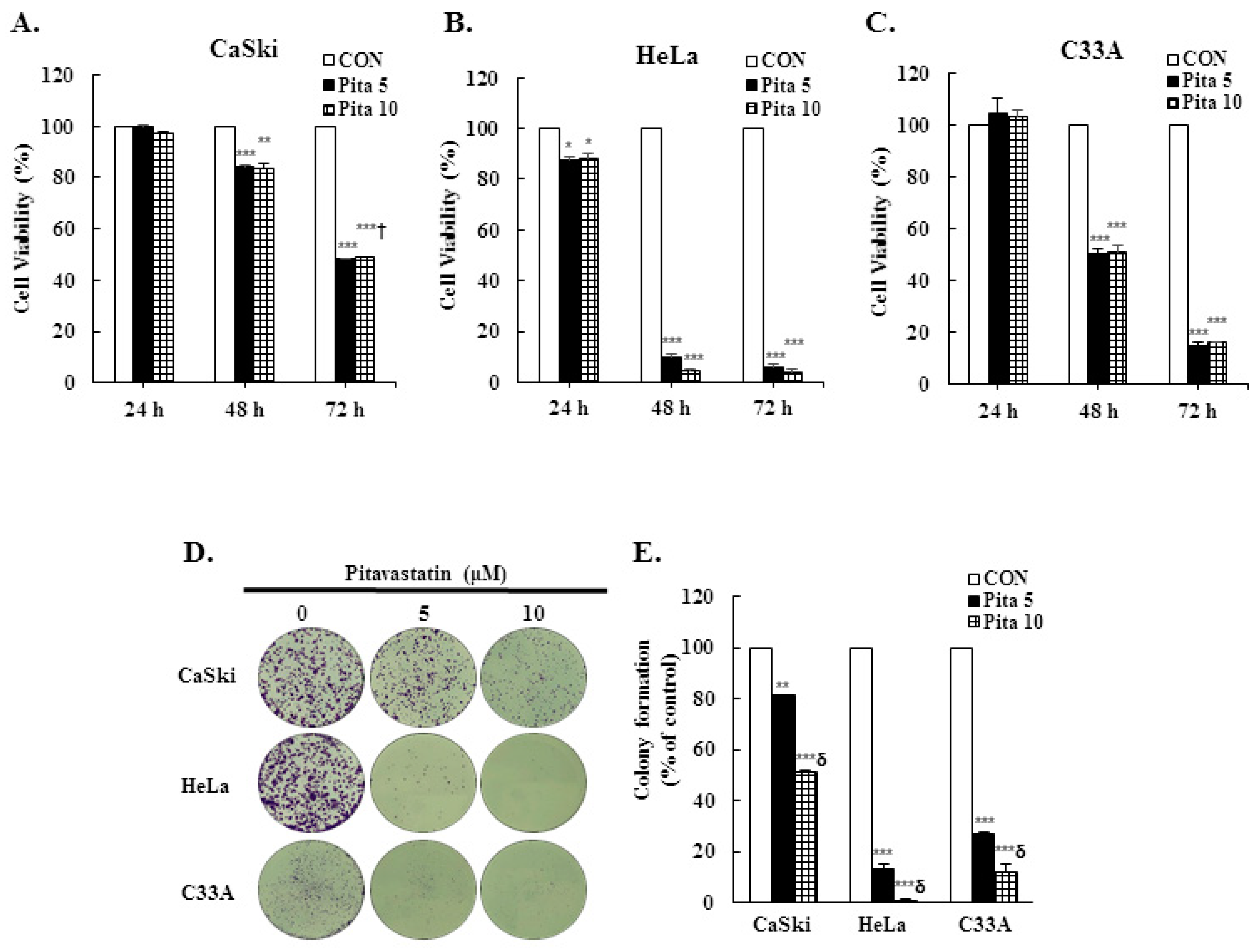
2.1. Pitavastatin Has a Dose-Dependent Effect on Suppressing Cell Growth and Colony Formation
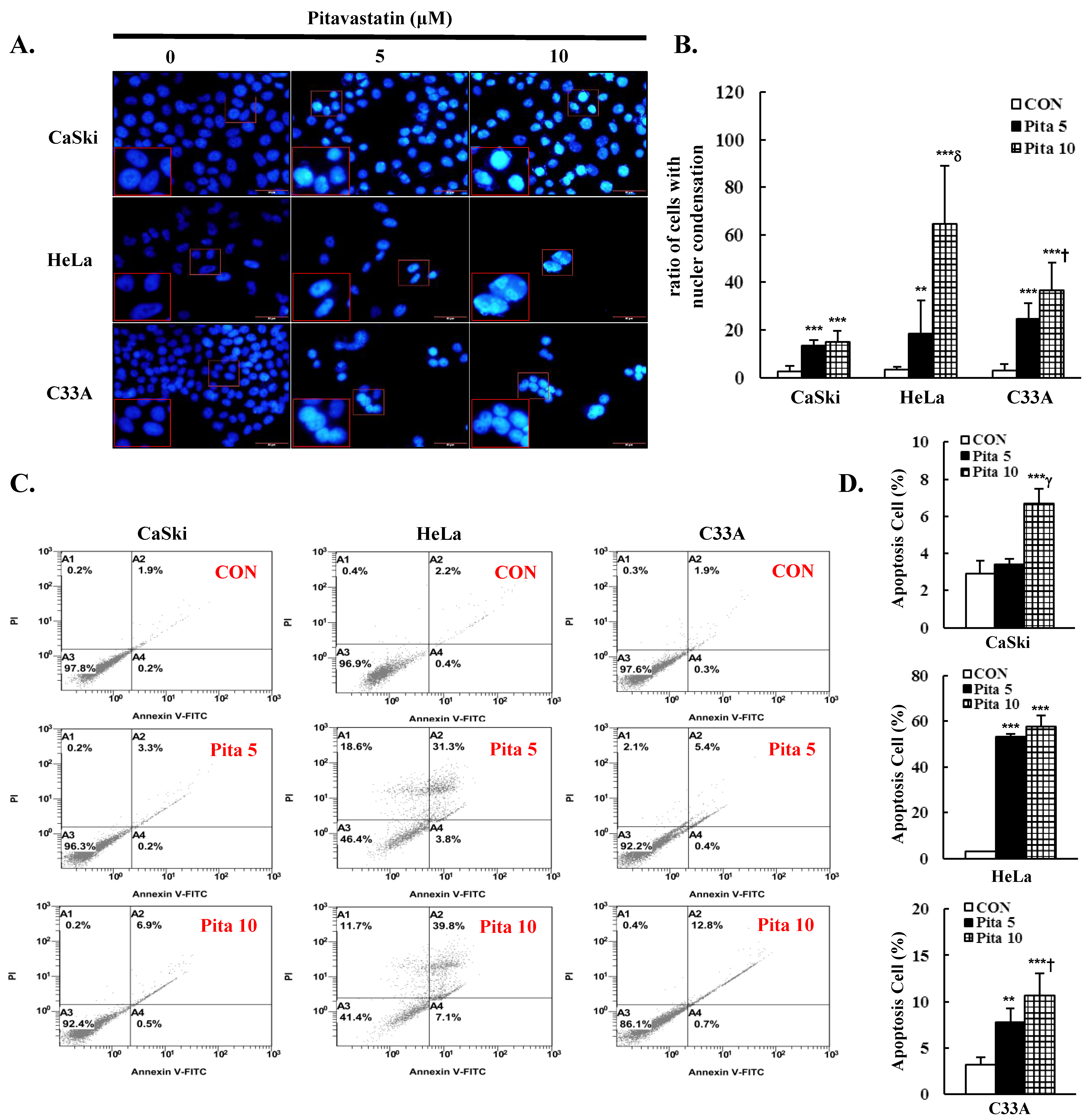
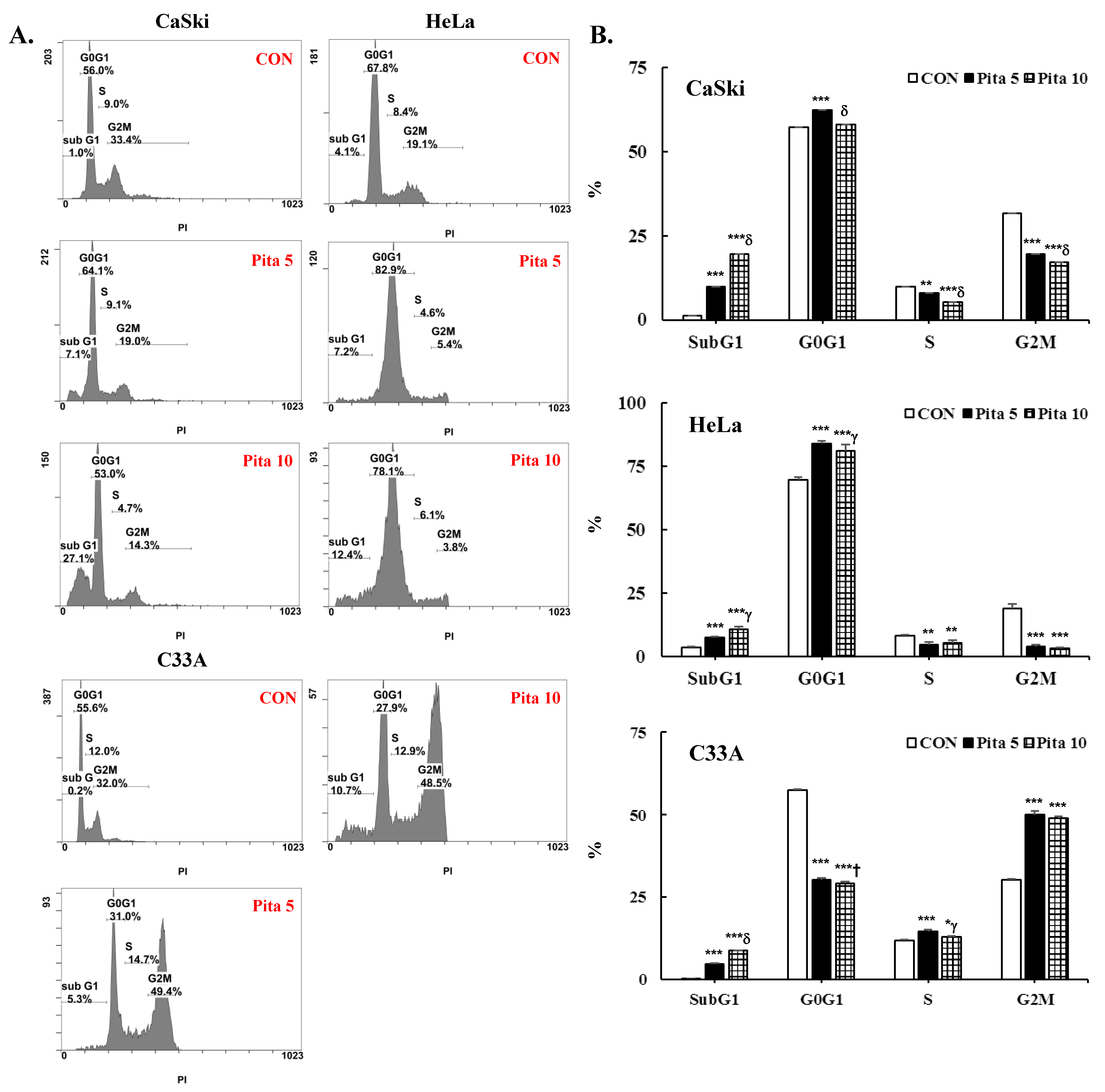
2.2. Pitavastatin Promotes Cell Apoptosis and Cell Cycle Arrest
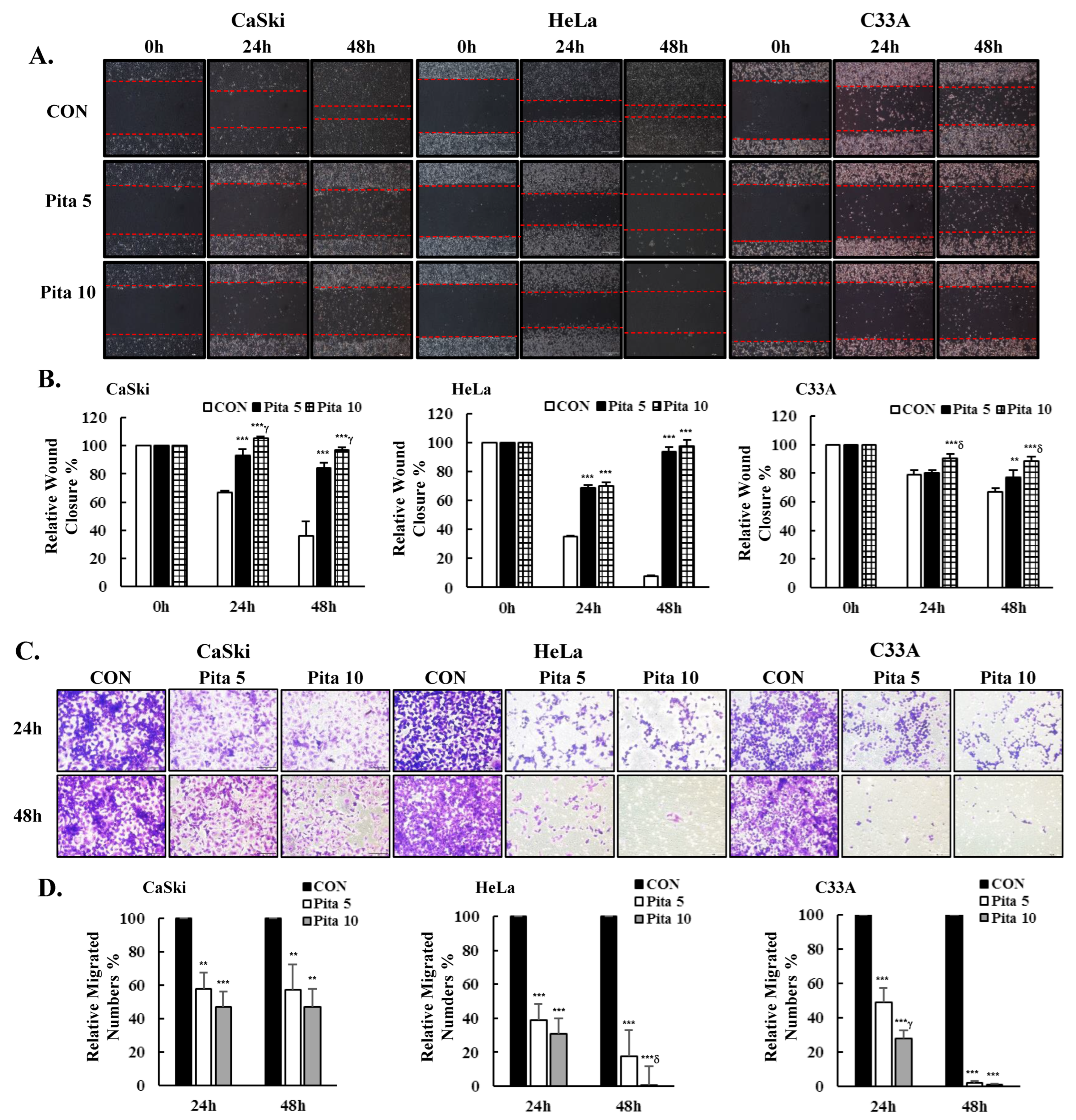
2.3. Pitavastatin Inhibits Cervical Cancer Cell Migration
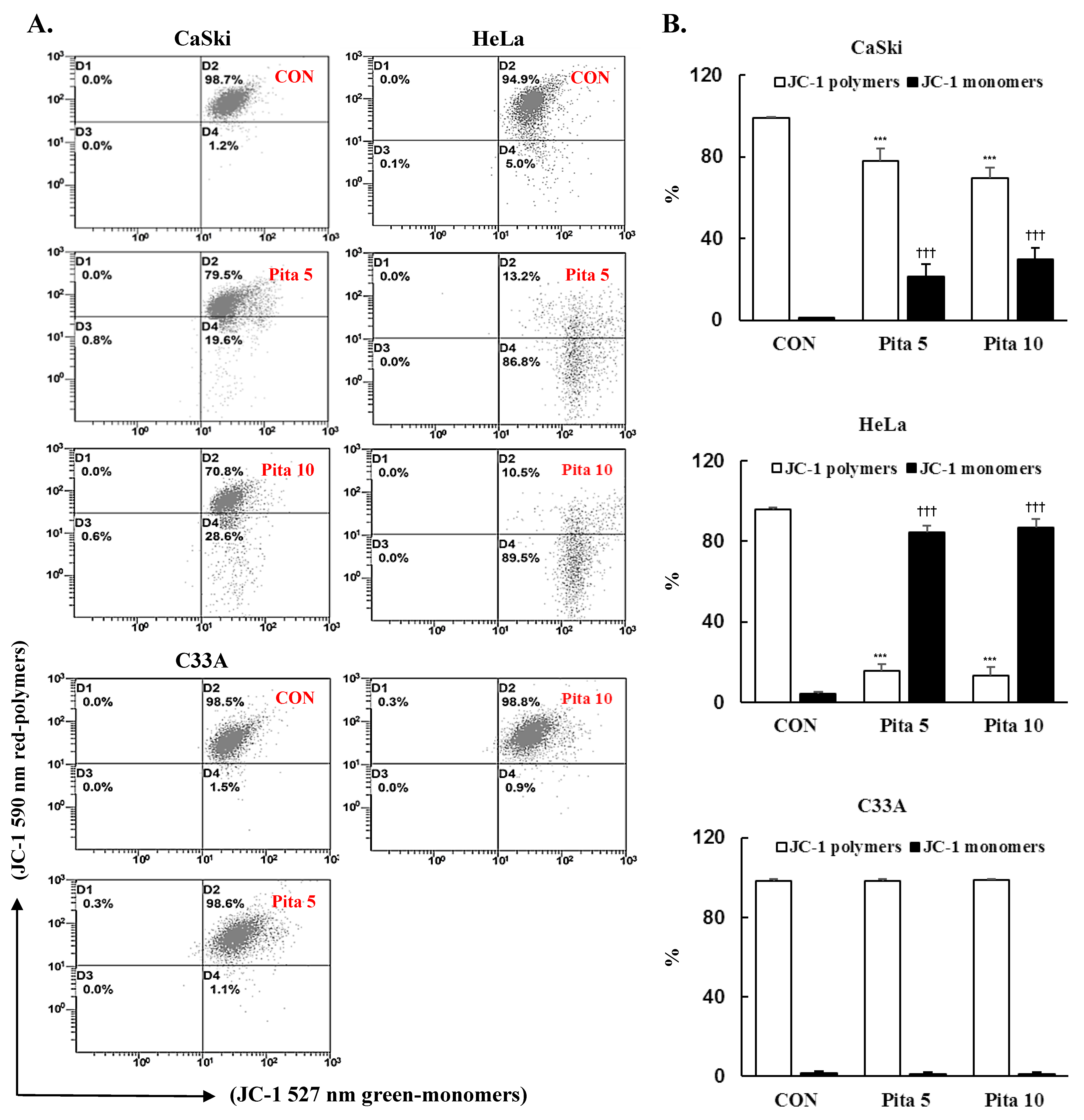
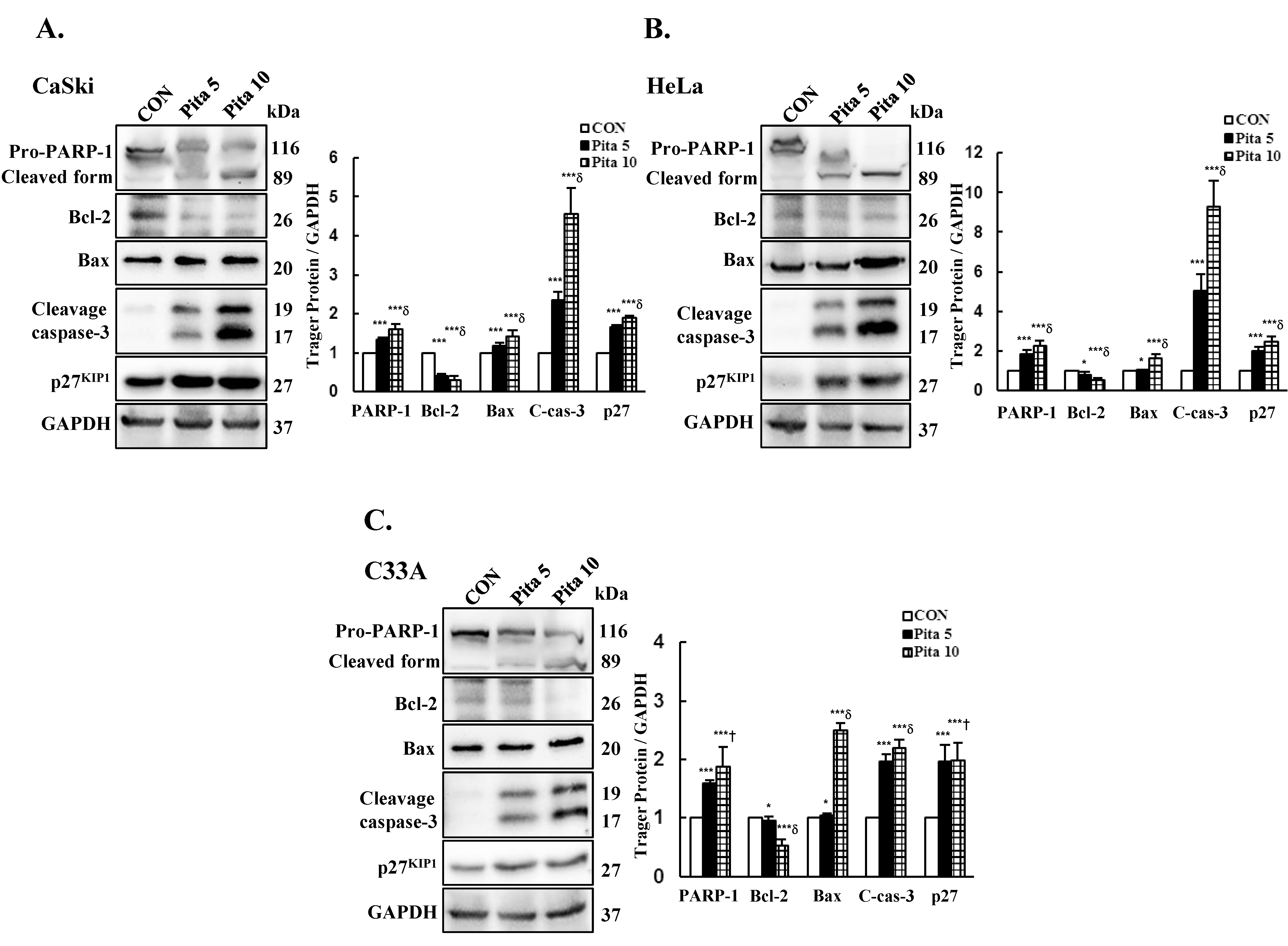
2.4. Pitavastatin Reduces the Mitochondrial Membrane Potential (∆ψm) and Activates Mitochondria-Mediated Apoptosis
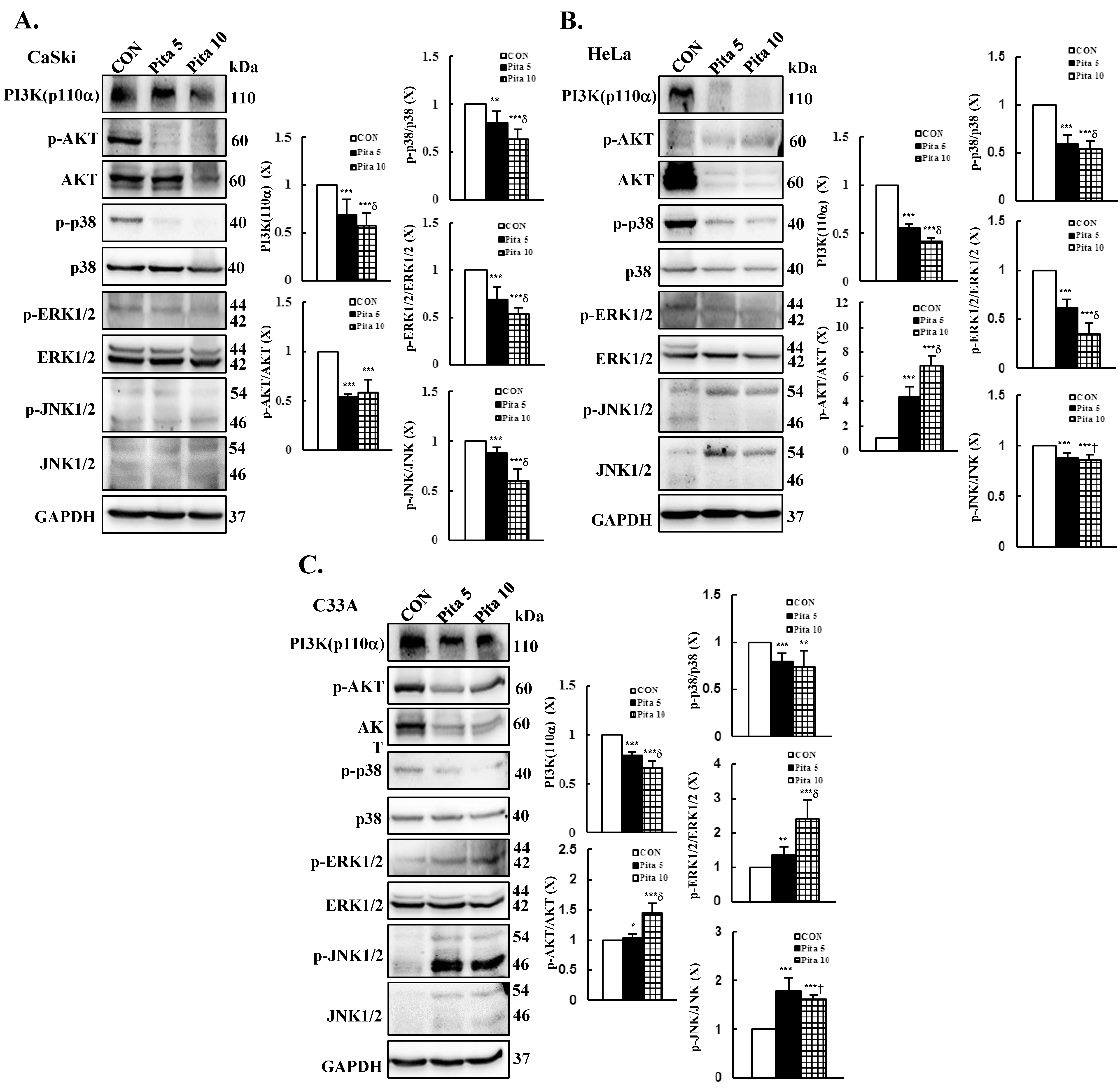
2.5. Pitavastatin-Induced Apoptosis Specifically Targets the AKT and MAPK Pathways
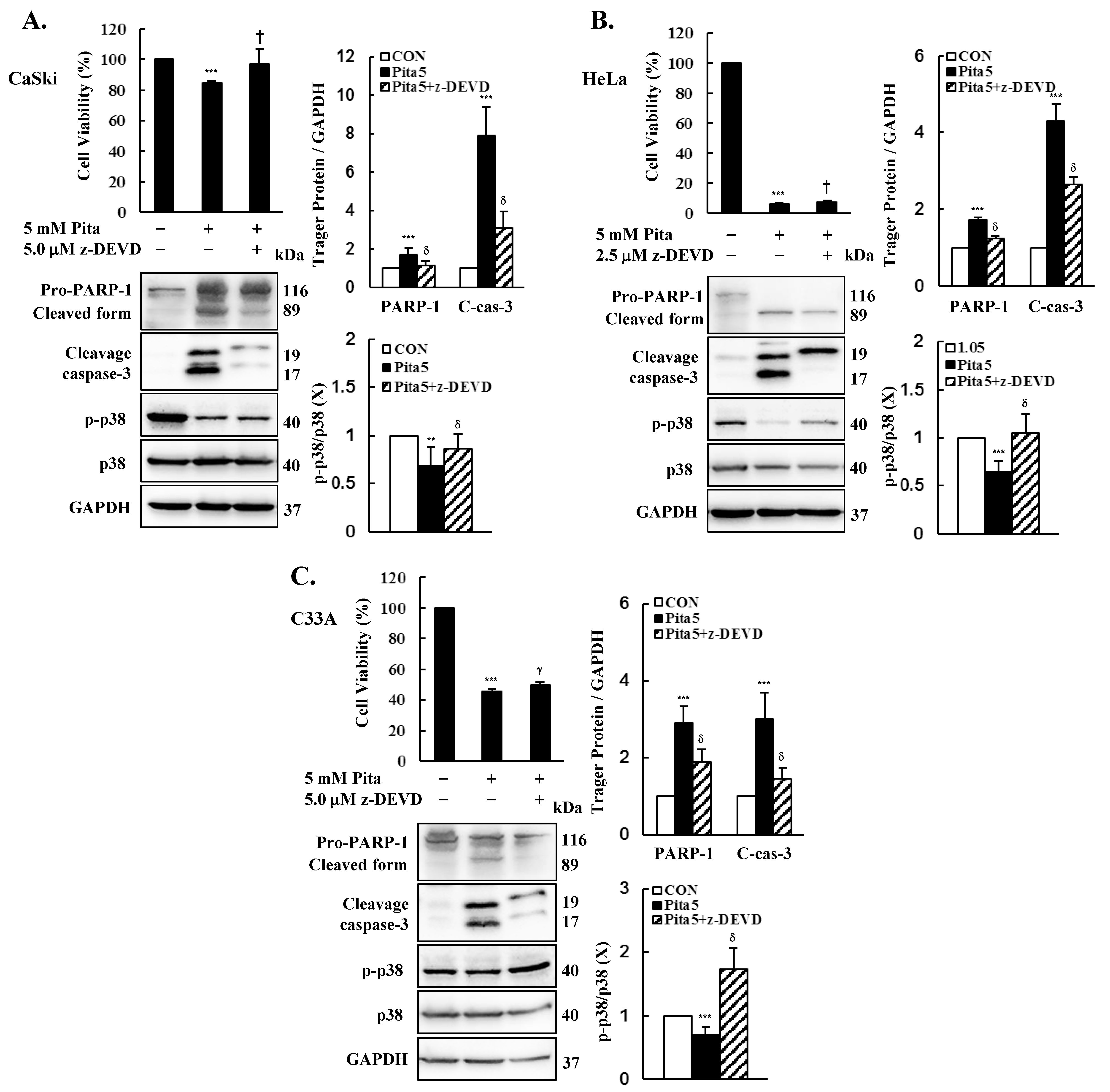
2.6. Caspase 3-Dependent Apoptosis Pathway Is Activated by Pitavastatin
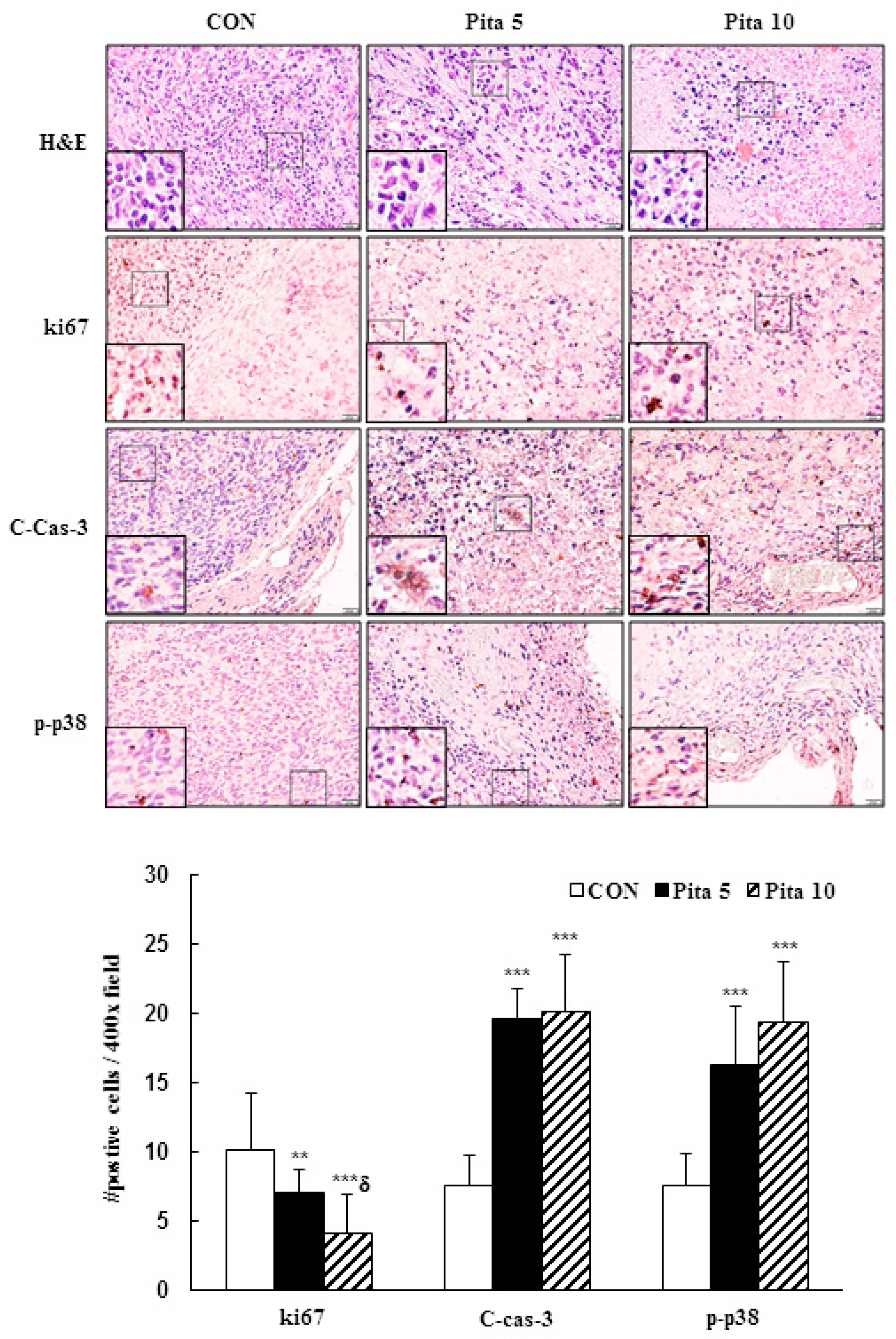
2.7. Pitavastatin Inhibits C-33 A Xenograft Tumor Growth
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture
4.3. Cell Cytotoxicity Assay
4.4. Colony Formation
4.5. DAPI Staining Assay
4.6. Apoptosis and Cell Cycle Analysis
4.7. Wound Healing and Migration Assay
4.8. Mitochondria Membrane Potential Measurement
4.9. Immunoblotting Analysis
4.10. Tumor Xenograft Mouse Model
4.11. Histology and Immunohistochemistry Analysis
4.12. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Basu, P.; Mittal, S.; Vale, D.B.; Kharaji, Y.C. Secondary prevention of cervical cancer. Best Pract. Res. Clin. Obstet. Gynaecol. 2018, 47, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Datta, N.R.; Stutz, E.; Liu, M.; Rogers, S.; Klingbiel, D.; Siebenhüner, A.; Singh, S.; Bodis, S. Concurrent chemoradiotherapy vs. radiotherapy alone in locally advanced cervix cancer: A systematic review and meta-analysis. Gynecol. Oncol. 2017, 145, 374–385. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Wang, D.; Li, H.; Wu, C.; Zhang, F.; Lin, Z.; Yao, T. Survival, treatment pattern, and treatment outcome in patients with cervical cancer metastatic to distant lymph nodes. Front. Oncol. 2022, 12, 952480. [Google Scholar] [CrossRef] [PubMed]
