
Targeting Mitochondrial Dysfunction and Reactive Oxygen Species for Neurodegenerative Disease Treatment


Abstract
1. Introduction
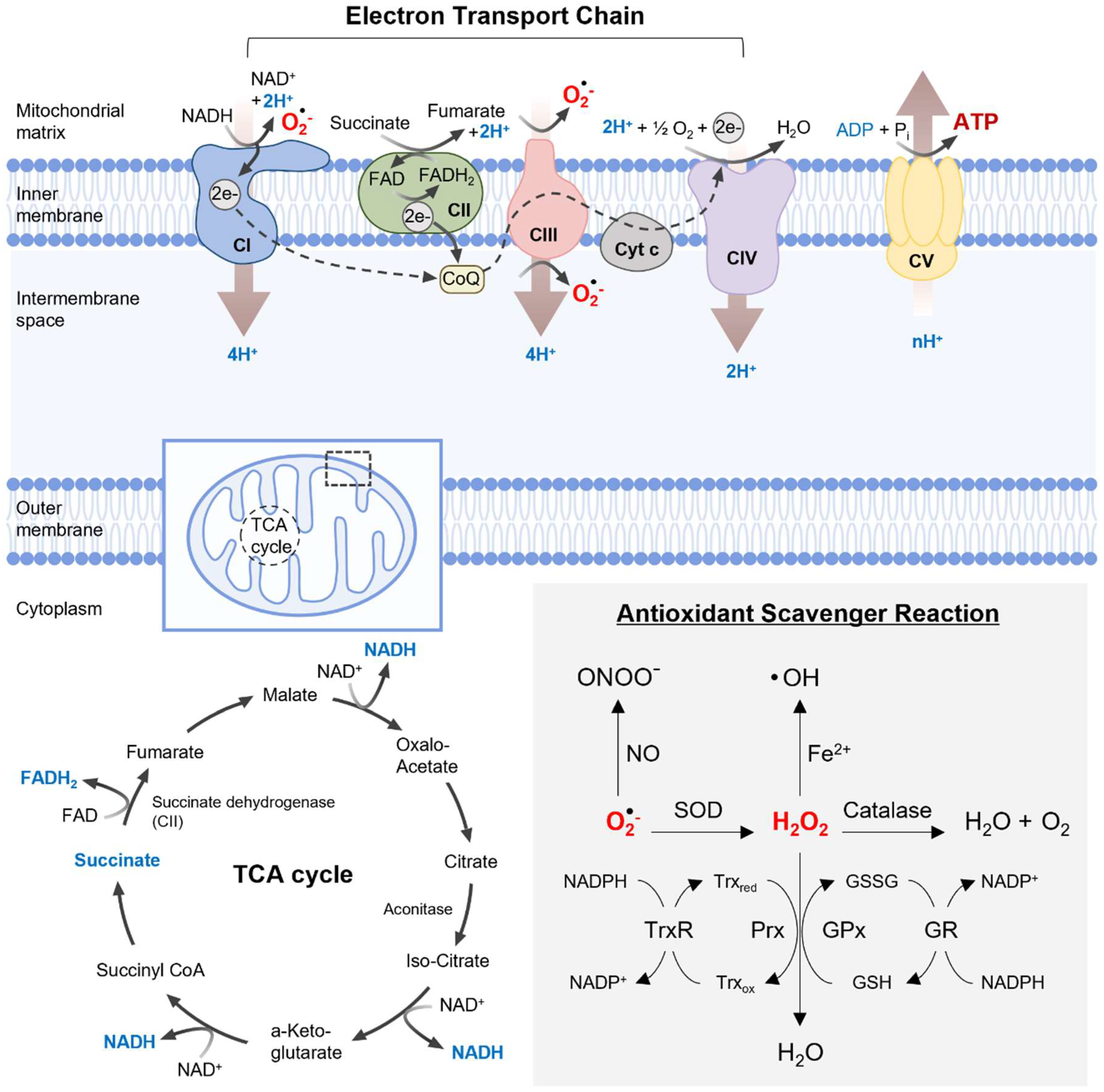
2. Mitochondrial Function and Dysfunction
2.1. Mitochondrial Energy Production and Oxidative Stress
2.2. Antioxidant Defense System
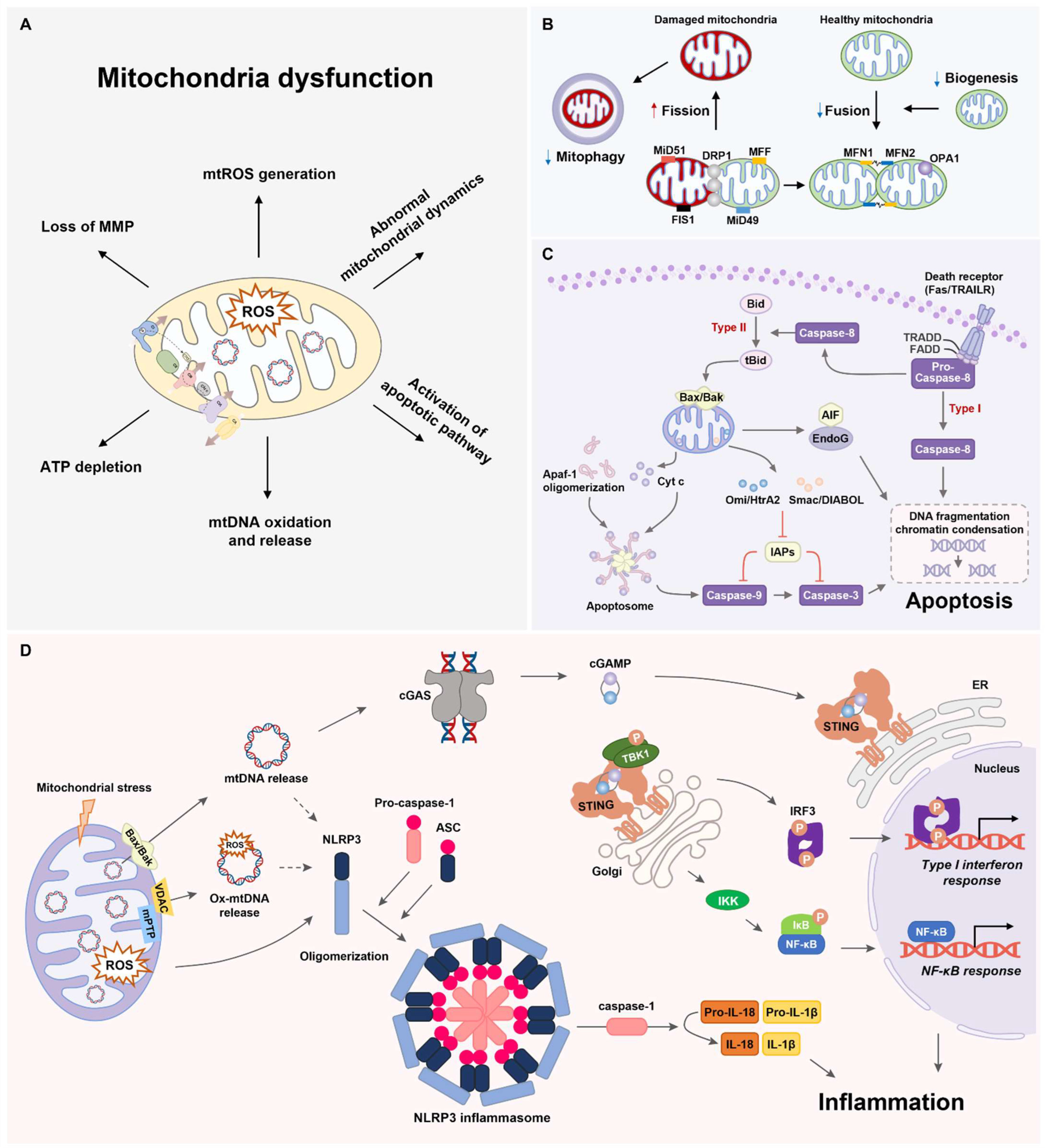
3. Mitochondrial Dynamics
3.1. Mitochondrial Fusion
3.2. Mitochondrial Fission
3.3. Mitophagy and Mitochondrial Biogenesis
3.4. Mitochondrial Dynamics and ROS
3.5. Mitochondrial Inheritance
3.6. Mitochondrial Distribution and Morphology
4. Mitochondrial Apoptosis Pathway
5. Mitochondria-Associated Inflammation
6. Mitochondria Dysfunction and Oxidative Stress in the Pathogenesis of Neurodegenerative Diseases
6.1. Alzheimer’s Disease
6.1.1. Oxidative Stress
6.1.2. Alterations in Mitochondrial Dynamics
6.1.3. Mitochondria-Driven Inflammation
6.2. Parkinson’s Disease
6.2.1. Oxidative Stress
6.2.2. Impaired Mitochondrial Dynamics and Function
6.2.3. Mitochondria-Activated Inflammatory Pathway
7. Mitochondrial Dysfunction and ROS as Therapeutic Targets for Neurodegenerative Diseases
7.1. Mitochondrial Fusion and Biogenesis Enhancers
7.2. Mitochondrial Fission Inhibitors
7.3. Antioxidants
7.4. Mitochondria-Driven Inflammation Inhibitors
8. Conclusions and Future Perspectives
Funding
Data Availability Statement
Conflicts of Interest
References
- Eaglesfield, R.; Tokatlidis, K. Targeting and Insertion of Membrane Proteins in Mitochondria. Front. Cell Dev. Biol. 2021, 9, 803205. [Google Scholar] [CrossRef] [PubMed]
- Giacomello, M.; Pyakurel, A.; Glytsou, C.; Scorrano, L. The cell biology of mitochondrial membrane dynamics. Nat. Rev. Mol. Cell Biol. 2020, 21, 204–224. [Google Scholar] [CrossRef]
- Boyman, L.; Karbowski, M.; Lederer, W.J. Regulation of Mitochondrial ATP Production: Ca2+ Signaling and Quality Control. Trends Mol. Med. 2020, 26, 21–39. [Google Scholar] [CrossRef]
- Palma, F.R.; Gantner, B.N.; Sakiyama, M.J.; Kayzuka, C.; Shukla, S.; Lacchini, R.; Cunniff, B.; Bonini, M.G. ROS production by mitochondria: Function or dysfunction? Oncogene 2024, 43, 295–303. [Google Scholar] [CrossRef]
- Ahola, S.; Langer, T.; MacVicar, T. Mitochondrial Proteolysis and Metabolic Control. Cold Spring Harb. Perspect. Biol. 2019, 11, a033936. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Hu, J. Mitochondrial Fusion: The Machineries In and Out. Trends Cell Biol. 2021, 31, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.J.; McIntyre, R.L.; Janssens, G.E.; Houtkooper, R.H. Mitochondrial fission and fusion: A dynamic role in aging and potential target for age-related disease. Mech. Ageing Dev. 2020, 186, 111212. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Fang, B.; Peng, B.; Wang, L.; Xue, Y.; Bai, H.; Lu, S.; Voelcker, N.H.; Li, L.; Fu, L.; et al. Recent Advances in Chemical Biology of Mitochondria Targeting. Front. Chem. 2021, 9, 683220. [Google Scholar] [CrossRef]
- Gao, X.-Y.; Yang, T.; Gu, Y.; Sun, X.-H. Mitochondrial Dysfunction in Parkinson’s Disease: From Mechanistic Insights to Therapy. Front. Aging Neurosci. 2022, 14, 885500. [Google Scholar] [CrossRef]
- Alqahtani, T.; Deore, S.L.; Kide, A.A.; Shende, B.A.; Sharma, R.; Dadarao Chakole, R.; Nemade, L.S.; Kishor Kale, N.; Borah, S.; Shrikant Deokar, S.; et al. Mitochondrial dysfunction and oxidative stress in Alzheimer’s disease, and Parkinson’s disease, Huntington’s disease and Amyotrophic Lateral Sclerosis—An updated review. Mitochondrion 2023, 71, 83–92. [Google Scholar] [CrossRef]
- Misrani, A.; Tabassum, S.; Yang, L. Mitochondrial Dysfunction and Oxidative Stress in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 617588. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-J.; Kim, Y.; Li, C.; Suh, J.; Sivapackiam, J.; Goncalves, T.M.; Jarad, G.; Zhao, G.; Urano, F.; Sharma, V.; et al. Blocking CHOP-dependent TXNIP shuttling to mitochondria attenuates albuminuria and mitigates kidney injury in nephrotic syndrome. Proc. Natl. Acad. Sci. USA 2022, 119, e2116505119. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Zhang, X.; Liu, C.; Liu, Q.; Chai, H.; Luo, Y.; Li, S. Role of Mitochondria in Neurodegenerative Diseases: From an Epigenetic Perspective. Front. Cell Dev. Biol. 2021, 9, 688789. [Google Scholar] [CrossRef] [PubMed]
- Habbane, M.; Montoya, J.; Rhouda, T.; Sbaoui, Y.; Radallah, D.; Emperador, S. Human Mitochondrial DNA: Particularities and Diseases. Biomedicines 2021, 9, 1364. [Google Scholar] [CrossRef]
