

Three-Dimensional Quantitative Structure–Activity Relationship Study of Transient Receptor Potential Vanilloid 1 Channel Antagonists Reveals Potential for Drug Design Purposes

Abstract
1. Introduction
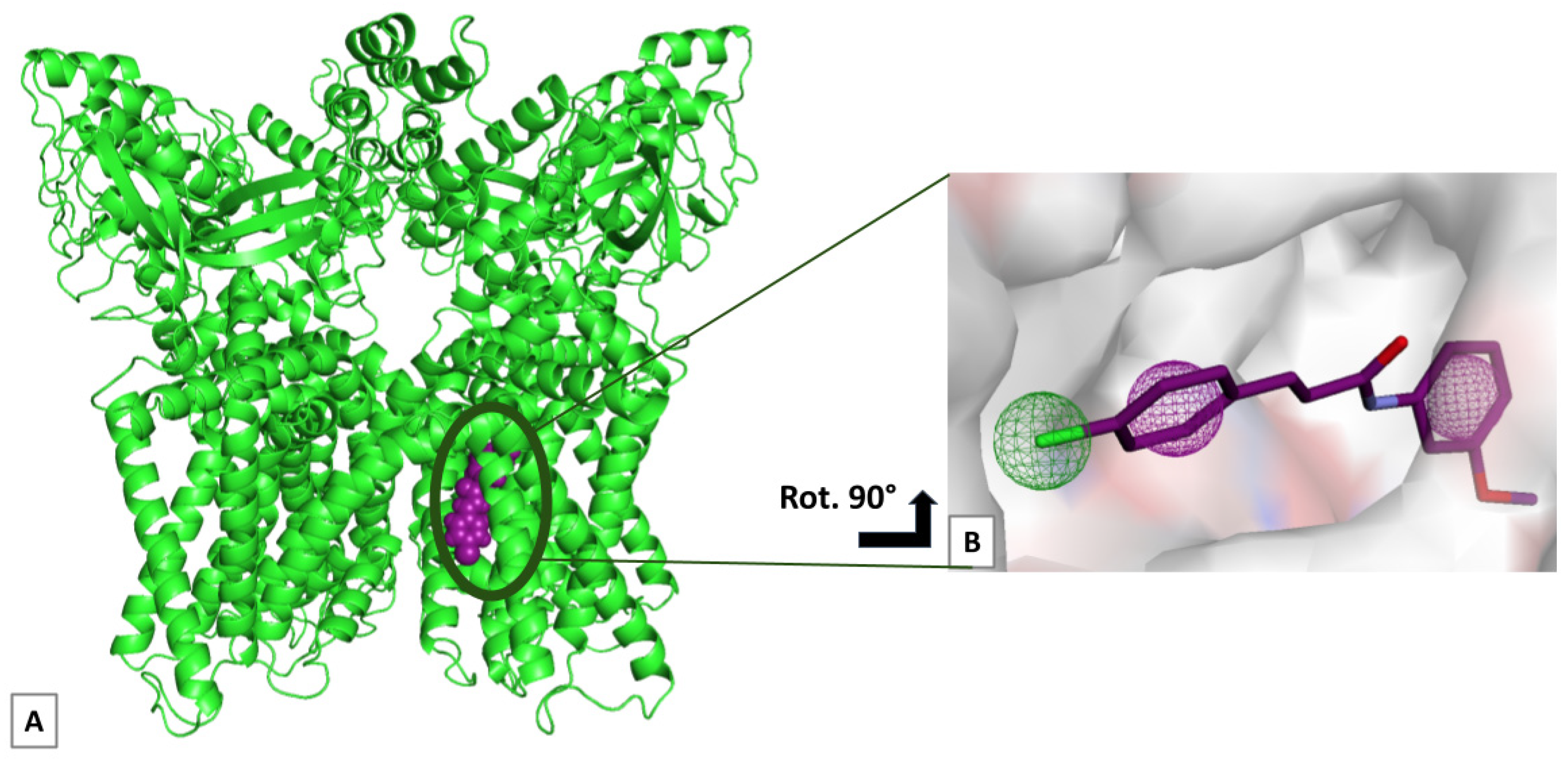
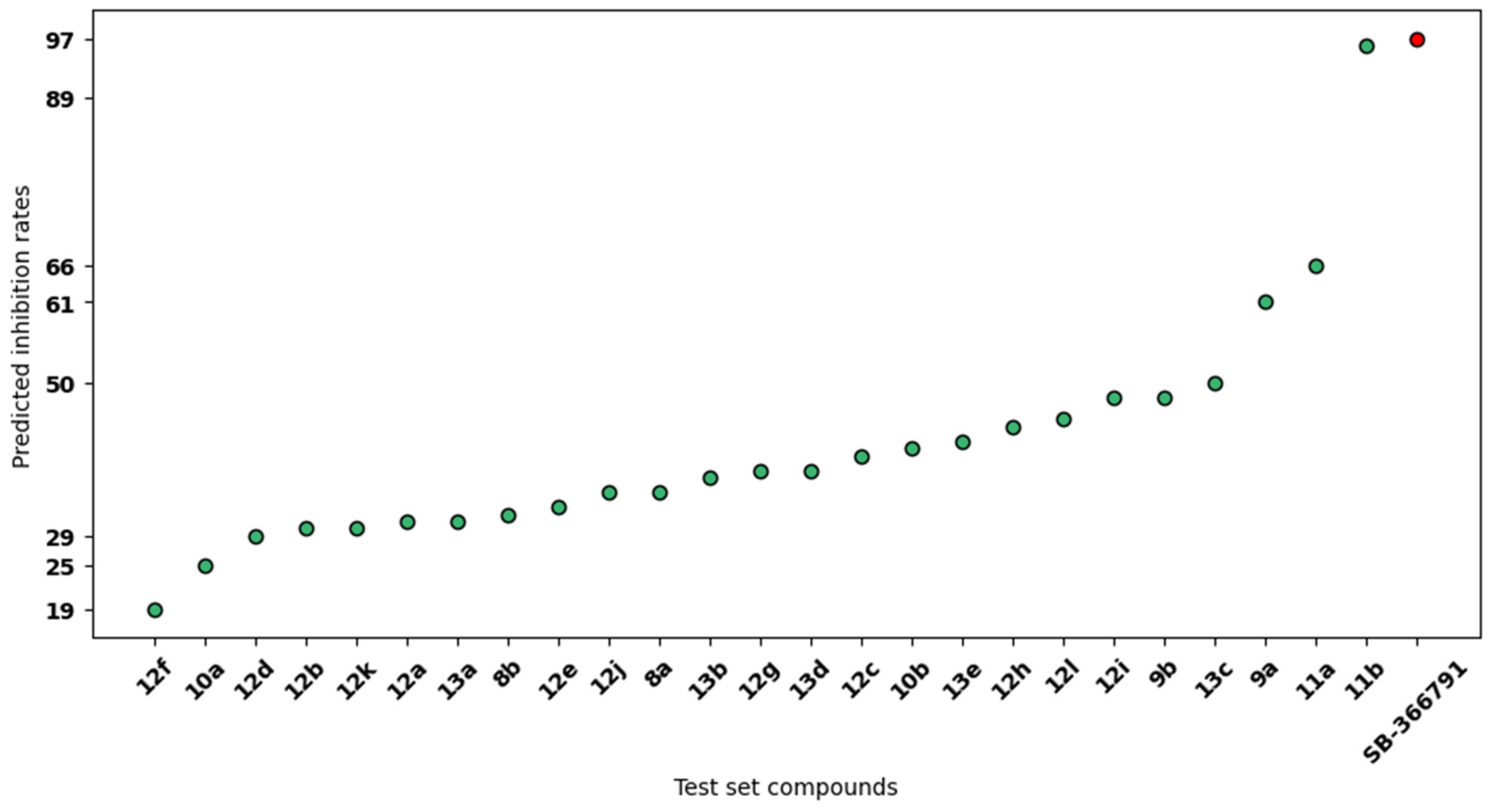
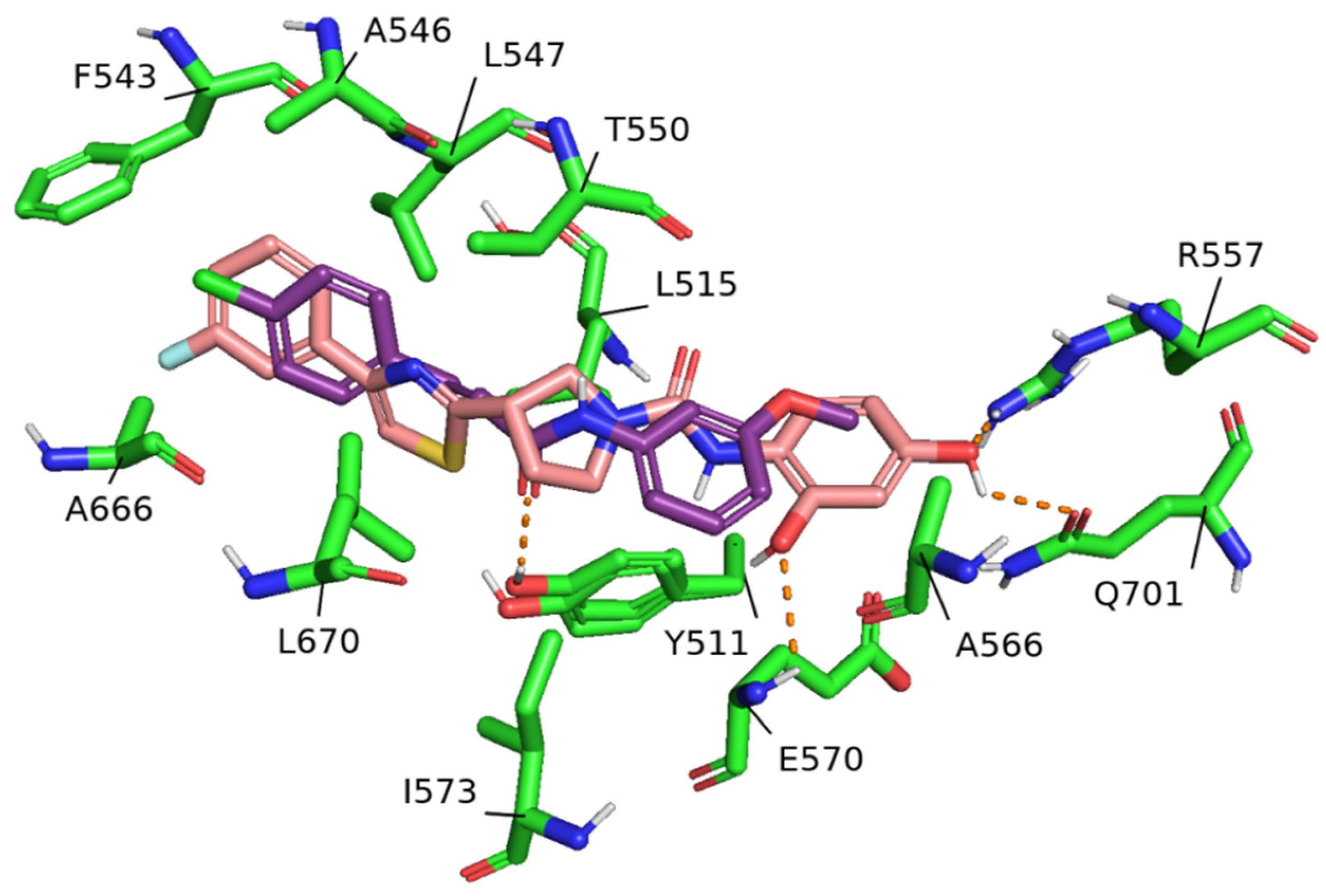
2. Results
3. Discussion
4. Materials and Methods
4.1. Pharmacophore Modeling
4.2. Three-Dimensional-QSAR Model Generation and Validation
4.3. Molecular Docking
4.4. BBB Permeability Prediction
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siniscalco, D.; Rossi, F.; Maione, S. Molecular approaches for neuropathic pain treatment. Curr. Med. Chem. 2007, 14, 1783–1787. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Carvajal, A.; Fernández-Ballester, G.; Ferrer-Montiel, A. TRPV1 in chronic pruritus and pain: Soft modulation as a therapeutic strategy. Front. Mol. Neurosci. 2022, 15, 930964. [Google Scholar] [CrossRef] [PubMed]
- Kerckhove, N.; Bodieu, L.; Ourties, G.; Bourdier, J.; Daulhac, L.; Eschalier, A.; Mallet, C. Ethosuximide improves chronic pain-induced anxiety- and depression-like behaviors. Eur. Neuropsychopharmacol. 2019, 29, 1419–1432. [Google Scholar] [CrossRef] [PubMed]
- Visibelli, A.; Peruzzi, L.; Poli, P.; Scocca, A.; Carnevale, S.; Spiga, O.; Santucci, A. Supporting Machine Learning Model in the Treatment of Chronic Pain. Biomedicines 2023, 11, 1776. [Google Scholar] [CrossRef] [PubMed]
- Paulus, D.J.; Rogers, A.H.; Bakhshaie, J.; Vowles, K.E.; Zvolensky, M.J. Pain severity and prescription opioid misuse among individuals with chronic pain: The moderating role of alcohol use severity. Drug Alcohol Depend. 2019, 204, 107456. [Google Scholar] [CrossRef] [PubMed]
- Koivisto, A.P.; Belvisi, M.G.; Gaudet, R.; Szallasi, A. Advances in TRP channel drug discovery: From target validation to clinical studies. Nat. Rev. Drug Discov. 2022, 21, 41–59. [Google Scholar] [CrossRef]