- Son, J.; Lin, H.Y.; Fu, S.; Biter, A.B.; Dumbrava, E.E.; Karp, D.D.; Naing, A.; Pant, S.; Piha-Paul, S.A.; Rodon, J. Predictors of Oncologic Outcome in Patients Receiving Phase I Investigational Therapy for Recurrent or Metastatic Cervical Cancer. J. Immunother. Precis. Oncol. 2023, 6, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Zaky, M.Y.; Fan, C.; Zhang, H.; Sun, X.-F. Unraveling the anticancer potential of statins: Mechanisms and clinical significance. Cancers 2023, 15, 4787. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.-B.; Hu, E.-D.; Fu, R.-Q. Statin use and cancer incidence in patients with type 2 diabetes mellitus: A network meta-analysis. Gastroenterol. Res. Pract. 2018, 2018, 8620682. [Google Scholar] [CrossRef]
- Almeida-Nunes, D.L.; Silvestre, R.; Dinis-Oliveira, R.J.; Ricardo, S. Enhancing Immunotherapy in Ovarian Cancer: The Emerging Role of Metformin and Statins. Int. J. Mol. Sci. 2023, 25, 323. [Google Scholar] [CrossRef]
- Gbelcová, H.; Rimpelová, S.; Jariabková, A.; Macášek, P.; Priščáková, P.; Ruml, T.; Šáchová, J.; Kubovčiak, J.; Kolář, M.; Vítek, L. Highly variable biological effects of statins on cancer, non-cancer, and stem cells in vitro. Sci. Rep. 2024, 14, 11830. [Google Scholar] [CrossRef]
- Duggan, S.T. Pitavastatin: A review of its use in the management of hypercholesterolaemia or mixed dyslipidaemia. Drugs 2012, 72, 565–584. [Google Scholar] [CrossRef]
- Yee, L.L.; Wright, E.A. Pitavastatin calcium: Clinical review of a new antihyperlipidemic medication. Clin. Ther. 2011, 33, 1023–1042. [Google Scholar] [CrossRef]
- Tang, W.-J.; Xu, D.; Liang, M.-X.; Wo, G.-Q.; Chen, W.-Q.; Tang, J.-H.; Zhang, W. Pitavastatin induces autophagy-dependent ferroptosis in MDA-MB-231 cells via the mevalonate pathway. Heliyon 2024, 10, e27084. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Tilija Pun, N.; Jang, W.J.; Bae, J.W.; Jeong, C.H. Pitavastatin induces apoptosis in oral squamous cell carcinoma through activation of FOXO3a. J. Cell. Mol. Med. 2020, 24, 7055–7066. [Google Scholar] [CrossRef] [PubMed]
- You, H.-Y.; Zhang, W.-J.; Xie, X.-M.; Zheng, Z.-H.; Zhu, H.-L.; Jiang, F.-Z. Pitavastatin suppressed liver cancer cells in vitro and in vivo. OncoTargets Ther. 2016, 9, 5383–5388. [Google Scholar]
- Hu, T.; Shen, H.; Huang, H.; Yang, Z.; Zhou, Y.; Zhao, G. Cholesterol-lowering drug pitavastatin targets lung cancer and angiogenesis via suppressing prenylation-dependent Ras/Raf/MEK and PI3K/Akt/mTOR signaling. Anti-Cancer Drugs 2020, 31, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-Y.; Zheng, S.-H.; Yang, W.-G.; Yang, C.; Yuan, W.-T. Targeting colon cancer stem cells with novel blood cholesterol drug pitavastatin. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 1226–1233. [Google Scholar] [PubMed]