- He, B.; Yu, H.; Liu, S.; Wan, H.; Fu, S.; Liu, S.; Yang, J.; Zhang, Z.; Huang, H.; Li, Q.; et al. Mitochondrial cristae architecture protects against mtDNA release and inflammation. Cell Rep. 2022, 41, 111774. [Google Scholar] [CrossRef]
- Urbani, A.; Prosdocimi, E.; Carrer, A.; Checchetto, V.; Szabò, I. Mitochondrial Ion Channels of the Inner Membrane and Their Regulation in Cell Death Signaling. Front. Cell Dev. Biol. 2020, 8, 620081. [Google Scholar] [CrossRef]
- Xian, H.; Liou, Y.-C. Functions of outer mitochondrial membrane proteins: Mediating the crosstalk between mitochondrial dynamics and mitophagy. Cell Death Differ. 2021, 28, 827–842. [Google Scholar] [CrossRef] [PubMed]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020, 37, 101674. [Google Scholar] [CrossRef]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef]
- Walker, M.A.; Tian, R. NAD(H) in mitochondrial energy transduction: Implications for health and disease. Curr. Opin. Physiol. 2018, 3, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.; Javadov, S. Elucidating the contribution of ETC complexes I and II to the respirasome formation in cardiac mitochondria. Sci. Rep. 2018, 8, 17732. [Google Scholar] [CrossRef] [PubMed]
- Tirichen, H.; Yaigoub, H.; Xu, W.; Wu, C.; Li, R.; Li, Y. Mitochondrial Reactive Oxygen Species and Their Contribution in Chronic Kidney Disease Progression Through Oxidative Stress. Front. Physiol. 2021, 12, 627837. [Google Scholar] [CrossRef] [PubMed]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Hu, X.; Shen, Q.; Xing, D. Mitochondria-specific drug release and reactive oxygen species burst induced by polyprodrug nanoreactors can enhance chemotherapy. Nat. Commun. 2019, 10, 1704. [Google Scholar] [CrossRef]
- Palma, F.R.; He, C.; Danes, J.M.; Paviani, V.; Coelho, D.R.; Gantner, B.N.; Bonini, M.G. Mitochondrial Superoxide Dismutase: What the Established, the Intriguing, and the Novel Reveal About a Key Cellular Redox Switch. Antioxid. Redox Signal. 2020, 32, 701–714. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, R.J. An Update on Mitochondrial Reactive Oxygen Species Production. Antioxidants 2020, 9, 472. [Google Scholar] [CrossRef] [PubMed]
- Sidarala, V.; Zhu, J.; Levi-D’ancona, E.; Pearson, G.L.; Reck, E.C.; Walker, E.M.; Kaufman, B.A.; Soleimanpour, S.A. Mitofusin 1 and 2 regulation of mitochondrial DNA content is a critical determinant of glucose homeostasis. Nat. Commun. 2022, 13, 2340. [Google Scholar] [CrossRef]
- Zaman, M.; Shutt, T.E. The Role of Impaired Mitochondrial Dynamics in MFN2-Mediated Pathology. Front. Cell Dev. Biol. 2022, 10, 858286. [Google Scholar] [CrossRef]
- Casellas-Díaz, S.; Larramona-Arcas, R.; Riqué-Pujol, G.; Tena-Morraja, P.; Müller-Sánchez, C.; Segarra-Mondejar, M.; Gavaldà-Navarro, A.; Villarroya, F.; Reina, M.; Martínez-Estrada, O.M.; et al. Mfn2 localization in the ER is necessary for its bioenergetic function and neuritic development. EMBO Rep. 2021, 22, e51954. [Google Scholar] [CrossRef]
- Gilkerson, R.; De La Torre, P.; Vallier, S.S. Mitochondrial OMA1 and OPA1 as Gatekeepers of Organellar Structure/Function and Cellular Stress Response. Front. Cell Dev. Biol. 2021, 9, 626117. [Google Scholar] [CrossRef]
- Moltedo, O.; Remondelli, P.; Amodio, G. The Mitochondria-Endoplasmic Reticulum Contacts and Their Critical Role in Aging and Age-Associated Diseases. Front. Cell Dev. Biol. 2019, 7, 172. [Google Scholar] [CrossRef]
- Zerihun, M.; Sukumaran, S.; Qvit, N. The Drp1-Mediated Mitochondrial Fission Protein Interactome as an Emerging Core Player in Mitochondrial Dynamics and Cardiovascular Disease Therapy. Int. J. Mol. Sci. 2023, 24, 5785. [Google Scholar] [CrossRef] [PubMed]
- Ihenacho, U.K.; Meacham, K.A.; Harwig, M.C.; Widlansky, M.E.; Hill, R.B. Mitochondrial Fission Protein 1: Emerging Roles in Organellar Form and Function in Health and Disease. Front. Endocrinol. 2021, 12, 660095. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Ysselstein, D.; Krainc, D. Mitochondria-lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature 2018, 554, 382–386. [Google Scholar] [CrossRef]
- Marchi, S.; Patergnani, S.; Missiroli, S.; Morciano, G.; Rimessi, A.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium 2018, 69, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Xie, P.; Dong, Y.; An, W. Inhibition of Drp1 SUMOylation by ALR protects the liver from ischemia-reperfusion injury. Cell Death Differ. 2021, 28, 1174–1192. [Google Scholar] [CrossRef]
- Chen, W.; Zhao, H.; Li, Y. Mitochondrial dynamics in health and disease: Mechanisms and potential targets. Signal Transduct. Target. Ther. 2023, 8, 333. [Google Scholar] [CrossRef]
