Abstract
Juvenile polyposis syndrome (JPS) is an inherited autosomal dominant condition that predisposes to the development of juvenile polyps throughout the gastrointestinal (GI) tract, and it poses an increased risk of GI malignancy. Germline causative variants were identified in the SMAD4 gene in a subset (20%) of JPS cases. Most SMAD4 germline genetic variants published to date are missense, nonsense, and frameshift mutations. SMAD4 germline alterations predicted to result in aberrant splicing have rarely been reported. Here, we report two unrelated Italian families harboring two different SMAD4 intronic variants, c.424+5G>A and c.425-9A>G, which are clinically associated with colorectal cancer and/or juvenile GI polyps. In silico prediction analysis, in vitro minigene assays, and RT-PCR showed that the identified variants lead to aberrant SMAD4 splicing via the exonization of intronic nucleotides, resulting in a premature stop codon. This is expected to cause the production of a truncated protein. This study expands the landscape of SMAD4 germline genetic variants associated with GI polyposis and/or cancer. Moreover, it emphasizes the importance of the functional characterization of SMAD4 splicing variants through RNA analysis, which can provide new insights into genetic disease variant interpretation, enabling tailored genetic counseling, management, and surveillance of patients with GI polyposis and/or cancer.
1. Introduction
Juvenile polyposis syndrome (JPS, OMIM 174900) is a rare autosomal dominant inherited cancer predisposition syndrome, with an estimated incidence varying from 1:16,000 to 1:100,000 [1]. It is characterized by the occurrence of multiple juvenile polyps in the upper and lower gastrointestinal (GI) tract, with a 9–50% increased lifetime risk of developing colorectal (CRC) and gastric cancer (GC) [2]. The diagnosis of JPS is suspected when one or more of the following clinical criteria are met: (i) at least five juvenile polyps in the colon or rectum; (ii) juvenile polyps throughout the GI tract; and (iii) any number of juvenile polyps in an individual with a family history of JPS [3]. JPS polyps vary in size, shape (sessile to pedunculated lesions), and number (from 1 to more than 100). Histologically, juvenile polyps are characterized by cystically dilated crypts with mucus-filled glands, prominent lamina propria, and inflammatory cell infiltration [4].
Germline loss-of-function alterations involving one of two tumor suppressor genes playing a pivotal role in the transforming growth factor-β and bone morphogenetic protein (TGF-β/BMP) signal transduction pathway, namely bone morphogenetic protein receptor type 1A (BMPR1A) and SMAD family member 4 (SMAD4), are known to be a molecular pathogenic mechanism in JPS [5].
Misregulation of the TGF-β/BMP signaling pathway plays a key role in carcinogenesis since it is involved in the regulation of different cellular mechanisms, including cell cycle progression, cell differentiation, cell death, and cell migration [6].
Overall, germline causative variants in SMAD4 and BMPR1A account for about 45–55% of cases of individuals with one or more clinical criteria of JPS, suggesting that genetic variants in other genes may contribute to JPS etiology.
JPS is characterized by a great variability of clinical manifestations in terms of number, location, and histology of GI polyps, extra-intestinal manifestations, and age at onset [7,8]. Moreover, JPS has incomplete penetrance, and approximately 20–60% of patients with JPS have no family history [5,9,10].
The vast majority of germline alterations affecting the SMAD4 gene consist of missense, nonsense, or frameshift variants, while germline SMAD4 large genomic deletions or SMAD4 alterations predicted to result in aberrant splicing have rarely been reported [11].
Germline characterization of JPS-related predisposing gene variants is extremely important for genetic counseling, clinical management, and tailored surveillance. In this study, we clinically and molecularly characterized two aberrant splicing variants, c.424+5G>A and c.425-9A>G, which were identified in two unrelated Italian families. Using various mRNA analysis approaches, we provide evidence of the pathogenicity of the identified SMAD4 splicing variant.
Moreover, we expanded our investigation by performing a literature review to explore all molecularly characterized SMAD4 aberrant splicing variants associated with the JPS clinical phenotype and cancer susceptibility.
2. Results
2.1. Clinical History and Genetic Findings
This study analyzes two unrelated non-consanguineous Italian pedigrees involving seven patients diagnosed with polyposis and CRC. In Family 1, the index case (Figure 1A, III:3) was a man who was diagnosed with and died of CRC at 62 years of age. The personal and familial clinical history of the index case was retrieved from our unit of medical genetics when the patient was already deceased.
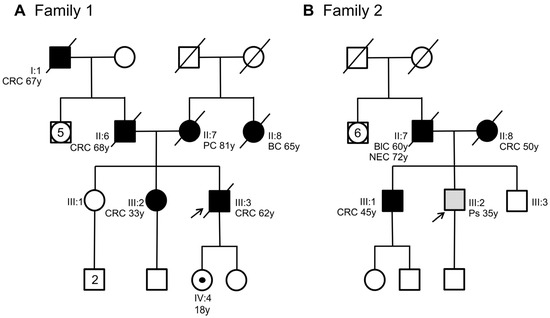
Figure 1.
Pedigree of the two families involved in this study. (A) Family 1 with members carrying the SMAD4 c.424+5G>A variant. (B) Family 2 with members carrying the SMAD4 c.425-9A>G variant. Squares indicate men and circles women. The arrow indicates the index case. Unfilled symbols indicate unaffected individuals. A central dot in a symbol denotes an asymptomatic variant carrier. Slashed symbols denote dead individuals. Black-filled symbols denote individuals with cancer, while grey-filled symbols correspond to patients with polyps. The following clinical manifestations are noted below each filled symbol: (CRC = colorectal cancer; PC = pancreatic cancer; BC = breast cancer; BlC = bladder cancer; NEC = neuroendocrine cancer; Ps = polyps), age at diagnosis (y = years).
The patient had a positive family history of cancer. His sister (Figure 1A, III:2), his father (Figure 1A, II:6), and his paternal grandfather (Figure 1A, I:1) developed CRC at 33, 68, and 67 years of age, respectively. The sister underwent genetic analyses of the MUTYH, APC, MLH1, MSH2, and MSH6 genes at another medical genetics center. This analysis did not reveal any germline alteration affecting one of these genes. Moreover, his mother (Figure 1A, II:7) died of pancreatic cancer at 81 years of age, and his maternal aunt died of breast cancer at 65 years of age. Considering the personal clinical history of CRC of the index case and his family history, including three relatives with CRC, the suspicion of hereditary CRC syndrome was raised. Next-generation sequencing (NGS) analysis using a panel of 25 hereditary cancer-related genes performed on the index case’s DNA identified a heterozygous variant in SMAD4 intron 2 (NM_005359.6: c.424+5G>A) (Figure 2A), which was confirmed by Sanger sequencing (Figure 2B).