- Tripathi, S.; Gupta, E.; Galande, S. Statins as anti-tumor agents: A paradigm for repurposed drugs. Cancer Rep. 2024, 7, e2078. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, C.M.; Singh, A.T. Apoptosis: A target for anticancer therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Sun, M.M.; Zhang, G.G.; Yang, J.; Chen, K.S.; Xu, W.W.; Li, B. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduct. Target. Ther. 2021, 6, 425. [Google Scholar] [CrossRef]
- Engelman, J.A. Targeting PI3K signalling in cancer: Opportunities, challenges and limitations. Nat. Rev. Cancer 2009, 9, 550–562. [Google Scholar] [CrossRef]
- Guo, Y.J.; Pan, W.W.; Liu, S.B.; Shen, Z.F.; Xu, Y.; Hu, L.L. ERK/MAPK signalling pathway and tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef] [PubMed]
- Braicu, C.; Buse, M.; Busuioc, C.; Drula, R.; Gulei, D.; Raduly, L.; Rusu, A.; Irimie, A.; Atanasov, A.G.; Slaby, O. A comprehensive review on MAPK: A promising therapeutic target in cancer. Cancers 2019, 11, 1618. [Google Scholar] [CrossRef] [PubMed]
- Nitulescu, G.M.; Van De Venter, M.; Nitulescu, G.; Ungurianu, A.; Juzenas, P.; Peng, Q.; Olaru, O.T.; Grădinaru, D.; Tsatsakis, A.; Tsoukalas, D. The Akt pathway in oncology therapy and beyond. Int. J. Oncol. 2018, 53, 2319–2331. [Google Scholar] [CrossRef] [PubMed]
- Sinkala, M.; Nkhoma, P.; Mulder, N.; Martin, D.P. Integrated molecular characterisation of the MAPK pathways in human cancers reveals pharmacologically vulnerable mutations and gene dependencies. Commun. Biol. 2021, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Park, N.J.-Y.; Le, T.M.; Lee, E.; Lee, D.; Nguyen, H.D.T.; Cho, J.; Park, J.-Y.; Han, H.S.; Chong, G.O. Immune Pathway And Gene Database (IMPAGT) revealed the immune dysregulation dynamics and overactivation of the PI3K/Akt pathway in tumor buddings of cervical cancer. Curr. Issues Mol. Biol. 2022, 44, 5139–5152. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y. KEGG Mapper for inferring cellular functions from protein sequences. Protein Sci. 2020, 29, 28–35. [Google Scholar] [CrossRef]