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N. Regulation of Mitochondrial Biogenesis as a Way for Active Longevity: Interaction Between the Nrf2 and PGC-1α Signaling Pathways. Front. Genet. 2019, 10, 435. [Google Scholar] [CrossRef]
- Eldeeb, M.A.; Bayne, A.N.; Fallahi, A.; Goiran, T.; MacDougall, E.J.; Soumbasis, A.; Zorca, C.E.; Tabah, J.-J.; Thomas, R.A.; Karpilovsky, N.; et al. Tom20 gates PINK1 activity and mediates its tethering of the TOM and TIM23 translocases upon mitochondrial stress. Proc. Natl. Acad. Sci. USA 2024, 121, e2313540121. [Google Scholar] [CrossRef]
- Magnani, N.D.; Marchini, T.; Calabró, V.; Alvarez, S.; Evelson, P. Role of Mitochondria in the Redox Signaling Network and Its Outcomes in High Impact Inflammatory Syndromes. Front. Endocrinol. 2020, 11, 568305. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-M.; Youn, S.-W.; Sudhahar, V.; Das, A.; Chandhri, R.; Cuervo Grajal, H.; Kweon, J.; Leanhart, S.; He, L.; Toth, P.T.; et al. Redox Regulation of Mitochondrial Fission Protein Drp1 by Protein Disulfide Isomerase Limits Endothelial Senescence. Cell Rep. 2018, 23, 3565–3578. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, T.; Ishihara, T.; Ichimura, A.; Ishihara, N. Mitochondrial dynamics define muscle fiber type by modulating cellular metabolic pathways. Cell Rep. 2023, 42, 112434. [Google Scholar] [CrossRef] [PubMed]
- Hinchy, E.C.; Gruszczyk, A.V.; Willows, R.; Navaratnam, N.; Hall, A.R.; Bates, G.; Bright, T.P.; Krieg, T.; Carling, D.; Murphy, M.P. Mitochondria-derived ROS activate AMP-activated protein kinase (AMPK) indirectly. J. Biol. Chem. 2018, 293, 17208–17217. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Chinnery, P.F. Inheritance of mitochondrial DNA in humans: Implications for rare and common diseases. J. Intern. Med. 2020, 287, 634–644. [Google Scholar] [CrossRef] [PubMed]
- Malina, C.; Larsson, C.; Nielsen, J. Yeast mitochondria: An overview of mitochondrial biology and the potential of mitochondrial systems biology. FEMS Yeast Res. 2018, 18, foy040. [Google Scholar] [CrossRef] [PubMed]
- Favaro, G.; Romanello, V.; Varanita, T.; Desbats, M.A.; Morbidoni, V.; Tezze, C.; Albiero, M.; Canato, M.; Gherardi, G.; De Stefani, D.; et al. DRP1-mediated mitochondrial shape controls calcium homeostasis and muscle mass. Nat. Commun. 2019, 10, 2576. [Google Scholar] [CrossRef] [PubMed]
- Helle, S.C.J.; Feng, Q.; Aebersold, M.J.; Hirt, L.; Gruter, R.R.; Vahid, A.; Sirianni, A.; Mostowy, S.; Snedeker, J.G.; Saric, A.; et al. Mechanical force induces mitochondrial fission. eLife 2017, 6, e30292. [Google Scholar] [CrossRef] [PubMed]
- Machiela, E.; Liontis, T.; Dues, D.J.; Rudich, P.D.; Traa, A.; Wyman, L.; Kaufman, C.; Cooper, J.F.; Lew, L.; Nadarajan, S.; et al. Disruption of mitochondrial dynamics increases stress resistance through activation of multiple stress response pathways. FASEB J. 2020, 34, 8475–8492. [Google Scholar] [CrossRef]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell. Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef]
- Itoh, K.; Murata, D.; Kato, T.; Yamada, T.; Araki, Y.; Saito, A.; Adachi, Y.; Igarashi, A.; Li, S.; Pletnikov, M.; et al. Brain-specific Drp1 regulates postsynaptic endocytosis and dendrite formation independently of mitochondrial division. eLife 2019, 8, e44739. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wang, H.; Ni, H.-M.; Xiong, A.; Wang, Z.; Sesaki, H.; Ding, W.-X.; Yang, L. Inhibition of Drp1 protects against senecionine-induced mitochondria-mediated apoptosis in primary hepatocytes and in mice. Redox Biol. 2017, 12, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Xiao, Y.; Hu, Z.; Gu, J.; Hua, R.; Hai, Z.; Chen, X.; Zhang, J.V.; Yu, Z.; Wu, T.; et al. MFN2 Deficiency Impairs Mitochondrial Functions and PPAR Pathway During Spermatogenesis and Meiosis in Mice. Front. Cell Dev. Biol. 2022, 10, 862506. [Google Scholar] [CrossRef] [PubMed]
- Hood, D.A.; Memme, J.M.; Oliveira, A.N.; Triolo, M. Maintenance of Skeletal Muscle Mitochondria in Health, Exercise, and Aging. Annu. Rev. Physiol. 2019, 81, 19–41. [Google Scholar] [CrossRef]
- Chen, X.; Ji, Y.; Liu, R.; Zhu, X.; Wang, K.; Yang, X.; Liu, B.; Gao, Z.; Huang, Y.; Shen, Y.; et al. Mitochondrial dysfunction: Roles in skeletal muscle atrophy. J. Transl. Med. 2023, 21, 503. [Google Scholar] [CrossRef] [PubMed]