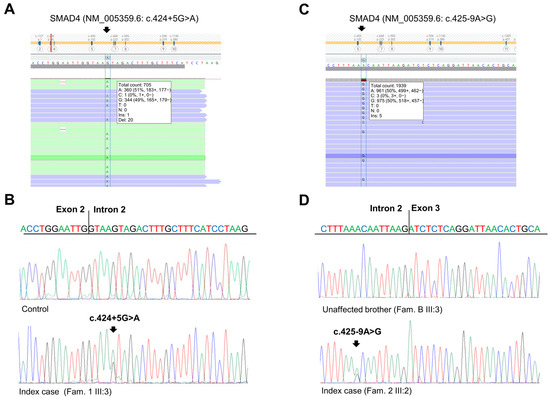
Figure 2.
Identification of SMAD4 germline variants. (A) Next-generation sequencing results showing the SMAD4 c.424+5G>A variant in the index case of Family 1. (B) Sequencing electropherograms of genomic DNA from a healthy control individual and the index case of Family 1, confirming the SMAD4 c.424+5G>A substitution. (C) Next-generation sequencing results showing the SMAD4 c.425-9A>G variant in the index case of Family 2. (D) Sequencing electropherograms of genomic DNA from an unaffected member and the index case of Family 2, confirming the SMAD4 c.425-9A>G substitution.
This variant was also found in the index case’s daughter who was 18 years old. The patient was immediately referred for clinical surveillance of the organs (stomach and colon) at risk for cancer (Figure 1A, IV:4). The other family members did not agree to undergo SMAD4 genetic analysis. To date, no other family members have undergone genetic testing.
In Family 2, the index case (Figure 1B, III:2) was a 35-year-old man with a pedunculated hamartomatous polyp and two small hyperplastic polyps in the large intestine. The patient had a positive family history for cancer and polyposis. His brother (Figure 1B, III:1) developed CRC at 45 years of age, while his father (Figure 1B, II:7) developed bladder cancer at 60 years and was diagnosed with and died of neuroendocrine cancer at 72 years of age. Moreover, the index case’s mother (Figure 1B, II:8) developed CRC and numerous polyps at 50 years of age. Histologically, these polyps were classified as likely hamartomatous, juvenile, and adenomatous, with a tubulovillous aspect and moderate or severe dysplasia. Interestingly, the index case’s mother carried a pathogenic variant in APC exon 15 (NM_00038.5: c.3336_3340del, p.Asn1113Serfs*4), which was previously identified by a different genetic diagnostic laboratory. This APC pathogenic variant was not detected in the index case nor in his CRC-affected brother (Figure 1B, III:2, III:1).
Considering the negative results of the APC genetic testing and the clinical manifestation in the index case and his brother, the clinical suspicion of an additional concurrent hereditary colon cancer predisposition syndrome in the Family 2 was advanced.
For these reasons, we expanded the genetic testing of the index case’s DNA by using a panel of 25 hereditary cancer-related genes. This NGS analysis revealed a heterozygous variant in SMAD4 intron 2 (NM_005359.6: c.425-9A>G) (Figure 2C), which was confirmed by Sanger sequencing (Figure 2D). This variant was also identified in the index case’s mother (Figure 1B, II:8) and CRC-affected brother (Figure 1B, III:1) but not in his healthy brother (Figure 1B, III:3).
The two identified SMAD4 variants were found to be rare in global population databases (1000 Genome, dbSNP, gnomAD, NHLBI ESP) [12,13,14,15]. Specifically, the SMAD4 c.425+5G>A variant was reported to have a global minor allele frequency (MAF) of less than 0.01, while the SMAD4 c.425-9A>G variant was not listed in these databases. In silico analysis using four splice site prediction algorithms (Splice Site Finder (SSF), MaxEntScan (MES), Splice Site Prediction by Neural Network (NNS), and Gene Splicer (GS)) integrated into Alamut VisualPlus version 1.7.2 (Sophia Genetics SAS; Bidart, France) revealed that the SMAD4 c.424+5G>A and c.425-9A>G variants may result in a splicing defect due to the abrogation of the canonical splice acceptor and donor sites at positions c.424 and c.425 of the SMAD4 gene, respectively (Figure S1A,B).
2.2. Analysis of mRNA by In Vitro Minigene Assay
To determine the effect of the SMAD4 c.424+5G>A and c.425-9A>G substitutions, we performed minigene assays using pSPL3 plasmid constructs (Figure 3A).
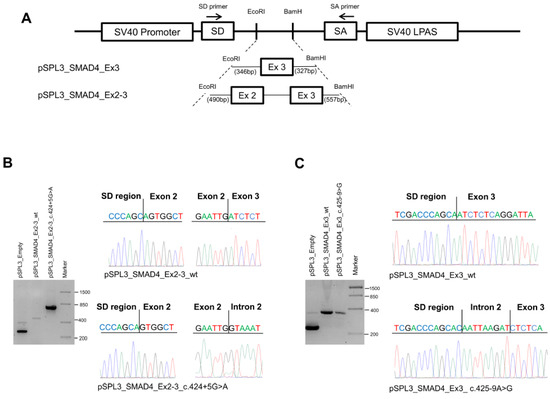
Figure 3.
Characterization of SMAD4 variants. (A) Schematic representation of the pSPL3 constructs and localization of the primers (indicated as arrows) used for RT-PCR experiments. (B) Agarose gel electrophoresis showing the RT-PCR products obtained in HEK-293 cells transfected with the pSPL3 empty vector (265 bp) or the pSPL3_SMAD4_Ex2-3_wt (467 bp) or pSPL3_SMAD4_Ex2-3_c.424+5G>A (about 800 bp) constructs (left). Sequencing electropherograms of the RT-PCR products originated from the wild-type and mutant cDNA splicing isoforms (right). (C) Agarose gel electrophoresis showing the RT-PCR products obtained from HEK-293 cells transfected with the pSPL3 empty vector or the pSPL3_SMAD4_Ex3_wt (295 bp) or pSPL3_SMAD4_Ex3_c.4245-9>G (302 bp) constructs (left). Sequencing electropherograms of the RT-PCR products originated from the wild-type and mutant cDNA splicing isoforms (right).
HEK-293 cells were transfected with the empty vector or with the pSPL3_SMAD4_Ex2-3_wt, pSPL3_SMAD4_Ex2-3_c.424+5G>A, pSPL3_SMAD4_Ex3_wt, or pSPL3_SMAD4_Ex3_c.425-9A>G constructs for 36 h; then, total RNA was extracted and amplified by RT-PCR using specific primers. Amplification of the pSPL3_SMAD4_Ex2-3_wt construct resulted in a 467 bp fragment containing exons 2 and 3, while amplification of the pSPL3_SMAD4_Ex2-3_c.424+5G>A construct produced a larger fragment of about 800 bp due to intron retention. The PCR products obtained were confirmed by Sanger sequencing (Figure 3B).