- Chen, Y.-H.; Wu, J.-X.; Yang, S.-F.; Chen, M.-L.; Chen, T.-H.; Hsiao, Y.-H. Metformin potentiates the anticancer effect of everolimus on cervical cancer in vitro and in vivo. Cancers 2021, 13, 4612. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-H.; Wu, J.-X.; Yang, S.-F.; Hsiao, Y.-H. Synergistic combination of luteolin and asiatic acid on cervical cancer in vitro and in vivo. Cancers 2023, 15, 548. [Google Scholar] [CrossRef]
- Lee, C.Y.; Chen, P.N.; Kao, S.H.; Wu, H.H.; Hsiao, Y.H.; Huang, T.Y.; Wang, P.H.; Yang, S.F. Deoxyshikonin triggers apoptosis in cervical cancer cells through p38 MAPK-mediated caspase activation. Environ. Toxicol. 2024. [Google Scholar] [CrossRef]
- Kharouba, M.; El-Kamel, A.; Mehanna, R.; Thabet, E.; Heikal, L. Pitavastatin-loaded bilosomes for oral treatment of hepatocellular carcinoma: A repurposing approach. Drug Deliv. 2022, 29, 2925–2944. [Google Scholar] [CrossRef]
- Chen, Y.-H.; Chen, Y.-C.; Lin, C.-C.; Hsieh, Y.-P.; Hsu, C.-S.; Hsieh, M.-C. Synergistic anticancer effects of gemcitabine with pitavastatin on pancreatic cancer cell line MIA PaCa-2 in vitro and in vivo. Cancer Manag. Res. 2020, 12, 4645–4665. [Google Scholar] [CrossRef]
- Chen, Y.H.; Huang, Y.C.; Yang, S.F.; Yen, H.H.; Tsai, H.D.; Hsieh, M.C.; Hsiao, Y.H. Pitavastatin and metformin synergistically activate apoptosis and autophagy in pancreatic cancer cells. Environ. Toxicol. 2021, 36, 1491–1503. [Google Scholar] [CrossRef] [PubMed]
- Jawad, M.J.; Ibrahim, S.; Kumar, M.; Burgert, C.; Li, W.-W.; Richardson, A. Identification of foods that affect the anti-cancer activity of pitavastatin in cells. Oncol. Lett. 2022, 23, 73. [Google Scholar] [CrossRef] [PubMed]
- Jawad, M.J.; Richardson, A. Ivermectin augments the anti-cancer activity of pitavastatin in ovarian cancer cells. Diseases 2023, 11, 49. [Google Scholar] [CrossRef] [PubMed]
- Piktel, D.; Nair, R.R.; Rellick, S.L.; Geldenhuys, W.J.; Martin, K.H.; Craig, M.D.; Gibson, L.F. Pitavastatin is anti-leukemic in a bone marrow microenvironment model of b-lineage acute lymphoblastic Leukemia. Cancers 2022, 14, 2681. [Google Scholar] [CrossRef] [PubMed]