- Brito, R.; Sheth, S.; Mukherjea, D.; Rybak, L.P.; Ramkumar, V. TRPV1: A Potential Drug Target for Treating Various Diseases. Cells 2014, 3, 517–545. [Google Scholar] [CrossRef] [PubMed]
- Nilius, B.; Szallasi, A. Transient receptor potential channels as drug targets: From the science of basic research to the art of medicine. Pharmacol. Rev. 2014, 66, 676–814. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Zheng, J. Understand spiciness: Mechanism of TRPV1 channel activation by capsaicin. Protein Cell 2017, 8, 169–177. [Google Scholar] [CrossRef]
- Treat, A.; Henri, V.; Liu, J.; Shen, J.; Gil-Silva, M.; Morales, A.; Rade, A.; Tidgewell, K.J.; Kolber, B.; Shen, Y. Novel TRPV1 Modulators with Reduced Pungency Induce Analgesic Effects in Mice. ACS Omega 2022, 7, 2929–2946. [Google Scholar] [CrossRef]
- Elokely, K.; Valisetty, P.; Delemotte, L.; Palovcak, E.; Klein, M.L.; Rohacs, T.; Carnevale, V. Understanding TRPV1 activation by ligands: Insights from the binding modes of capsaicin and resiniferatoxin. Proc. Natl. Acad. Sci. USA 2016, 113, E137–E145. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.H.; Jung, S.H.; Sethi, G.; Ahn, K.S. Pleiotropic Pharmacological Actions of Capsazepine, a Synthetic Analogue of Capsaicin, against Various Cancers and Inflammatory Diseases. Molecules 2019, 24, 995. [Google Scholar] [CrossRef] [PubMed]
- Rami, H.K.; Thompson, M.; Stemp, G.; Fell, S.; Jerman, J.C.; Stevens, A.J.; Smart, D.; Sargent, B.; Sanderson, D.; Randall, A.D.; et al. Discovery of SB-705498: A potent, selective and orally bioavailable TRPV1 antagonist suitable for clinical development. Bioorg. Med. Chem. Lett. 2006, 16, 3287–3291. [Google Scholar] [CrossRef] [PubMed]