On the other hand, amplification of the pSPL3_SMAD4_Ex3_wt construct resulted in a 295 bp fragment containing exon 3, whereas amplification of the pSPL3_SMAD4_Ex3_c.425-9A>G construct produced a larger fragment (303 bp) due to the retention of the 8-nucleotide CAATTAAG sequence before the exon 3 splice donor, as confirmed by Sanger sequencing (Figure 3C).
2.3. Analysis of Patients’ Processed Transcripts
To corroborate the results obtained in the minigene assays, the effect of the SMAD4 c.424+5G>A and c.425-9A>G substitutions was further analyzed in the identified families. To this end, total RNA was isolated from peripheral blood of both carrier (Figure 1A, IV:4; Figure 1B, III:2) and non-carrier individuals. Then, specific SMAD4 exon transcripts were amplified by RT-PCR, and the obtained fragments were sequenced (Figure 4A,C).
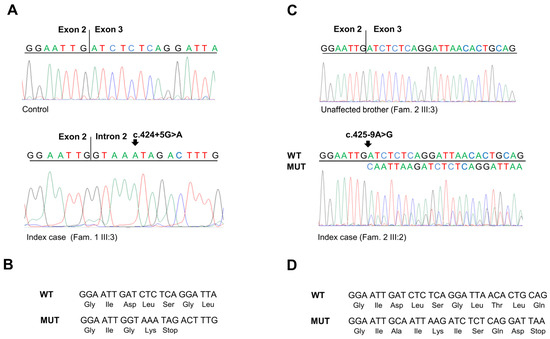
Figure 4.
(A) Sequencing electropherograms showing the SMAD4 RT-PCR products obtained from the mRNA extracted from peripheral blood of a healthy control individual and of the index case of Family 1 carrying the SMAD4 c.424+5 G>A variant. (B) Predicted translation products from the SMAD4 mutated allele with c.424+5 G>A variant (MUT) compared to the SMAD4 wild-type allele (WT). (C) Sequencing electropherograms showing the SMAD4 RT-PCR products obtained from the mRNA extracted from peripheral blood of an unaffected member and of the index case of Family 2 carrying the SMAD4 c.425-9A>G variant. (D) Predicted translation products from the SMAD4 mutated allele with c.425-9 A>G variant (MUT) compared to the SMAD4 wild-type allele (WT).
Amplification and sequencing of the SMAD4 exon 2–3 transcript confirmed that the c.424+5G>A substitution results in the exonization of the intronic sequence after the exon 2 splice donor site. This splicing alteration causes a frameshift in the coding region and the creation of a premature stop codon (p.Asp142Glyfs*2) (Figure 4A,B).
Similarly, amplification and sequencing of the SMAD4 exon 3 transcript confirmed that the c.425-9A>G substitution results in the insertion of eight nucleotides (CAATTAAG) before the canonical splice donor sequence in exon 3, making it a weak splicing signal. Moreover, this generates a new upstream splice donor site that produces a frameshift in the coding region and the creation of a premature stop codon (p.Asp142Alafs*7) (Figure 4C,D).
2.4. Literature Review of SMAD4 Splicing Variants
Next, we performed a literature review of SMAD4 splicing variants to explore the association between genetic alterations that affect SMAD4 splicing and the JPS clinical phenotype and cancer susceptibility. So far, 12 unique SMAD4 splicing variants, including those characterized in the present study, have been described in 24 patients with JPS or a personal and familial history of cancer (Figure S2, Table S1) [16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31]. This analysis revealed that a significant proportion of these variants (4/12, 33.3%) are located in SMAD4 intron 2. Among these, the SMAD4 c.424+5G>A variant, which we molecularly characterized in the present study, was mostly identified in patients with a personal and/or family history of cancer. The other SMAD4 variants located in intron 2 (i.e., c.425-9A>G, which was identified and molecularly characterized in the present study, c.424+1G>A, and c.425-6A>G) were detected in patients with a family history of JPS. A single SMAD4 splicing variant (c.667+3G>A) was identified in intron 4. It was found in a patient with breast cancer. Several other SMAD4 splicing variants (5/12, 41.6%) that occur in intron 8 were identified in patients with a personal and/or family history of JPS. Two of these variants (c.1139G>A and c.1139+1G>A) were previously characterized at the mRNA level and were reported to cause a reading frameshift [16,27]. The remaining SMAD4 splicing variants (c.1308+1G>A and c.1447+1G>A) identified so far were located in intron 9 and 10, and they were detected in patients with a personal and/or family history of JPS (Figure S2, Table S1).
3. Discussion
Genetic variants that disrupt normal patterns of mRNA splicing impair protein synthesis and/or alter protein structure, playing a determining role in disease etiology [32]. It has been reported that a relevant fraction of disease-causative genetic variants (15%–60%) have the potential to alter normal splicing mechanisms [33]. However, the majority of variants involving splice site genomic regions are classified as variants of uncertain significance (VUSs) due to the lack of functional studies on mRNA [34]. As such, investigating the molecular effect of genetic variants that affect the splicing process is crucial to gain insights into disease pathogenesis.
In this study, we expanded the landscape of SMAD4 splicing variants acting as causative events in JPS etiology and cancer. In particular, we demonstrated that both the c.424+5G>A and the c.425-9A>G SMAD4 variants, which were identified in two unrelated Italian families, induce aberrant pre-mRNA splicing by creating new cryptic splicing sites and promote the exonization of intronic sequences.
The SMAD4 gene is located at 18q21.1 and consists of 12 exons and 10 introns, encoding a protein with 552 amino acids and a molecular weight of 60 kDa [35]. The SMAD4 protein contains three domains: the Mad homology 1 (MH1) domain at the N-terminal (aa 14–138), the Mad homology 2 (MH2) domain at the C-terminal (aa 323–552), and the linker region located between the MH1 and MH2 domains (aa 139–322) [35]. SMAD4 has an important role in the TGF-β/SMAD signaling pathway and interacts with other transcription factors to regulate cell proliferation, growth, and differentiation [36].
Loss-of-function somatic alterations of the SMAD4 gene have been linked to a wide range of tumor types, including pancreatic, colorectal, gastric, breast, and thyroid cancer, and germline genetic alterations, which have been associated with JPS [37,38,39,40,41]. Overall, the majority of SMAD4 germline variants are frameshift, nonsense, and missense variants, while only a small proportion consists of single or multiexon deletions and splicing variants [11].