- Piktel, D.; Moore, J.C.; Nesbit, S.; Sprowls, S.A.; Craig, M.D.; Rellick, S.L.; Nair, R.R.; Meadows, E.; Hollander, J.M.; Geldenhuys, W.J. Chemotherapeutic activity of pitavastatin in vincristine resistant B-cell acute lymphoblastic leukemia. Cancers 2023, 15, 707. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, M.I.; Abed, M.N.; Richardson, A. Inhibition of the mevalonate pathway augments the activity of pitavastatin against ovarian cancer cells. Sci. Rep. 2017, 7, 8090. [Google Scholar] [CrossRef] [PubMed]
- Hussein, B.H.; Kasabri, V.; Al-Hiari, Y.; Arabiyat, S.; Ikhmais, B.; Alalawi, S.; Al-Qirim, T. Selected statins as dual antiproliferative-antiinflammatory compounds. Asian Pac. J. Cancer Prev. APJCP 2022, 23, 4047–4062. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Mukthavaram, R.; Chao, Y.; Nomura, N.; Bharati, I.; Fogal, V.; Pastorino, S.; Teng, D.; Cong, X.; Pingle, S. In vitro and in vivo anticancer effects of mevalonate pathway modulation on human cancer cells. Br. J. Cancer 2014, 111, 1562–1571. [Google Scholar] [CrossRef]
- Otahal, A.; Aydemir, D.; Tomasich, E.; Minichsdorfer, C. Delineation of cell death mechanisms induced by synergistic effects of statins and erlotinib in non-small cell lung cancer cell (NSCLC) lines. Sci. Rep. 2020, 10, 959. [Google Scholar] [CrossRef]
- Dewidar, S.A.; Hamdy, O.; Soliman, M.M.; El Gayar, A.M.; El-Mesery, M. Enhanced therapeutic efficacy of doxorubicin/cyclophosphamide in combination with pitavastatin or simvastatin against breast cancer cells. Med. Oncol. 2023, 41, 7. [Google Scholar] [CrossRef] [PubMed]
- Crescencio, M.E.; Rodríguez, E.; Páez, A.; Masso, F.A.; Montaño, L.F.; López-Marure, R. Statins inhibit the proliferation and induce cell death of human papilloma virus positive and negative cervical cancer cells. Int. J. Biomed. Sci. IJBS 2009, 5, 411–420. [Google Scholar] [PubMed]
- Paškevičiūtė, M.; Petrikaitė, V. Differences of statin activity in 2D and 3D pancreatic cancer cell cultures. Drug Des. Dev. Ther. 2017, 11, 3273–3280. [Google Scholar] [CrossRef] [PubMed]
- Al-Qatati, A.; Aliwaini, S. Combined pitavastatin and dacarbazine treatment activates apoptosis and autophagy resulting in synergistic cytotoxicity in melanoma cells. Oncol. Lett. 2017, 14, 7993–7999. [Google Scholar] [CrossRef] [PubMed]