- Gavva, N.R.; Bannon, A.W.; Surapaneni, S.; Hovland, D.J.; Lehto, S.G.; Gore, A.; Juan, T.; Deng, H.; Han, B.; Klionsky, L.; et al. The vanilloid receptor TRPV1 is tonically activated in vivo and involved in body temperature regulation. J. Neurosci. 2007, 27, 3366–3374. [Google Scholar] [CrossRef] [PubMed]
- Ann, J.; Kim, H.S.; Thorat, S.A.; Kim, H.; Ha, H.J.; Choi, K.; Kim, Y.H.; Kim, M.; Hwang, S.W.; Pearce, L.V.; et al. Discovery of Nonpungent Transient Receptor Potential Vanilloid 1 (TRPV1) Agonist as Strong Topical Analgesic. J. Med. Chem. 2020, 63, 418–424. [Google Scholar] [CrossRef]
- Qiao, Y.; Zhang, Y.; Qiao, Z.; He, W.; Chen, Y.; Song, D.; Wang, G.; Guo, N.; Shao, L.; Tian, Z.; et al. Discovery of (S)-N-(3-isopropylphenyl)-2-(5-phenylthiazol-2-yl)pyrrolidine-1-carboxamide as potent and brain-penetrant TRPV1 antagonist. Eur. J. Med. Chem. 2022, 233, 114191. [Google Scholar] [CrossRef] [PubMed]
- Gunthorpe, M.J.; Rami, H.K.; Jerman, J.C.; Smart, D.; Gill, C.H.; Soffin, E.M.; Hannan, S.L.; Lappin, S.C.; Egerton, J.; Smith, G.D.; et al. Identification and characterisation of SB-366791, a potent and selective vanilloid receptor (VR1/TRPV1) antagonist. Neuropharmacology 2004, 46, 133–149. [Google Scholar] [CrossRef] [PubMed]
- Neuberger, A.; Oda, M.; Nikolaev, Y.A.; Nadezhdin, K.D.; Gracheva, E.O.; Bagriantsev, S.N.; Sobolevsky, A.I. Human TRPV1 structure and inhibition by the analgesic SB-366791. Nat. Commun. 2023, 14, 2451. [Google Scholar] [CrossRef]
- Zankov, D.V.; Matveieva, M.; Nikonenko, A.V.; Nugmanov, R.I.; Baskin, I.I.; Varnek, A.; Polishchuk, P.; Madzhidov, T.I. QSAR Modeling Based on Conformation Ensembles Using a Multi-Instance Learning Approach. J. Chem. Inf. Model. 2021, 61, 4913–4923. [Google Scholar] [CrossRef]
- Orosz, A.; Héberger, K.; Rácz, A. Comparison of Descriptor- and Fingerprint Sets in Machine Learning Models for ADME-Tox Targets. Front. Chem. 2022, 10, 852893. [Google Scholar] [CrossRef]