The literature review carried out in this study revealed that 12 unique SMAD4 splicing variants have been reported to date in 24 patients with cancer and/or JPS. Of these variants, only two have been characterized at the mRNA level. In this regard, it is worth noting that the characterization of the molecular effects of SMAD4 splicing variants may support the clinical diagnosis and management of patients with a personal and/or family history of cancer and/or polyposis.
The SMAD4 splicing variant c.424+5G>A is located five nucleotides downstream of the splice donor site of intron 2. It was previously described in patients with familial intestinal GC [19], familial pancreatic cancer [21], familial breast cancer [23], and familial colon polyposis/cancer [22]. The patient phenotypes described in these reports do not seem to fulfill the clinical diagnostic criteria of JPS. This variant was also identified in a patient diagnosed with one juvenile polyp and one tubular adenoma of the colon [20]. Due to the lack of sufficient evidence and functional studies investigating its effect at the mRNA level, the clinical assessment of the SMAD4 c.424+5G>A variant remained inconclusive. Recently, in an effort to identify genomic variants that cause splicing changes using transcriptome sequencing data available in public sequence repositories, Shiraishi et al. considered the SMAD4 c.424+5G>A variant as a putative pathogenic intron retention associated variant (ppIRAV) [42]. Using in vitro splicing analyses, the authors showed that this variant induces the retention of SMAD4 intron 2 [42].
In the present study, we identified the SMAD4 c.424+5G>A variant in a male patient with a personal and family history of CRC. This variant was also detected in his daughter, who reported no clinical manifestations at the time of clinical ascertainment, most likely due to her young age. We experimentally confirmed that the SMAD4 c.424+5G>A variant promotes the retention of SMAD4 intron 2 by performing an RT-PCR analysis of patient-derived RNA and an in vitro minigene assay. Moreover, we found that this splicing alteration leads to a reading frameshift and may result in the production of a truncated protein. According to the American College of Medical Genetics and Genomics and Association for Molecular Pathology (ACMG/AMP) guidelines for variant classification, the SMAD4 c.424+5G>A variant can be classified as pathogenic due to its low frequency in population databases—due to its deleterious effect on the splicing process as predicted by computational tools and its clinical and molecular characterization as performed in the present study—satisfying the following ACMG/AMP pathogenic criteria (PS1, PM2, PM4, PP3, PP4) [43]. Clinically, the phenotype of the patients harboring the SMAD4 c.424+5G>A variant, as reported in the scientific literature and the present study, does not seem to fulfill the clinical criteria of JPS. A possible explanation as to why this happens may be the lack of an endoscopic evaluation (i.e., small bowel video capsule endoscopy, esophagogastroduodenoscopy) of all the anatomical regions classically involved in JPS. As a result, it might not have been possible to ascertain whether these patients’ colon or stomach tumors originated from juvenile polyps. Anyway, it has recently been reported that not all of the patients carrying a pathogenic variant involving the SMAD4 or BMPR1A genes and submitted to an endoscopic investigation meet the clinical criteria for JPS diagnosis [9]. Current guidelines recommend surveillance programs for patients who fulfill JPS clinical criteria or harbor a germline disease-causative variant in the SMAD4 or BMPR1A genes [44,45]. However, the detection of a VUS in addition to a personal and/or family history of cancer adds to the complexity of clinical surveillance for these patients [46]. Indeed, based on the National Comprehensive Cancer Network guidelines, germline VUSs identified in the SMAD4 or BMPR1A genes in patients without clinical criteria of JPS should not be used to guide medical decision making [45].
The SMAD4 splicing variant c.425-9A>G is located nine nucleotides upstream of the splice acceptor site of intron 3. To our knowledge, this variant has never been described in the literature before. Based on the ClinVar database, it was previously reported in a patient with a SMAD4-related clinical phenotype. In the present study, this variant was identified in a family with clinical manifestations of JPS. In particular, it was detected in two affected brothers, one of which had a pedunculated hamartomatous polyp of the large intestine, while the other developed CRC at 45 years of age. The two brothers inherited the SMAD4 c.425-9A>G variant from their mother, who also harbored an APC pathogenic variant and developed CRC and numerous colon polyps with different histological classifications (hamartomatous, juvenile, and adenomatous). Thus, it can be hypothesized that the clinical phenotype of the affected mother characterized by a colon mixed polyposis was due to the additive effect of the APC and SMAD4 genetic variants, which are associated with the development of adenomatous colon polyps and juvenile colon polyps, respectively. To ascertain the impact of the SMAD4 c.425-9A>G variant on splicing, total RNA was isolated from the index case, and the SMAD4 transcript between exon 1 and 4 was amplified by RT-PCR and sequenced. This analysis revealed the retention of eight intronic nucleotides, with a consequent frameshift and the potential production of a truncated protein (p.Asp142Alafs*7). An in vitro minigene assay provided consistent results and confirmed that the SMAD4 c.425-9A>G variant affects the splicing process. According to ACMG/AMP criteria, our clinical and molecular characterization of this variant provides evidence of pathogenicity, satisfying the following ACMG/AMP pathogenic criteria (PS2, PM2, PM4, PP1, PP3, PP4).
The two SMAD4 splicing variants identified and characterized in the present study potentially affect the linker domain of the SMAD4 protein. The other two SMAD4 splicing variants that have been previously characterized at the mRNA level (i.e., c.1308+1G>A and c.1447+1G>A) are located in intron 9 and 10, and they potentially affect the MH2 protein domain [29,31]. Based on previous reports, these are located in the MH2 domain in most patients harboring SMAD4 pathogenic variants, while they occur in the MH1 and linker domains in a small proportion of patients [9]. However, investigations on genotype–phenotype associations did not identify a clear correlation between genetic variants affecting specific SMAD4 protein domains and clinical heterogeneity in SMAD4 mutation carriers [11]. In future studies, in-depth molecular characterization of SMAD4 splicing variants may enable a better understanding of the clinical phenotypes associated with these alterations.
4. Materials and Methods
4.1. Patient Recruitment
The patients underwent genetic testing after providing informed consent. Molecular testing carried out in this study was based on the routine clinical diagnostic assessment performed at our institute. Written informed consent to perform genetic testing and further studies was obtained from the patients using a form approved by the competent ethics committee, in line with the principles of the Declaration of Helsinki and any other applicable local ethical and legal requirements (protocol code No. 170, date of approval: 31 October 2016).