- Mertens, B.; Nogueira, T.; Stranska, R.; Naesens, L.; Andrei, G.; Snoeck, R. Cidofovir is active against human papillomavirus positive and negative head and neck and cervical tumor cells by causing DNA damage as one of its working mechanisms. Oncotarget 2016, 7, 47302. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Rieckmann, T.; Tribius, S.; Grob, T.J.; Meyer, F.; Busch, C.-J.; Petersen, C.; Dikomey, E.; Kriegs, M. HNSCC cell lines positive for HPV and p16 possess higher cellular radiosensitivity due to an impaired DSB repair capacity. Radiother. Oncol. 2013, 107, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, C.A.; Rodríguez, E.; Varela, E.; Zapata, E.; Paez, A.; Massó, F.A.; Montaño, L.F.; Lopez-Marure, R. Statin-induced inhibition of MCF-7 breast cancer cell proliferation is related to cell cycle arrest and apoptotic and necrotic cell death mediated by an enhanced oxidative stress. Cancer Investig. 2008, 26, 698–707. [Google Scholar] [CrossRef]
- Hacıseyitoğlu, A.Ö.; Doğan, T.Ç.; Dilsiz, S.A.; Canpınar, H.; Eken, A.; Bucurgat, Ü.Ü. Pitavastatin induces caspase-mediated apoptotic death through oxidative stress and DNA damage in combined with cisplatin in human cervical cancer cell line. J. Appl. Toxicol. 2024, 44, 623–640. [Google Scholar] [CrossRef]
- Zhu, Y.Y.; Huang, H.Y.; Wu, Y.L. Anticancer and apoptotic activities of oleanolic acid are mediated through cell cycle arrest and disruption of mitochondrial membrane potential in HepG2 human hepatocellular carcinoma cells. Mol. Med. Rep. 2015, 12, 5012–5018. [Google Scholar] [CrossRef]
- Zhang, J.; Feng, Z.; Wang, C.; Zhou, H.; Liu, W.; Kanchana, K.; Dai, X.; Zou, P.; Gu, J.; Cai, L. Curcumin derivative WZ35 efficiently suppresses colon cancer progression through inducing ROS production and ER stress-dependent apoptosis. Am. J. Cancer Res. 2017, 7, 275. [Google Scholar]
- Li, J.; Li, T.-x.; Ma, Y.; Zhang, Y.; Li, D.-y.; Xu, H.-r. Bursopentin (BP5) induces G1 phase cell cycle arrest and endoplasmic reticulum stress/mitochondria-mediated caspase-dependent apoptosis in human colon cancer HCT116 cells. Cancer Cell Int. 2019, 19, 130. [Google Scholar] [CrossRef] [PubMed]
- King, D.; Yeomanson, D.; Bryant, H.E. PI3King the lock: Targeting the PI3K/Akt/mTOR pathway as a novel therapeutic strategy in neuroblastoma. J. Pediatr. Hematol./Oncol. 2015, 37, 245–251. [Google Scholar] [CrossRef]
- Alzahrani, A.S. PI3K/Akt/mTOR inhibitors in cancer: At the bench and bedside. In Seminars in Cancer Biology; Elsevier: Amsterdam, The Netherlands, 2019; pp. 125–132. [Google Scholar]