- Bahia, M.S.; Kaspi, O.; Touitou, M.; Binayev, I.; Dhail, S.; Spiegel, J.; Senderowitz, H. A comparison between 2D and 3D descriptors in QSAR modeling based on bio-active conformations. Mol. Inform. 2023, 42, 2200186. [Google Scholar] [CrossRef] [PubMed]
- Ekins, S.; Bravi, G.; Binkley, S.; Gillespie, J.S.; Ring, B.J.; Wikel, J.H.; Wrighton, S.A. Three- and four-dimensional-quantitative structure activity relationship (3D/4D-QSAR) analyses of CYP2C9 inhibitors. Drug Metab. Dispos. 2000, 28, 994–1002. [Google Scholar] [PubMed]
- Vedani, A.; Dobler, M. 5D-QSAR: The key for simulating induced fit? J. Med. Chem. 2002, 45, 2139–2149. [Google Scholar] [CrossRef] [PubMed]
- Vedani, A.; Dobler, M.; Lill, M.A. Combining protein modeling and 6D-QSAR. Simulating the binding of structurally diverse ligands to the estrogen receptor. J. Med. Chem. 2005, 48, 3700–3703. [Google Scholar] [CrossRef] [PubMed]
- Lovrić, M.; Duričić, T.; Tran, H.T.; Hussain, H.; Lacić, E.; Rasmussen, M.A.; Kern, R. Should We Embed in Chemistry? A Comparison of Unsupervised Transfer Learning with PCA, UMAP, and VAE on Molecular Fingerprints. Pharmaceuticals 2021, 14, 758. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.; Bae, H.; Jo, J.; Yoon, S. Comprehensive ensemble in QSAR prediction for drug discovery. BMC Bioinform. 2019, 20, 521. [Google Scholar] [CrossRef] [PubMed]
- Cramer, R.D.; Patterson, D.E.; Bunce, J.D. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc. 1988, 110, 5959–5967. [Google Scholar] [CrossRef] [PubMed]
- Tosco, P.; Balle, T. Open3DQSAR: A new open-source software aimed at high-throughput chemometric analysis of molecular interaction fields. J. Mol. Model. 2011, 17, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Morales-Lázaro, S.L.; LLorente, I.; Sierra-Ramírez, A.E.; López-Romero, M.; Ortíz-Rentería, M.; Serrano-Flores, B.; Simon, S.A.; Islas, L.I.; Rosenbaum, T. Inhibition of TRPV1 channels by a naturally occurring omega-9 fatty acid reduces pain and itch. Nat. Commun. 2016, 7, 13092. [Google Scholar] [CrossRef]