- Vringer, E.; Tait, S.W.G. Mitochondria and cell death-associated inflammation. Cell Death Differ. 2023, 30, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Jan, R.; Chaudhry, G.-E. Understanding Apoptosis and Apoptotic Pathways Targeted Cancer Therapeutics. Adv. Pharm. Bull. 2019, 9, 205–218. [Google Scholar] [CrossRef]
- Xu, F.; Ren, L.; Song, M.; Shao, B.; Han, Y.; Cao, Z.; Li, Y. Fas- and Mitochondria-Mediated Signaling Pathway Involved in Osteoblast Apoptosis Induced by AlCl3. Biol. Trace Element Res. 2018, 184, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Pena-Blanco, A.; Garcia-Saez, A.J. Bax, Bak and beyond-mitochondrial performance in apoptosis. FEBS J. 2018, 285, 416–431. [Google Scholar] [CrossRef]
- Cosentino, K.; Hertlein, V.; Jenner, A.; Dellmann, T.; Gojkovic, M.; Peña-Blanco, A.; Dadsena, S.; Wajngarten, N.; Danial, J.S.; Thevathasan, J.V.; et al. The interplay between BAX and BAK tunes apoptotic pore growth to control mitochondrial-DNA-mediated inflammation. Mol. Cell 2022, 82, 933–949.e9. [Google Scholar] [CrossRef]
- Chertkova, R.V.; Brazhe, N.A.; Bryantseva, T.V.; Nekrasov, A.N.; Dolgikh, D.A.; Yusipovich, A.I.; Sosnovtseva, O.; Maksimov, G.V.; Rubin, A.B.; Kirpichnikov, M.P. New insight into the mechanism of mitochondrial cytochrome c function. PLoS ONE 2017, 12, e0178280. [Google Scholar] [CrossRef] [PubMed]
- Shao, G.; Wang, L.; Wang, X.; Fu, C. Apaf-1/caspase-4 pyroptosome: A mediator of mitochondrial permeability transition-triggered pyroptosis. Signal Transduct. Target. Ther. 2021, 6, 116. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.K.; Shteinfer-Kuzmine, A.; Chalifa-Caspi, V.; Shoshan-Barmatz, V. Non-apoptotic activity of the mitochondrial protein SMAC/Diablo in lung cancer: Novel target to disrupt survival, inflammation, and immunosuppression. Front. Oncol. 2022, 12, 992260. [Google Scholar] [CrossRef] [PubMed]
- Sevrioukova, I.F. Apoptosis-Inducing Factor: Structure, Function, and Redox Regulation. Antioxid. Redox Signal. 2011, 14, 2545–2579. [Google Scholar] [CrossRef] [PubMed]
- Bauer, T.M.; Murphy, E. Role of Mitochondrial Calcium and the Permeability Transition Pore in Regulating Cell Death. Circ. Res. 2020, 126, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Czabotar, P.E.; Garcia-Saez, A.J. Mechanisms of BCL-2 family proteins in mitochondrial apoptosis. Nat. Rev. Mol. Cell Biol. 2023, 24, 732–748. [Google Scholar] [CrossRef] [PubMed]
- Cavalcante, G.C.; Schaan, A.P.; Cabral, G.F.; Santana-Da-Silva, M.N.; Pinto, P.; Vidal, A.F.; Ribeiro-Dos-Santos, A. A Cell’s Fate: An Overview of the Molecular Biology and Genetics of Apoptosis. Int. J. Mol. Sci. 2019, 20, 4133. [Google Scholar] [CrossRef] [PubMed]
- Kalkavan, H.; Green, D.R. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 2018, 25, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Guilbaud, E.; Tait, S.W.G.; Yamazaki, T.; Galluzzi, L. Mitochondrial control of inflammation. Nat. Rev. Immunol. 2023, 23, 159–173. [Google Scholar] [CrossRef]
- Decout, A.; Katz, J.D.; Venkatraman, S.; Ablasser, A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 2021, 21, 548–569. [Google Scholar] [CrossRef]
- Kemmoku, H.; Takahashi, K.; Mukai, K.; Mori, T.; Hirosawa, K.M.; Kiku, F.; Uchida, Y.; Kuchitsu, Y.; Nishioka, Y.; Sawa, M.; et al. Single-molecule localization microscopy reveals STING clustering at the trans-Golgi network through palmitoylation-dependent accumulation of cholesterol. Nat. Commun. 2024, 15, 220. [Google Scholar] [CrossRef] [PubMed]
- Xian, H.; Watari, K.; Sanchez-Lopez, E.; Offenberger, J.; Onyuru, J.; Sampath, H.; Ying, W.; Hoffman, H.M.; Shadel, G.S.; Karin, M. Oxidized DNA fragments exit mitochondria via mPTP- and VDAC-dependent channels to activate NLRP3 inflammasome and interferon signaling. Immunity 2022, 55, 1370–1385.e8. [Google Scholar] [CrossRef]
- Chen, Q.-L.; Yin, H.-R.; He, Q.-Y.; Wang, Y. Targeting the NLRP3 inflammasome as new therapeutic avenue for inflammatory bowel disease. Biomed. Pharmacother. 2021, 138, 111442. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Hu, D.; Wu, C.; Feng, Y.; Li, A.; Liu, W.; Wang, Y.; Chen, K.; Tian, M.; Xiao, F.; et al. STING promotes NLRP3 localization in ER and facilitates NLRP3 deubiquitination to activate the inflammasome upon HSV-1 infection. PLoS Pathog. 2020, 16, e1008335. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhang, Z.; Sandhu, G.; Ma, X.; Yang, X.; Geiger, J.D.; Kong, J. Evidence of oxidative stress-induced BNIP3 expression in amyloid beta neurotoxicity. Brain Res. 2007, 1138, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Swomley, A.M.; Butterfield, D.A. Oxidative stress in Alzheimer disease and mild cognitive impairment: Evidence from human data provided by redox proteomics. Arch. Toxicol. 2015, 89, 1669–1680. [Google Scholar] [CrossRef] [PubMed]