4.2. Genetic Analysis
Genomic DNA was extracted from peripheral blood with the MagCore® Genomic DNA Whole Blood Kit (Amerigo Scientific, New York, NY, USA) according to the manufacturer’s instructions. Then, genomic DNA samples of the index cases of Family 1 and Family 2 were subjected to genetic testing by NGS using a commercial Ion AmpliSeq™ BRCA Reflex—Hereditary Cancer Research Panel (Life Technologies, Carlsbad, CA, USA), which includes 25 hereditary cancer-related genes (APC, ATM, BARD1, BMPR1A, BRIP1, CDH1, CDK4, CDKN2A, CHEK2, EPCAM, MLH1, MRE11A, MSH2, MSH6, MUTYH, NBN, PALB2, PMS2, PTEN, RAD50, RAD51C, RAD51D, SMAD4, STK11, and TP53). The analysis was performed as previously described [47]. The clinical classification of the identified genetic variants was performed according to the ACMG/AMP guidelines [43]. The identified genetic variants putatively responsible for the clinical phenotype of the index cases were validated using Sanger sequencing. Segregation analysis was subsequently performed on family members willing to be tested. The genomic region of the SMAD4 gene was screened for genetic alterations using previously published primer sequences [48]. PCR products were isolated by agarose gel electrophoresis, purified, and sequenced using the BigDye Terminator Cycle sequencing kit (Thermo Fisher, Foster City, CA, USA). All samples were analyzed on a SeqStudio Genetic Analyzer (Thermo Fisher, Foster City, CA, USA). The global population frequency of the identified SMAD4 variants was retrieved from the 1000 Genome [12], dbSNP [13], gnomAD [49], and NHLBI Exome Sequencing Project [15] databases. Moreover, the HGMD [50], LitVar 2.0 [51], and Clinvar [52] databases were interrogated to assess the pathogenicity of the identified variant.
To evaluate the effect of the SMAD4 c.424+5G>A and c.425-9A>G variants on RNA splicing, four splice site prediction algorithms integrated into Alamut Visual version 2.15 (Sophia Genetics SAS; Bidart, France) were interrogated simultaneously: Splice Site Finder (SSF), MaxEntScan (MES), Splice Site Prediction by Neural Network (NNS), and Gene Splicer (GS). The default thresholds of each tool were used for the analysis. A variation of more than 10% in at least two algorithms was considered indicative of an effect on the splicing process.
4.3. RT-PCR and mRNA Analysis
Total RNA from peripheral blood was extracted with the QIAamp RNA Blood Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. One microgram of RNA was reverse-transcribed to cDNA using the Maxima H Minus First Strand cDNA Synthesis Kit (Life Technologies, Carlsbad, CA, USA). The flanking regions of the SMAD4 variant sites (NM_005359.6: c.424+5G>A and c.425-9A>G) were amplified using the DreamTaq mastermix (Life Technologies, Carlsbad, CA, USA) and the following primers (10 pmol each): SMAD4_Ex1-2_F AGGCTTCAGGTGGCTGGTC and SMAD4_Ex3-4_R CTTGATGGAGCATTACTCTGCAGT. PCR amplification was carried out at 95 °C for 30 s, followed by 35 cycles at 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s, with a final elongation at 72 °C for 5 min. PCR products were loaded onto 2% agarose gel in 0.5X TBE and visualized using SYBR Safe DNA Gel Stain (Life Technologies, Carlsbad, CA, USA). Sequencing and capillary electrophoresis were performed on a SeqStudio Genetic Analyzer.
4.4. Cell Line
The HEK-293 cell line was purchased from ATCC and cultured in DMEM high glucose (HG) without pyruvate (Life Technologies, Carlsbad, CA, USA) and with 10% FBS (Life Technologies, Carlsbad, CA, USA), 1% pyruvate (Life Technologies, Carlsbad, CA, USA), 1% NEAA (Life Technologies, Carlsbad, CA, USA), and 100 U/mL penicillin-streptomycin (Life Technologies, Carlsbad, CA, USA) in a 37 °C and 5% CO2 incubator. The cell line was tested to be mycoplasma-free using the Venor®GeM Advance kit (Minerva Biolabs, Berlin, Germany) multiple times throughout this study.
4.5. Plasmid Constructs
The pSPL3_SMAD4_Ex2-3_wt and pSPL3_SMAD4_Ex2-3_c.424+5G>A constructs were obtained by amplifying fragments containing SMAD4 exon 2-3 (NM_005359.6), flanked by upstream (490 nt) and downstream (557 nt) intronic sequences, from the wild-type or mutant allele. Amplification was carried out using the following primers: Cloning_SMAD4_Ex2-3_F_EcoRI CCCATGAATTCGCCCAAGCTGGAGTGCAGT and Cloning_SMAD4__Ex2-3_R_BamHI ACCGATGGATCCAGCTGTGATGGGCATATGCT. The pSPL3_SMAD4_Ex3_wt and pSPL3_SMAD4_Ex3_c.425-9A>G constructs were obtained by amplifying fragments containing SMAD4 exon 3 (NM_005359.6), flanked by upstream (346 nt) and downstream (327 nt) intronic sequences, from the wild-type or mutant allele. Amplification was carried out using the following primers: Cloning_SMAD4_Ex3_F_EcoRI CCCATGAATTCGGAAGATAGCCCGCGACT and Cloning_SMAD4_Ex3_R_BamHI ACCGATGGATCCTGGCTCTGTGAGATTTTCTTCA. The obtained fragments were cloned into the pSPL3 splicing vector linearized with EcoRI and BamHI. All constructs were confirmed by direct sequencing.
4.6. Minigene Assay
HEK-293 cells were transfected for 36 h using Lipofectamine 3000 (Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s instructions. Cells were harvested, and total RNA was extracted with the PureLink™ RNA Mini Kit (Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s instructions and used for RT-PCR to confirm splicing patterns. cDNA was synthesized as described above and used as a template for PCR amplification with the following vector-specific primers: SD6_ FW GTCTGAGTCACCTGGACAACC and SA2_ RV GATCTCAGTGGTATTTGTGAGC. PCR amplification was carried out with Phusion High-Fidelity DNA Polymerase at 98 °C for 30 s, followed by 35 cycles at 98 °C for 10 s, 52 °C for 10 s, and 72 °C for 15 s, with a final elongation at 72 °C for 5 min. PCR products were loaded onto 2% agarose gel in 0.5X TBE and visualized using SYBR Safe DNA Gel Stain. Sequencing and capillary electrophoresis were performed on the SeqStudio Genetic Analyzer.
4.7. Literature Review
This literature review was performed using curated databases (HGMD and LitVar 2.0) [50,51] that integrate data from PubMed, PubMed Central Open Access Subset, dbSNP [13], and ClinVar [52], and that enable an accurate search for genetic variants and related human inherited diseases. We searched for any previously identified SMAD4 splicing variant listed in the ClinVar database. We retrieved all the articles matching the search criteria from the aforementioned databases and collected relevant clinical information (i.e., specific SMAD4 splicing variant identified; patient genders; age at diagnosis; number of colorectal or gastric polyposis; presence of colorectal, gastric, or other cancer; and family history). Studies including patients without clinical information were excluded.