- Gavva, N.R.; Klionsky, L.; Qu, Y.; Shi, L.; Tamir, R.; Edenson, S.; Zhang, T.J.; Viswanadhan, V.N.; Toth, A.; Pearce, L.V.; et al. Molecular determinants of vanilloid sensitivity in TRPV1. J. Biol. Chem. 2004, 279, 20283–20295. [Google Scholar] [CrossRef]
- Shaker, B.; Yu, M.-S.; Song, J.S. LightBBB: Computational prediction model of blood-brain-barrier penetration based on LightGBM. Bioinformatics 2021, 37, 1135–1139. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, L.; Su, W.; Pei, R. AlzPlatform: An Alzheimer’s Disease Domain-Specific Chemogenomics Knowledgebase for Polypharmacology and Target Identification Research. J. Chem. Inf. Model. 2014, 54, 1050–1060. [Google Scholar] [CrossRef] [PubMed]
- Rautio, J.; Laine, K.; Gynther, M.; Savolainen, J. Prodrug approaches for CNS delivery. AAPS J. 2008, 10, 92–102. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Puranen, J.S.; Vainio, M.J.; Johnson, M.S. Accurate conformation-dependent molecular electrostatic potentials for high-throughput in silico drug discovery. J. Comput. Chem. 2010, 31, 1722–1732. [Google Scholar] [CrossRef] [PubMed]
- Vainio, M.J.; Johnson, M.S. Generating conformer ensembles using a multiobjective genetic algorithm. J. Chem. Inf. Model. 2007, 47, 2462–2474. [Google Scholar] [CrossRef]
- Sunseri, J.; Koes, D.R. Pharmit: Interactive exploration of chemical space. Nucleic Acids Res. 2016, 44, W442–W448. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Wu, E.L.; Cheng, X.; Jo, S.; Rui, H.; Song, K.C.; Dávila-Contreras, E.M.; Qi, Y.; Lee, J.; Monje-Galvan, V.; Venable, R.M.; et al. CHARMM-GUI Membrane Builder Toward Realistic Biological Membrane Simulations. J. Comput. Chem. 2014, 35, 1997–2004. [Google Scholar] [CrossRef]