- Shukla, D.; Mandal, P.K.; Tripathi, M.; Vishwakarma, G.; Mishra, R.; Sandal, K. Quantitation of in vivo brain glutathione conformers in cingulate cortex among age-matched control, MCI, and AD patients using MEGA-PRESS. Hum. Brain Mapp. 2020, 41, 194–217. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.-W.; Kim, S.J.; Kim, M.-S. Oxidative stress with tau hyperphosphorylation in memory impaired 1,2-diacetylbenzene-treated mice. Toxicol. Lett. 2017, 279, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef]
- Kim, D.I.; Lee, K.H.; Gabr, A.A.; Choi, G.E.; Kim, J.S.; Ko, S.H.; Han, H.J. Abeta-Induced Drp1 phosphorylation through Akt activation promotes excessive mitochondrial fission leading to neuronal apoptosis. Biochim. Biophys. Acta 2016, 1863, 2820–2834. [Google Scholar] [CrossRef]
- Cho, D.H.; Nakamura, T.; Fang, J.; Cieplak, P.; Godzik, A.; Gu, Z.; Lipton, S.A. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science 2009, 324, 102–105. [Google Scholar] [CrossRef]
- Manczak, M.; Reddy, P.H. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: Implications for mitochondrial dysfunction and neuronal damage. Hum. Mol. Genet. 2012, 21, 2538–2547. [Google Scholar] [CrossRef]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Jiang, S.; Nandy, P.; Wang, W.; Ma, X.; Hsia, J.; Wang, C.; Wang, Z.; Niu, M.; Siedlak, S.L.; Torres, S.; et al. Mfn2 ablation causes an oxidative stress response and eventual neuronal death in the hippocampus and cortex. Mol. Neurodegener. 2018, 13, 5. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wang, X.; Hu, Y.; Zhao, J.-N.; Huang, C.-H.; Li, T.; Zhang, B.-G.; He, Y.; Wu, Y.-Q.; Zhang, Z.-J.; et al. Acetylated tau exacerbates learning and memory impairment by disturbing with mitochondrial homeostasis. Redox Biol. 2023, 62, 102697. [Google Scholar] [CrossRef]
- Sharma, N.; Banerjee, R.; Davis, R.L. Early Mitochondrial Defects in the 5xFAD Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2023, 91, 1323–1338. [Google Scholar] [CrossRef] [PubMed]
- Lonnemann, N.; Hosseini, S.; Marchetti, C.; Skouras, D.B.; Stefanoni, D.; D’alessandro, A.; Dinarello, C.A.; Korte, M. The NLRP3 inflammasome inhibitor OLT1177 rescues cognitive impairment in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2020, 117, 32145–32154. [Google Scholar] [CrossRef]
- Xie, X.; Ma, G.; Li, X.; Zhao, J.; Zhao, Z.; Zeng, J. Activation of innate immune cGAS-STING pathway contributes to Alzheimer’s pathogenesis in 5xFAD mice. Nat. Aging 2023, 3, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Flores, J.; Noël, A.; Foveau, B.; Beauchet, O.; LeBlanc, A.C. Pre-symptomatic Caspase-1 inhibitor delays cognitive decline in a mouse model of Alzheimer disease and aging. Nat. Commun. 2020, 11, 4571. [Google Scholar] [CrossRef]
- Udeochu, J.C.; Amin, S.; Huang, Y.; Fan, L.; Torres, E.R.S.; Carling, G.K.; Liu, B.; McGurran, H.; Coronas-Samano, G.; Kauwe, G.; et al. Tau activation of microglial cGAS-IFN reduces MEF2C-mediated cognitive resilience. Nat. Neurosci. 2023, 26, 737–750. [Google Scholar] [CrossRef]
- Zhou, C.; Wang, L.; Cheng, W.; Lv, J.; Guan, X.; Guo, T.; Wu, J.; Zhang, W.; Gao, T.; Liu, X.; et al. Two distinct trajectories of clinical and neurodegeneration events in Parkinson’s disease. NPJ Park. Dis. 2023, 9, 111. [Google Scholar] [CrossRef] [PubMed]
- MacMahon Copas, A.N.; McComish, S.F.; Fletcher, J.M.; Caldwell, M.A. The Pathogenesis of Parkinson’s Disease: A Complex Interplay Between Astrocytes, Microglia, and T Lymphocytes? Front. Neurol. 2021, 12, 666737. [Google Scholar] [CrossRef] [PubMed]
- Bloomingdale, P.; Karelina, T.; Ramakrishnan, V.; Bakshi, S.; Véronneau-Veilleux, F.; Moye, M.; Sekiguchi, K.; Meno-Tetang, G.; Mohan, A.; Maithreye, R.; et al. Hallmarks of neurodegenerative disease: A systems pharmacology perspective. CPT Pharmacomet. Syst. Pharmacol. 2022, 11, 1399–1429. [Google Scholar] [CrossRef]
- Behl, T.; Kumar, S.; Althafar, Z.M.; Sehgal, A.; Singh, S.; Sharma, N.; Badavath, V.N.; Yadav, S.; Bhatia, S.; Al-Harrasi, A.; et al. Exploring the Role of Ubiquitin-Proteasome System in Parkinson’s Disease. Mol. Neurobiol. 2022, 59, 4257–4273. [Google Scholar] [CrossRef]
- González-Rodríguez, P.; Zampese, E.; Stout, K.A.; Guzman, J.N.; Ilijic, E.; Yang, B.; Tkatch, T.; Stavarache, M.A.; Wokosin, D.L.; Gao, L.; et al. Disruption of mitochondrial complex I induces progressive parkinsonism. Nature 2021, 599, 650–656. [Google Scholar] [CrossRef]