5. Conclusions
In the present work, we provide evidence for the clinical classification of two SMAD4 splicing variants as pathogenic by using various mRNA analysis approaches. One of these variants (c.424+5G>A) was identified in several patients with a personal and/or family history of cancer. The previous inconclusive classification of this variant may have limited the access to targeted surveillance programs for affected patients while also preventing an in-depth evaluation of the associated phenotypic features. Our study emphasizes that the functional characterization of SMAD4 splicing variants by means of an RNA analysis is a valuable approach in genetic disease variant interpretation. An accurate classification of SMAD4 variants based on clinical and molecular evidence is expected to improve the management of patients and their families, leading to more effective screening and/or clinical surveillance strategies. This in turn may help to better decipher the clinical phenotypes associated with these alterations. In conclusion, this study supports the potential of RNA analyses in the clinical classification of SMAD4 splicing variants in order to ensure tailored genetic counseling, management, and surveillance for patients with a suspected genetic predisposition to GI polyposis and/or cancer.
Supplementary Materials
The supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms25147939/s1.
Author Contributions
Conceptualization, V.D. and C.S.; data curation, P.S.; formal analysis, A.L.B., F.C., A.M. and A.F.G.; funding acquisition, C.F., P.S., M.L.S., V.G., V.D. and C.S.; investigation, G.F. and V.D.; methodology, G.F. and V.D.; software, C.F.; visualization, M.L.S. and K.D.M.; writing—original draft, G.F., V.D. and C.S.; writing—review and editing, G.F., V.G., V.D. and C.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Italian Ministry of Health “Ricerca Corrente 2024–2026” to C.F. and V.D.; by “Ricerca Corrente 2023–2025” to V.G.; by “Ricerca Corrente 2024–2026” to C.S.; by the “Starting Grant” SG-2019-12371540 to P.S.; by the Italian Association for Cancer Research (AIRC) IG-23794 to C.S.
Institutional Review Board Statement
This study was conducted in accordance with the guidelines of the Declaration of Helsinki and approved by the Institutional Ethics Committee of IRCCS Istituto Tumori “Giovanni Paolo II” (protocol code No. 170, date of approval: 31 October 2016).
Informed Consent Statement
Informed consent was obtained from all subjects involved in this study.
Data Availability Statement
The data presented in this study are available in this article.
Acknowledgments
We thank Francesco Paolo Jori for his helpful discussion during the preparation of this manuscript and for providing editorial assistance.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Latchford, A.R.; Neale, K.; Phillips, R.K.S.; Clark, S.K. Juvenile Polyposis Syndrome: A Study of Genotype, Phenotype, and Long-Term Outcome. Dis. Colon Rectum 2012, 55, 1038–1043. [Google Scholar] [CrossRef] [PubMed]
- Blatter, R.; Tschupp, B.; Aretz, S.; Bernstein, I.; Colas, C.; Evans, D.G.; Genuardi, M.; Hes, F.J.; Hüneburg, R.; Järvinen, H.; et al. Disease Expression in Juvenile Polyposis Syndrome: A Retrospective Survey on a Cohort of 221 European Patients and Comparison with a Literature-Derived Cohort of 473 SMAD4/BMPR1A Pathogenic Variant Carriers. Genet. Med. 2020, 22, 1524–1532. [Google Scholar] [CrossRef] [PubMed]
- Jass, J.R.; Williams, C.B.; Bussey, H.J.; Morson, B.C. Juvenile Polyposis—A Precancerous Condition. Histopathology 1988, 13, 619–630. [Google Scholar] [CrossRef] [PubMed]
- van Hattem, W.A.; Langeveld, D.; de Leng, W.W.J.; Morsink, F.H.; van Diest, P.J.; Iacobuzio-Donahue, C.A.; Giardiello, F.M.; Offerhaus, G.J.A.; Brosens, L.A.A. Histological Variations in Juvenile Polyp Phenotype Correlate with Genetic Defect Underlying Juvenile Polyposis. Am. J. Surg. Pathol. 2011, 35, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Chow, E.; Macrae, F. A Review of Juvenile Polyposis Syndrome. J. Gastroenterol. Hepatol. 2005, 20, 1634–1640. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Fan, T.; Xiao, C.; Tian, H.; Zheng, Y.; Li, C.; He, J. TGF-β Signaling in Health, Disease, and Therapeutics. Signal Transduct. Target. Ther. 2024, 9, 61. [Google Scholar] [CrossRef] [PubMed]
- Rosner, G.; Petel-Galil, Y.; Laish, I.; Levi, Z.; Kariv, R.; Strul, H.; Gilad, O.; Gluck, N. Adenomatous Polyposis Phenotype in BMPR1A and SMAD4 Variant Carriers. Clin. Transl. Gastroenterol. 2022, 13, e00527. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.H.; Li, J.; Zhao, Z.Y.; Xu, X.D.; Du, Y.Q.; Yan, H.L.; Liu, L.J.; Bai, C.G.; Zhang, W. Juvenile Polyposis Syndrome Might Be Misdiagnosed as Familial Adenomatous Polyposis: A Case Report and Literature Review. BMC Gastroenterol. 2020, 20, 167. [Google Scholar] [CrossRef]
- Jelsig, A.M.; van Overeem Hansen, T.; Gede, L.B.; Qvist, N.; Christensen, L.-L.; Lautrup, C.K.; Ljungmann, K.; Christensen, L.T.; Rønlund, K.; Tørring, P.M.; et al. Whole Genome Sequencing and Disease Pattern in Patients with Juvenile Polyposis Syndrome: A Nationwide Study. Fam. Cancer 2023, 22, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Shen, N.; Wang, X.; Lu, Y.; Xiao, F.; Xiao, J. Importance of Early Detection of Juvenile Polyposis Syndrome: A Case Report and Literature Review. Medicine 2020, 99, e23494. [Google Scholar] [CrossRef]