- Chen, Y.-H.; Yang, S.-F.; Yang, C.-K.; Tsai, H.-D.; Chen, T.-H.; Chou, M.-C.; Hsiao, Y.-H. Metformin induces apoptosis and inhibits migration by activating the AMPK/p53 axis and suppressing PI3K/AKT signaling in human cervical cancer cells. Mol. Med. Rep. 2021, 23, 88. [Google Scholar] [CrossRef]
- Chen, Y.-H.; Wu, J.-X.; Yang, S.-F.; Yang, C.-K.; Chen, T.-H.; Hsiao, Y.-H. Anticancer effects and molecular mechanisms of apigenin in cervical cancer cells. Cancers 2022, 14, 1824. [Google Scholar] [CrossRef] [PubMed]
- Elbaset, M.A.; Mohamed, B.M.; Moustafa, P.E.; Esatbeyoglu, T.; Afifi, S.M.; Hessin, A.F.; Abdelrahman, S.S.; Fayed, H.M. Renoprotective Effect of Pitavastatin against TAA-Induced Renal Injury: Involvement of the miR-93/PTEN/AKT/mTOR Pathway. Adv. Pharmacol. Pharm. Sci. 2024, 2024, 6681873. [Google Scholar] [CrossRef] [PubMed]
- Elbaset, M.A.; Mohamed, B.M.; Hessin, A.; Abd El-Rahman, S.S.; Esatbeyoglu, T.; Afifi, S.M.; Fayed, H.M. Nrf2/HO-1, NF-κB and PI3K/Akt signalling pathways decipher the therapeutic mechanism of pitavastatin in early phase liver fibrosis in rats. J. Cell. Mol. Med. 2024, 28, e18116. [Google Scholar] [CrossRef]
- Kobayashi, N.; Takeshima, H.; Fukushima, H.; Koguchi, W.; Mamada, Y.; Hirata, H.; Machida, Y.; Shinoda, M.; Suzuki, N.; Yokotsuka, F. Cardioprotective effects of pitavastatin on cardiac performance and remodeling in failing rat hearts. Am. J. Hypertens. 2009, 22, 176–182. [Google Scholar] [CrossRef]
- Malik, S.; Sharma, A.K.; Bharti, S.; Nepal, S.; Bhatia, J.; Nag, T.C.; Narang, R.; Arya, D.S. In Vivo Cardioprotection by Pitavastatin From Ischemic-reperfusion Injury Through Suppression of IKK/NF-κB and Upregulation of pAkt–e-NOS. J. Cardiovasc. Pharmacol. 2011, 58, 199–206. [Google Scholar] [CrossRef]
- Nagaoka, K.; Matoba, T.; Mao, Y.; Nakano, Y.; Ikeda, G.; Egusa, S.; Tokutome, M.; Nagahama, R.; Nakano, K.; Sunagawa, K. A new therapeutic modality for acute myocardial infarction: Nanoparticle-mediated delivery of pitavastatin induces cardioprotection from ischemia-reperfusion injury via activation of PI3K/Akt pathway and anti-inflammation in a rat model. PLoS ONE 2015, 10, e0132451. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.-F.; Gao, Y.-X.; Lian, J.-J.; Guo, D.-D.; Liu, T.-T.; Xie, Y.-F.; Wang, L.; Yang, H.