- Maegawa, H.; Niwa, H. Generation of Mitochondrial Toxin Rodent Models of Parkinson’s Disease Using 6-OHDA, MPTP, and Rotenone. Methods Mol. Biol. 2021, 2322, 95–110. [Google Scholar]
- Taib, C.N.M.; Mustapha, M. MPTP-induced mouse model of Parkinson’s disease: A promising direction of therapeutic strategies. Bosn. J. Basic Med. Sci. 2021, 21, 422–433. [Google Scholar] [CrossRef]
- Funayama, M.; Nishioka, K.; Li, Y.; Hattori, N. Molecular genetics of Parkinson’s disease: Contributions and global trends. J. Hum. Genet. 2023, 68, 125–130. [Google Scholar] [CrossRef]
- Houlden, H.; Singleton, A.B. The genetics and neuropathology of Parkinson’s disease. Acta Neuropathol. 2012, 124, 325–338. [Google Scholar] [CrossRef]
- Harper, J.W.; Ordureau, A.; Heo, J.-M. Building and decoding ubiquitin chains for mitophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 93–108. [Google Scholar] [CrossRef]
- Imai, Y. PINK1-Parkin signaling in Parkinson’s disease: Lessons from Drosophila. Neurosci. Res. 2020, 159, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Zambon, F.; Cherubini, M.; Fernandes, H.J.R.; Lang, C.; Ryan, B.J.; Volpato, V.; Bengoa-Vergniory, N.; Vingill, S.; Attar, M.; Booth, H.D.E.; et al. Cellular alpha-synuclein pathology is associated with bioenergetic dysfunction in Parkinson’s iPSC-derived dopamine neurons. Hum. Mol. Genet. 2019, 28, 2001–2013. [Google Scholar] [CrossRef]
- Zhou, Z.D.; Lee, J.C.T.; Tan, E.K. Pathophysiological mechanisms linking F-box only protein 7 (FBXO7) and Parkinson’s disease (PD). Mutat. Res. Rev. Mutat. Res. 2018, 778, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Noda, S.; Sato, S.; Fukuda, T.; Ueno, S.; Tada, N.; Hattori, N. Impaired mitochondrial accumulation and Lewy pathology in neuron-specific FBXO7-deficient mice. Mol. Brain 2022, 15, 54. [Google Scholar] [CrossRef] [PubMed]
- Aasly, J.O. Long-Term Outcomes of Genetic Parkinson’s Disease. J. Mov. Disord. 2020, 13, 81–96. [Google Scholar] [CrossRef]
- Di Maio, R.; Hoffman, E.K.; Rocha, E.M.; Keeney, M.T.; Sanders, L.H.; De Miranda, B.R.; Zharikov, A.; Van Laar, A.; Stepan, A.F.; Lanz, T.A.; et al. LRRK2 activation in idiopathic Parkinson’s disease. Sci. Transl. Med. 2018, 10, aar5429. [Google Scholar] [CrossRef]
- Ludtmann, M.H.R.; Kostic, M.; Horne, A.; Gandhi, S.; Sekler, I.; Abramov, A.Y. LRRK2 deficiency induced mitochondrial Ca(2+) efflux inhibition can be rescued by Na(+)/Ca(2+)/Li(+) exchanger upregulation. Cell. Death Dis. 2019, 10, 265. [Google Scholar] [CrossRef]
- Zhou, W.; Ma, D.; Tan, E.-K. Mitochondrial CHCHD2 and CHCHD10: Roles in Neurological Diseases and Therapeutic Implications. Neuroscientist 2020, 26, 170–184. [Google Scholar] [CrossRef]
- Ren, Y.L.; Jiang, Z.; Wang, J.Y.; He, Q.; Li, S.X.; Gu, X.J.; Qi, Y.R.; Zhang, M.; Yang, W.J.; Cao, B.; et al. Loss of CHCHD2 Stability Coordinates with C1QBP/CHCHD2/CHCHD10 Complex Impairment to Mediate PD-Linked Mitochondrial Dysfunction. Mol. Neurobiol. 2024. [Google Scholar] [CrossRef]
- Jiang, T.; Wang, Y.; Wang, X.; Xu, J. CHCHD2 and CHCHD10: Future therapeutic targets in cognitive disorder and motor neuron disorder. Front. Neurosci. 2022, 16, 988265. [Google Scholar] [CrossRef]
- Tio, M.; Wen, R.; Choo, C.N.; Bin Tan, J.; Chua, A.; Xiao, B.; Sundaram, J.R.; Chan, C.H.S.; Tan, E.-K. Genetic and pharmacologic p32-inhibition rescue CHCHD2-linked Parkinson’s disease phenotypes in vivo and in cell models. J. Biomed. Sci. 2024, 31, 24. [Google Scholar] [CrossRef]
- Gordon, R.; Albornoz, E.A.; Christie, D.C.; Langley, M.R.; Kumar, V.; Mantovani, S.; Robertson, A.A.B.; Butler, M.S.; Rowe, D.B.; O’neill, L.A.; et al. Inflammasome inhibition prevents α-synuclein pathology and dopaminergic neurodegeneration in mice. Sci. Transl. Med. 2018, 10, aah4066. [Google Scholar] [CrossRef]
- Amo-Aparicio, J.; Daly, J.; Højen, J.F.; Dinarello, C.A. Pharmacologic inhibition of NLRP3 reduces the levels of α-synuclein and protects dopaminergic neurons in a model of Parkinson’s disease. J. Neuroinflamm. 2023, 20, 147. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Ng, W.L.; Goh, S.Y.; Gulam, M.Y.; Wang, L.-F.; Tan, E.-K.; Ahn, M.; Chao, Y.-X. Targeting the inflammasome in Parkinson’s disease. Front. Aging Neurosci. 2022, 14, 957705. [Google Scholar] [CrossRef] [PubMed]
- Trudler, D.; Nazor, K.L.; Eisele, Y.S.; Grabauskas, T.; Dolatabadi, N.; Parker, J.; Sultan, A.; Zhong, Z.; Goodwin, M.S.; Levites, Y.; et al. Soluble alpha-synuclein-antibody complexes activate the NLRP3 inflammasome in hiPSC-derived microglia. Proc. Natl. Acad. Sci. USA 2021, 118, e2025847118. [Google Scholar] [CrossRef]