- Cao, K.; Plazzer, J.-P.; Macrae, F. SMAD4 Variants and Its Genotype-Phenotype Correlations to Juvenile Polyposis Syndrome. Hered. Cancer Clin. Pract. 2023, 21, 27. [Google Scholar] [CrossRef] [PubMed]
- 1000 Genomes Project Consortium; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A Global Reference for Human Genetic Variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef]
- Smigielski, E.M.; Sirotkin, K.; Ward, M.; Sherry, S.T. dbSNP: A Database of Single Nucleotide Polymorphisms. Nucleic Acids Res. 2000, 28, 352–355. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The Mutational Constraint Spectrum Quantified from Variation in 141,456 Humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Auer, P.L.; Reiner, A.P.; Wang, G.; Kang, H.M.; Abecasis, G.R.; Altshuler, D.; Bamshad, M.J.; Nickerson, D.A.; Tracy, R.P.; Rich, S.S.; et al. Guidelines for Large-Scale Sequence-Based Complex Trait Association Studies: Lessons Learned from the NHLBI Exome Sequencing Project. Am. J. Hum. Genet. 2016, 99, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Aretz, S.; Stienen, D.; Uhlhaas, S.; Stolte, M.; Entius, M.M.; Loff, S.; Back, W.; Kaufmann, A.; Keller, K.-M.; Blaas, S.H.; et al. High Proportion of Large Genomic Deletions and a Genotype Phenotype Update in 80 Unrelated Families with Juvenile Polyposis Syndrome. J. Med. Genet. 2007, 44, 702–709. [Google Scholar] [CrossRef] [PubMed]
- Calva-Cerqueira, D.; Chinnathambi, S.; Pechman, B.; Bair, J.; Larsen-Haidle, J.; Howe, J.R. The Rate of Germline Mutations and Large Deletions of SMAD4 and BMPR1A in Juvenile Polyposis. Clin. Genet. 2009, 75, 79–85. [Google Scholar] [CrossRef]
- Woodford-Richens, K.L.; Rowan, A.J.; Poulsom, R.; Bevan, S.; Salovaara, R.; Aaltonen, L.A.; Houlston, R.S.; Wright, N.A.; Tomlinson, I.P. Comprehensive Analysis of SMAD4 Mutations and Protein Expression in Juvenile Polyposis: Evidence for a Distinct Genetic Pathway and Polyp Morphology in SMAD4 Mutation Carriers. Am. J. Pathol. 2001, 159, 1293–1300. [Google Scholar] [CrossRef]
- Carvalho, J.; Oliveira, P.; Senz, J.; São José, C.; Hansford, S.; Teles, S.P.; Ferreira, M.; Corso, G.; Pinheiro, H.; Lemos, D.; et al. Redefinition of Familial Intestinal Gastric Cancer: Clinical and Genetic Perspectives. J. Med. Genet. 2021, 58, 1–11. [Google Scholar] [CrossRef]
- Jelsig, A.M.; Brusgaard, K.; Hansen, T.P.; Qvist, N.; Larsen, M.; Bojesen, A.; Nielsen, C.B.; Ousager, L.B. Germline Variants in Hamartomatous Polyposis Syndrome-Associated Genes from Patients with One or Few Hamartomatous Polyps. Scand. J. Gastroenterol. 2016, 51, 1118–1125. [Google Scholar] [CrossRef]
- Chaffee, K.G.; Oberg, A.L.; McWilliams, R.R.; Majithia, N.; Allen, B.A.; Kidd, J.; Singh, N.; Hartman, A.-R.; Wenstrup, R.J.; Petersen, G.M. Prevalence of Germline Mutations in Cancer Genes among Pancreatic Cancer Patients with Positive Family History. Genet. Med. 2018, 20, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Yurgelun, M.B.; Allen, B.; Kaldate, R.R.; Bowles, K.R.; Judkins, T.; Kaushik, P.; Roa, B.B.; Wenstrup, R.J.; Hartman, A.-R.; Syngal, S. Identification of a Variety of Mutations in Cancer Predisposition Genes in Patients With Suspected Lynch Syndrome. Gastroenterology 2015, 149, 604–613.e20. [Google Scholar] [CrossRef] [PubMed]
- Tung, N.; Battelli, C.; Allen, B.; Kaldate, R.; Bhatnagar, S.; Bowles, K.; Timms, K.; Garber, J.E.; Herold, C.; Ellisen, L.; et al. Frequency of Mutations in Individuals with Breast Cancer Referred for BRCA1 and BRCA2 Testing Using Next-Generation Sequencing with a 25-Gene Panel. Cancer 2015, 121, 25–33. [Google Scholar] [CrossRef]
- Tsaousis, G.N.; Papadopoulou, E.; Apessos, A.; Agiannitopoulos, K.; Pepe, G.; Kampouri, S.; Diamantopoulos, N.; Floros, T.; Iosifidou, R.; Katopodi, O.; et al. Analysis of Hereditary Cancer Syndromes by Using a Panel of Genes: Novel and Multiple Pathogenic Mutations. BMC Cancer 2019, 19, 535. [Google Scholar] [CrossRef] [PubMed]
- Tung, N.; Lin, N.U.; Kidd, J.; Allen, B.A.; Singh, N.; Wenstrup, R.J.; Hartman, A.-R.; Winer, E.P.; Garber, J.E. Frequency of Germline Mutations in 25 Cancer Susceptibility Genes in a Sequential Series of Patients With Breast Cancer. J. Clin. Oncol. 2016, 34, 1460–1468. [Google Scholar] [CrossRef] [PubMed]
- Mafficini, A.; Brosens, L.A.A.; Piredda, M.L.; Conti, C.; Mattiolo, P.; Turri, G.; Mastrosimini, M.G.; Cingarlini, S.; Crinò, S.F.; Fassan, M.; et al. Juvenile Polyposis Diagnosed with an Integrated Histological, Immunohistochemical and Molecular Approach Identifying New SMAD4 Pathogenic Variants. Fam. Cancer 2022, 21, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Micolonghi, C.; Piane, M.; Germani, A.; Sadeghi, S.; Libi, F.; Savio, C.; Fabiani, M.; Mancini, R.; Ranieri, D.; Pizzuti, A.; et al. A New SMAD4 Splice Site Variant in a Three-Generation Italian Family with Juvenile Polyposis Syndrome. Diagnostics 2022, 12, 2684. [Google Scholar] [CrossRef] [PubMed]
- Kager, L.M.; Meijer, S.L.; Bastiaansen, B.A.J. A Full Stomach. Gastroenterology 2014, 147, 974–976. [Google Scholar] [CrossRef] [PubMed]
- Wain, K.E.; Ellingson, M.S.; McDonald, J.; Gammon, A.; Roberts, M.; Pichurin, P.; Winship, I.; Riegert-Johnson, D.L.; Weitzel, J.N.; Lindor, N.M. Appreciating the Broad Clinical Features of SMAD4 Mutation Carriers: A Multicenter Chart Review. Genet. Med. 2014, 16, 588–593. [Google Scholar] [CrossRef]