- Jo, S.; Lim, J.B.; Klauda, J.B.; Im, W. CHARMM-GUI Membrane Builder for Mixed Bilayers and Its Application to Yeast Membranes. Biophys. J. 2009, 97, 50–58. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Im, W. Automated Builder and Database of Protein/Membrane Complexes for Molecular Dynamics Simulations. PLoS ONE 2007, 2, e880. [Google Scholar] [CrossRef] [PubMed]
- Bauer, P.; Hess, B.; Lindahl, E. GROMACS 2022.5 Manual; Zenodo: Geneva, Switzerland, 2023. [Google Scholar]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Ravindranath, P.A.; Forli, S.; Goodsell, D.S.; Olson, A.J.; Sanner, M.F. AutoDockFR: Advances in Protein-Ligand Docking with Explicitly Specified Binding Site Flexibility. PLoS Comput. Biol. 2015, 11, e1004586. [Google Scholar] [CrossRef] [PubMed]
- Koebel, M.R.; Schmadeke, G.; Posner, R.G.; Sirimulla, S. AutoDock VinaXB: Implementation of XBSF, new empirical halogen bond scoring function, into AutoDock Vina. J. Cheminform. 2016, 8, 27. [Google Scholar] [CrossRef]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein-ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Compounds | R1 | R2 | A | X |
| 8a | phenyl | 4-pyrimidinyl | S | N |
| 8b | phenyl | 4-(2-Cl-pyrimidinyl) | S | N |
| 9a | phenyl | 4-pyrimidinyl | N | S |
| 9b | phenyl | phenyl | N | O |
| 10a | p-F-phenyl | 4-pyridinyl | S | N |
| 10b | p-F-phenyl | 2-thiophenyl | S | N |
| 11a | m-F-phenyl | 5-imidazolyl | S | N |
| 11b | m-F-phenyl | 2,4-dihydroxy-phenyl | S | N |
| 12a | m-Me-phenyl | m-iPr-phenyl | S | N |
| 12b | 3-F-4-Me-phenyl | m-iPr-phenyl | S | N |
| 12c | phenyl | 5-(3-iPr-pyridinyl) | S | N |
| 12d | phenyl | 4-(2-iPr-pyridinyl) | S | N |
| 12e | 3-(1,2,5-triMe-pyrrolyl | m-iPr-phenyl | S | N |
| 12f | 3-(2,5-diMe-thiophenyl) | m-iPr-phenyl | S | N |
| 12g | 3-thiophenyl | m-iPr-phenyl | S | N |