- Yang, L.; Zhao, K.; Calingasan, N.Y.; Luo, G.; Szeto, H.H.; Beal, M.F. Mitochondria Targeted Peptides Protect Against 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Neurotoxicity. Antioxid. Redox Signal. 2009, 11, 2095–2104. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Mao, P.; Calkins, M.J.; Cornea, A.; Reddy, A.P.; Murphy, M.P.; Szeto, H.H.; Park, B.; Reddy, P.H. Mitochondria-targeted antioxidants protect against amyloid-beta toxicity in Alzheimer’s disease neurons. J. Alzheimers Dis. 2010, 20 (Suppl. 2), S609–S631. [Google Scholar] [CrossRef]
- Reddy, P.H.; Manczak, M.; Kandimalla, R. Mitochondria-targeted small molecule SS31: A potential candidate for the treatment of Alzheimer’s disease. Hum. Mol. Genet. 2017, 26, 1597. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.L.; Wang, W.; Han, N.; Sun, H.L.; Dong, F.M.; Song, Y.X.; Feng, R.F.; Wang, J.H. The mitochondria-targeted small molecule SS31 delays progression of behavioral deficits by attenuating beta-amyloid plaque formation and mitochondrial/synaptic deterioration in APP/PS1 mice. Biochem. Biophys. Res. Commun. 2023, 658, 36–43. [Google Scholar] [CrossRef]
- Bido, S.; Soria, F.N.; Fan, R.Z.; Bezard, E.; Tieu, K. Mitochondrial division inhibitor-1 is neuroprotective in the A53T-α-synuclein rat model of Parkinson’s disease. Sci. Rep. 2017, 7, 7495. [Google Scholar] [CrossRef]
- Qi, X.; Qvit, N.; Su, Y.-C.; Mochly-Rosen, D. A Novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J. Cell Sci. 2013, 126 Pt 3, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Filichia, E.; Hoffer, B.; Qi, X.; Luo, Y. Inhibition of Drp1 mitochondrial translocation provides neural protection in dopaminergic system in a Parkinson’s disease model induced by MPTP. Sci. Rep. 2016, 6, 32656. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Johnson, M.; Renna, H.A.; Sheehan, K.M.; Ahmed, S.; Palaia, T.; Pinkhasov, A.; Gomolin, I.H.; De Leon, J.; Reiss, A.B. Therapeutic Potential of P110 Peptide: New Insights into Treatment of Alzheimer’s Disease. Life 2023, 13, 2156. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-N.; Zimmerman, M.; Milledge, G.Z.; Hou, X.-L.; Cheng, J.; Wang, Z.-H.; Li, P.A. Water-Soluble Coenzyme Q10 Reduces Rotenone-Induced Mitochondrial Fission. Neurochem. Res. 2017, 42, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- Komaki, H.; Faraji, N.; Komaki, A.; Shahidi, S.; Etaee, F.; Raoufi, S.; Mirzaei, F. Investigation of protective effects of coenzyme Q10 on impaired synaptic plasticity in a male rat model of Alzheimer’s disease. Brain Res. Bull. 2019, 147, 14–21. [Google Scholar] [CrossRef]
- Olajide, O.J.; La Rue, C.; Bergdahl, A.; Chapman, C.A. Inhibiting amyloid beta (1–42) peptide-induced mitochondrial dysfunction prevents the degradation of synaptic proteins in the entorhinal cortex. Front. Aging Neurosci. 2022, 14, 960314. [Google Scholar] [CrossRef]
- Xi, Y.; Feng, D.; Tao, K.; Wang, R.; Shi, Y.; Qin, H.; Murphy, M.P.; Yang, Q.; Zhao, G. MitoQ protects dopaminergic neurons in a 6-OHDA induced PD model by enhancing Mfn2-dependent mitochondrial fusion via activation of PGC-1alpha. Biochim. Biophys. Acta Mol. Basis. Dis. 2018, 1864 Pt B, 2859–2870. [Google Scholar] [CrossRef]
- Szabo, A.; Sumegi, K.; Fekete, K.; Hocsak, E.; Debreceni, B.; Setalo, G., Jr.; Kovacs, K.; Deres, L.; Kengyel, A.; Kovacs, D.; et al. Activation of mitochondrial fusion provides a new treatment for mitochondria-related diseases. Biochem. Pharmacol. 2018, 150, 86–96. [Google Scholar] [CrossRef] [PubMed]
- MacMullen, C.; Davis, R.L. High-Throughput Phenotypic Assay for Compounds That Influence Mitochondrial Health Using iPSC-Derived Human Neurons. SLAS Discov. 2021, 26, 811–822. [Google Scholar] [CrossRef]
- Varkuti, B.H.; Liu, Z.; Kepiro, M.; Pacifico, R.; Gai, Y.; Kamenecka, T.; Davis, R.L. High-Throughput Small Molecule Screen Identifies Modulators of Mitochondrial Function in Neurons. iScience 2020, 23, 101364. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Name | Target | Disease Models | Mechanism |
|---|---|---|---|
| Mitochondrial fusion and biogenesis enhancers | |||
| SS−31, | Mitochondrial dysfunction | MPTP−injected mice, MPP+−treated SN4741 cells | Restores oxygen consumption rates and ATP levels, and prevents mitochondrial swelling and apoptosis [116] |
| Aβ−incubated N2a cells | Prevents neurotoxicity by Increasing ATP levels, cytochrome oxidase activity, MMP, and the expression levels of MFN2, OPA1, and Prxs [117] | ||