- Pyatt, R.E.; Pilarski, R.; Prior, T.W. Mutation Screening in Juvenile Polyposis Syndrome. J. Mol. Diagn. 2006, 8, 84–88. [Google Scholar] [CrossRef][Green Version]
- Schwenter, F.; Ratjen, F.; Berk, T.; Gallinger, S.; Gryfe, R.; Gradinger, A.B.; Faughnan, M.E.; Durno, C.A. Juvenile Polyposis Syndrome, SMAD4 Mutations, and Hereditary Hemorrhagic Telangiectasia. J. Pediatr. Gastroenterol. Nutr. 2012, 54, 120–122. [Google Scholar] [CrossRef] [PubMed]
- Scotti, M.M.; Swanson, M.S. RNA Mis-Splicing in Disease. Nat. Rev. Genet. 2016, 17, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.-S.; Cooper, T.A. Splicing in Disease: Disruption of the Splicing Code and the Decoding Machinery. Nat. Rev. Genet. 2007, 8, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Federici, G.; Soddu, S. Variants of Uncertain Significance in the Era of High-Throughput Genome Sequencing: A Lesson from Breast and Ovary Cancers. J. Exp. Clin. Cancer Res. 2020, 39, 46. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Mishra, L.; Deng, C.-X. The Role of TGF-β/SMAD4 Signaling in Cancer. Int. J. Biol. Sci. 2018, 14, 111–123. [Google Scholar] [CrossRef]
- Yang, G.; Yang, X. Smad4-Mediated TGF-Beta Signaling in Tumorigenesis. Int. J. Biol. Sci. 2010, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Racu, M.-L.; Lebrun, L.; Schiavo, A.A.; Van Campenhout, C.; De Clercq, S.; Absil, L.; Minguijon Perez, E.; Maris, C.; Decaestecker, C.; Salmon, I.; et al. The Role of SMAD4 Inactivation in Epithelial–Mesenchymal Plasticity of Pancreatic Ductal Adenocarcinoma: The Missing Link? Cancers 2022, 14, 973. [Google Scholar] [CrossRef] [PubMed]
- Woodford-Richens, K.L.; Rowan, A.J.; Gorman, P.; Halford, S.; Bicknell, D.C.; Wasan, H.S.; Roylance, R.R.; Bodmer, W.F.; Tomlinson, I.P.M. SMAD4 Mutations in Colorectal Cancer Probably Occur before Chromosomal Instability, but after Divergence of the Microsatellite Instability Pathway. Proc. Natl. Acad. Sci. USA 2001, 98, 9719–9723. [Google Scholar] [CrossRef]
- Wang, L.-H.; Kim, S.-H.; Lee, J.H.; Choi, Y.-L.; Kim, Y.C.; Park, T.S.; Hong, Y.-C.; Wu, C.-F.; Shin, Y.K. Inactivation of SMAD4 Tumor Suppressor Gene During Gastric Carcinoma Progression. Clin. Cancer Res. 2007, 13, 102–110. [Google Scholar] [CrossRef]
- Liu, N.; Yu, C.; Shi, Y.; Jiang, J.; Liu, Y. SMAD4 Expression in Breast Ductal Carcinoma Correlates with Prognosis. Oncol. Lett. 2015, 10, 1709–1715. [Google Scholar] [CrossRef]
- D’Inzeo, S.; Nicolussi, A.; Ricci, A.; Mancini, P.; Porcellini, A.; Nardi, F.; Coppa, A. Role of Reduced Expression of SMAD4 in Papillary Thyroid Carcinoma. J. Mol. Endocrinol. 2010, 45, 229–244. [Google Scholar] [CrossRef]
- Shiraishi, Y.; Okada, A.; Chiba, K.; Kawachi, A.; Omori, I.; Mateos, R.N.; Iida, N.; Yamauchi, H.; Kosaki, K.; Yoshimi, A. Systematic Identification of Intron Retention Associated Variants from Massive Publicly Available Transcriptome Sequencing Data. Nat. Commun. 2022, 13, 5357. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, M.G.; British Society for Gastroenterology; Association of Coloproctology for Great Britain and Ireland. Guidance on Gastrointestinal Surveillance for Hereditary Non-Polyposis Colorectal Cancer, Familial Adenomatous Polypolis, Juvenile Polyposis, and Peutz-Jeghers Syndrome. Gut 2002, 51 (Suppl. 5), V21–V27. [Google Scholar] [CrossRef]
- Weiss, J.M.; Gupta, S.; Burke, C.A.; Axell, L.; Chen, L.-M.; Chung, D.C.; Clayback, K.M.; Dallas, S.; Felder, S.; Gbolahan, O.; et al. NCCN Guidelines® Insights: Genetic/Familial High-Risk Assessment: Colorectal, Version 1.2021. J. Natl. Compr. Cancer Netw. 2021, 19, 1122–1132. [Google Scholar] [CrossRef]
- Kelly, P.; Mahon, S.; Friend, P. When Germline Genetic Testing Results Are Unclear: Highlighting Variants of Uncertain Significance. J. Adv. Pract. Oncol. 2023, 14, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Disciglio, V.; Sanese, P.; Fasano, C.; Lotesoriere, C.; Valentini, A.M.; Forte, G.; Lepore Signorile, M.; De Marco, K.; Grossi, V.; Lolli, I.; et al. Identification and Somatic Characterization of the Germline PTEN Promoter Variant Rs34149102 in a Family with Gastrointestinal and Breast Tumors. Genes 2022, 13, 644. [Google Scholar] [CrossRef] [PubMed]
- Houlston, R.; Bevan, S.; Williams, A.; Young, J.; Dunlop, M.; Rozen, P.; Eng, C.; Markie, D.; Woodford-Richens, K.; Rodriguez-Bigas, M.A.; et al. Mutations in DPC4 (SMAD4) Cause Juvenile Polyposis Syndrome, but Only Account for a Minority of Cases. Hum. Mol. Genet. 1998, 7, 1907–1912. [Google Scholar] [CrossRef] [PubMed]
- Soussi, T.; Leroy, B.; Devir, M.; Rosenberg, S. High Prevalence of Cancer-Associated TP53 Variants in the gnomAD Database: A Word of Caution Concerning the Use of Variant Filtering. Hum. Mutat. 2019, 40, 516–524. [Google Scholar] [CrossRef]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Evans, K.; Hayden, M.; Heywood, S.; Hussain, M.; Phillips, A.D.; Cooper, D.N. The Human Gene Mutation Database: Towards a Comprehensive Repository of Inherited Mutation Data for Medical Research, Genetic Diagnosis and next-Generation Sequencing Studies. Hum. Genet. 2017, 136, 665–677. [Google Scholar] [CrossRef]
- Allot, A.; Wei, C.-H.; Phan, L.; Hefferon, T.; Landrum, M.; Rehm, H.L.; Lu, Z. Tracking Genetic Variants in the Biomedical Literature Using LitVar 2.0. Nat. Genet. 2023, 55, 901–903. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public Archive of Relationships among Sequence Variation and Human Phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).