| 12h | 2,2′-diMe-phenyl | m-iPr-phenyl | S | N |
| 12i | m-F-phenyl | 2-OMe-4-hydroxy-phenyl | S | N |
| 12j | m-F-phenyl | 1-naphtalyl | S | N |
| 12k | m-F-phenyl | 2,3-diMe-phenyl | S | N |
| 12l | m-F-phenyl | 2,3-diMe-4-hydroxy-phenyl | S | N |
| 13a | phenyl | 4-(2-pyridonyl) | S | N |
| 13b | phenyl | 3-(2-pyridonyl) | S | N |
| 13c | m-F-phenyl | 3-(2-pyridonyl) | S | N |
| 13d | 4-(2-pyridonyl) | m-iPr-phenyl | S | N |
| 13e | 5-(2-pyridonyl) | m-iPr-phenyl | S | N |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gianibbi, B.; Visibelli, A.; Spinsanti, G.; Spiga, O. Three-Dimensional Quantitative Structure–Activity Relationship Study of Transient Receptor Potential Vanilloid 1 Channel Antagonists Reveals Potential for Drug Design Purposes. Int. J. Mol. Sci. 2024, 25, 7951. https://doi.org/10.3390/ijms25147951
Gianibbi B, Visibelli A, Spinsanti G, Spiga O. Three-Dimensional Quantitative Structure–Activity Relationship Study of Transient Receptor Potential Vanilloid 1 Channel Antagonists Reveals Potential for Drug Design Purposes. International Journal of Molecular Sciences. 2024; 25(14):7951. https://doi.org/10.3390/ijms25147951
Chicago/Turabian StyleGianibbi, Beatrice, Anna Visibelli, Giacomo Spinsanti, and Ottavia Spiga. 2024. "Three-Dimensional Quantitative Structure–Activity Relationship Study of Transient Receptor Potential Vanilloid 1 Channel Antagonists Reveals Potential for Drug Design Purposes" International Journal of Molecular Sciences 25, no. 14: 7951. https://doi.org/10.3390/ijms25147951
APA StyleGianibbi, B., Visibelli, A., Spinsanti, G., & Spiga, O. (2024). Three-Dimensional Quantitative Structure–Activity Relationship Study of Transient Receptor Potential Vanilloid 1 Channel Antagonists Reveals Potential for Drug Design Purposes. International Journal of Molecular Sciences, 25(14), 7951. https://doi.org/10.3390/ijms25147951