| APP transgenic mice (Tg2576) | Enhances PGC−1α/Nrf1/Tfam signaling−induced mitochondrial biogenesis and MFN1/2−mediated mitochondrial fusion, and suppresses H2O2 production and lipid peroxidation [118] | ||
| APP/PS1 mice | Improves mitochondrial homeostasis by blocking DRP1 expression and inhibits apoptosis by decreasing Aβ and ROS levels [119] | ||
| BGP−15 | Mitochondrial dysfunction | Primary hippocampal neurons induced by acetylated tau (TauKQ) overexpression | Reduces fragmented mitochondria and mtROS levels through the activation of PGC−1α/Nrf1/Tfam pathway and MFN1/MFN2/OPA1 signaling, preventing ATP and MMP reduction [85] |
| Mitochondrial fission inhibitors | |||
| Mdivi−1 | DRP1 | Aβ−treated SK−NK−SH cells | Attenuates ROS levels, MMP disruption, and apoptosis by inhibiting caspase−9/−3 activation [80] |
| hA53T−α-syn−induced rat | Decreases mitochondrial fragmentation, lipid peroxidation, and α-synuclein aggregates, and enhances mitochondrial respiratory capacity [120] | ||
| P110 | DRP1 activity DRP1-FIS1 interation | MPP+−treated SH−SY5Y cells | Inhibits DRP1 translocation to the mitochondria, mitochondrial fission, and mtROS, and improves MMP and mitochondrial integrity [121] |
| MPTP−injected mice | Suppresses pro-apoptotic Bax/PUMA expression by inhibiting DRP1-dependent p53 mitochondrial translocation [122] | ||
| SH−SY5Y cells | Increases Tfam/Nrf1 expression and mitochondrial area, and decreases mitochondrial fragmentation and mtROS production [123] | ||
| Antioxidants | |||
| CoQ10 | ROS | Rotenone-treated HT22 cells | Prevents DRP1/FIS1−induced mitochondrial fragmentation and cell death [124] |
| Aβ-injected adult rats | Protects hippocampal synaptic plasticity by decreasing serum levels of malondialdehyde and total oxidant [125] | ||
| MitoQ | mtROS | APP transgenic mice (Tg2576) | Reduces mitochondrial damage by inhibiting cyclophilin D expression and enhances neurite outgrowth [117] |
| hAβ1–42-incubated entorhinal cortex slices | Blocks the reduction of SOD2/mitochondrial Cyt c and synaptic proteins PSD95/synaptophysin expression [126] | ||
| 6-OHDA-treated SN4741 cells and mice | Promotes MFN2−dependent mitochondrial fusion by activating PGC−1α, resulting in the suppression of mitochondrial fragmentation and neuronal apoptosis [127] | ||
| Mitochondria-driven inflammation inhibitors | |||
| MCC950 | ATPase activity of NLRP3 | 6-OHDA administered mice, MitoPark mice, PFF-injected mice | Inhibits the activation and release of caspase−1/IL−1β/ASC, α-synuclein accumulation, and dopaminergic degeneration [112] |
| OLT1177 | ATPase activity of NLRP3 | APP/PS1 mice | Reduces the levels of proinflammatory cytokines IL−1β, IL−6, and TNF−α, microglia activation, and the number of Aβ plaques [87] |
| MPTP-injected mice | Induces the clearance of α-synuclein by increasing LC3−mediate autophagy and suppresses the levels of IL−1β, IL−18, IL−6, and IL−17A and microgliosis reactivity [113] | ||
| VX−765 | Caspase−1 activity | APPSw/Ind mutant J20 mice | Decreases Iba1−positive microglial inflammation and cleaved IL−1β expression, but does not affect Aβ accumulation [89] |
| TDI−6570 | cGAS | P301S tau transgenic mice | Restores synaptic integrity and plasticity by reducing IFN−stimulated gene expression and enhancing MEF2C transcriptional network [90] |
| H−151 | STING | 5×FAD mice | Suppresses the activation of the TBK1/p65/IRF3 pathway, the expression of neuroinflammatory genes, and Aβ42 levels [88] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, E.-H.; Kim, M.-H.; Park, S.-J. Targeting Mitochondrial Dysfunction and Reactive Oxygen Species for Neurodegenerative Disease Treatment. Int. J. Mol. Sci. 2024, 25, 7952. https://doi.org/10.3390/ijms25147952
Choi E-H, Kim M-H, Park S-J. Targeting Mitochondrial Dysfunction and Reactive Oxygen Species for Neurodegenerative Disease Treatment. International Journal of Molecular Sciences. 2024; 25(14):7952. https://doi.org/10.3390/ijms25147952
Chicago/Turabian StyleChoi, Eui-Hwan, Mi-Hye Kim, and Sun-Ji Park. 2024. "Targeting Mitochondrial Dysfunction and Reactive Oxygen Species for Neurodegenerative Disease Treatment" International Journal of Molecular Sciences 25, no. 14: 7952. https://doi.org/10.3390/ijms25147952
APA StyleChoi, E.-H., Kim, M.-H., & Park, S.-J. (2024). Targeting Mitochondrial Dysfunction and Reactive Oxygen Species for Neurodegenerative Disease Treatment. International Journal of Molecular Sciences, 25(14), 7952. https://doi.org/10.3390/ijms25147952