-J.; Wang, M.; Feng, Z.-W. Anti-inflammatory effects of pitavastatin in interleukin-1β-induced SW982 human synovial cells. Int. Immunopharmacol. 2017, 50, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Tomar, A.; Puthanmadhom Narayanan, S.; Nag, T.C.; Arya, D.S.; Bhatia, J. Pitavastatin attenuates cisplatin-induced renal injury by targeting MAPK and apoptotic pathways. J. Pharm. Pharmacol. 2019, 71, 1072–1081. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, A.; Takemura, G.; Mikami, A.; Aoyama, T.; Ohno, T.; Maruyama, R.; Nakagawa, M.; Minatoguchi, S.; Fujiwara, H. A therapeutic dose of the lipophilic statin pitavastatin enhances oxidant-induced apoptosis in human vascular smooth muscle cells. J. Cardiovasc. Pharmacol. 2006, 48, 160–165. [Google Scholar]
- Kim, K.-I.; Kim, S.-M.; Lee, Y.-Y.; Lee, Y.; Kim, C.-D.; Yoon, T.-J. Pitavastatin Induces Apoptosis of Cutaneous Squamous Cell Carcinoma Cells through Geranylgeranyl Pyrophosphate-Dependent c-Jun N-Terminal Kinase Activation. Ann. Dermatol. 2023, 35, 116. [Google Scholar] [CrossRef] [PubMed]
- Pater, M.M.; Pater, A. Human papillomavirus types 16 and 18 sequences in carcinoma cell lines of the cervix. Virology 1985, 145, 313–318. [Google Scholar] [CrossRef]
- Donat, U.; Rother, J.; Schäfer, S.; Hess, M.; Härtl, B.; Kober, C.; Langbein-Laugwitz, J.; Stritzker, J.; Chen, N.G.; Aguilar, R.J. Characterization of metastasis formation and virotherapy in the human C33A cervical cancer model. PLoS ONE 2014, 9, e98533. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.-H.; Wu, J.-X.; Yang, S.-F.; Wu, Y.-C.; Hsiao, Y.-H. Molecular Mechanisms Underlying the Anticancer Properties of Pitavastatin against Cervical Cancer Cells. Int. J. Mol. Sci. 2024, 25, 7915. https://doi.org/10.3390/ijms25147915
Chen Y-H, Wu J-X, Yang S-F, Wu Y-C, Hsiao Y-H. Molecular Mechanisms Underlying the Anticancer Properties of Pitavastatin against Cervical Cancer Cells. International Journal of Molecular Sciences. 2024; 25(14):7915. https://doi.org/10.3390/ijms25147915
Chicago/Turabian StyleChen, Ya-Hui, Jyun-Xue Wu, Shun-Fa Yang, Yun-Chia Wu, and Yi-Hsuan Hsiao. 2024. "Molecular Mechanisms Underlying the Anticancer Properties of Pitavastatin against Cervical Cancer Cells" International Journal of Molecular Sciences 25, no. 14: 7915. https://doi.org/10.3390/ijms25147915