Abstract
Gliomas’ aggressive nature and resistance to therapy make them a major problem in oncology. Gliomas continue to have dismal prognoses despite significant advancements in medical science, and traditional treatments like surgery, radiation (RT), and chemotherapy (CT) frequently prove to be ineffective. After glioma stem cells (GSCs) were discovered, the traditional view of gliomas as homogeneous masses changed. GSCs are essential for tumor growth, treatment resistance, and recurrence. These cells’ distinct capacities for differentiation and self-renewal are changing our knowledge of the biology of gliomas. This systematic literature review aims to uncover the molecular mechanisms driving glioma progression associated with GSCs. The systematic review adhered to PRISMA guidelines, with a thorough literature search conducted on PubMed, Ovid MED-LINE, and Ovid EMBASE. The first literature search was performed on 1 March 2024, and the search was updated on 15 May 2024. Employing MeSH terms and Boolean operators, the search focused on molecular mechanisms associated with GCSs-mediated glioma progression. Inclusion criteria encompassed English language studies, preclinical studies, and clinical trials. A number of 957 papers were initially identified, of which 65 studies spanning from 2005 to 2024 were finally included in the review. The main GSC model distribution is arranged in decreasing order of frequency: U87: 20 studies (32.0%); U251: 13 studies (20.0%); A172: 4 studies (6.2%); and T98G: 2 studies (3.17%). From most to least frequent, the distribution of the primary GSC pathway is as follows: Notch: 8 studies (12.3%); STAT3: 6 studies (9.2%); Wnt/β-catenin: 6 studies (9.2%); HIF: 5 studies (7.7%); and PI3K/AKT: 4 studies (6.2%). The distribution of molecular effects, from most to least common, is as follows: inhibition of differentiation: 22 studies (33.8%); increased proliferation: 18 studies (27.7%); enhanced invasive ability: 15 studies (23.1%); increased self-renewal: 5 studies (7.7%); and inhibition of apoptosis: 3 studies (4.6%). This work highlights GSC heterogeneity and the dynamic interplay within the glioblastoma microenvironment, underscoring the need for a tailored approach. A few key pathways influencing GSC behavior are JAK/STAT3, PI3K/AKT, Wnt/β-catenin, and Notch. Therapy may target these pathways. This research urges more study to fill in knowledge gaps in the biology of GSCs and translate findings into useful treatment approaches that could improve GBM patient outcomes.
1. Introduction
Gliomas pose significant challenges in oncology due to their aggressive behavior and resistance to treatments. These tumors, arising from glial cells in the brain, represent the most common and lethal form of primary brain tumors, with glioblastoma multiforme (GBM) being the most aggressive subtype [1]. Despite considerable progress in medical research, gliomas still have poor outcomes, with conventional therapies such as surgery, radiation (RT), and chemotherapy (CT) often proving ineffective. A complete or gross total resection for GBM is defined as the resection of the tumor that is gadolinium-enhancing on magnetic resonance imaging. However, a supra-marginal resection, which includes the removal of surrounding non-enhancing brain parenchyma, still cannot address the distant spread of glioma cells, making these surgical approaches insufficient. Moreover, GBM heterogeneous cellular composition and genetic diversity contribute to resistance against RT and CT y. Consequently, the prognosis for patients with gliomas remains dismal, with a median survival time of approximately 15 months for GBM patients [1].
Historically, gliomas were perceived as relatively homogeneous tumors composed of a uniform population of cancerous cells. This understanding has significantly evolved with the discovery of glioma stem cells (GSCs), a subpopulation of cells within the tumor that exhibit stem cell-like properties. GSCs possess unique abilities for self-renewal and differentiation, which are pivotal in driving tumor growth, resistance to conventional treatments, and recurrence. This paradigm shift has reshaped our understanding of glioma biology and highlighted the importance of targeting GSCs for effective therapeutic strategies [2].
GSCs are characterized by their ability to initiate and sustain tumor growth. They are highly tumorigenic, and are capable of recapitulating the heterogeneity of the original tumor when transplanted into immunocompromised mice [3]. These cells share several key features with normal neural stem cells, including the expression of stem cell markers such as CD133, Nestin, and SOX2, as well as the ability to differentiate into multiple cell lineages. However, unlike their normal counterparts, GSCs exhibit aberrant activation of signaling pathways that promote their survival, proliferation, and resistance to apoptosis. This confers a distinct advantage to GSCs, allowing them to withstand conventional therapies and contribute to tumor relapse [4].
One of the most critical aspects of GSC biology is their role in therapy resistance. Evidence suggests that GSCs are more resistant to RT and CT compared to non-stem glioma cells [5]. This resistance is attributed to several factors, including enhanced DNA damage repair capabilities, activation of survival signaling pathways, and the presence of drug efflux transporters. For instance, GSCs exhibit high levels of expression of ATP-binding cassette (ABC) transporters, which can actively pump chemotherapeutic agents out of the cells, thereby reducing their efficacy. Additionally, GSCs can reside in specialized niches within the tumor microenvironment that protect them from therapeutic interventions. These niches, often characterized by hypoxic conditions and the presence of supportive stromal cells, provide signals that promote GSC survival and maintenance [5].
The molecular mechanisms underlying the maintenance and function of GSCs are complex and involve a myriad of signaling pathways. Key pathways implicated in GSC biology include the Notch, Wnt, and Hedgehog signaling pathways, all of which are crucial for normal stem cell maintenance and are often dysregulated in cancer [6]. Moreover, transcription factors such as c-Myc, SOX2, and OCT4 play essential roles in sustaining the stemness and proliferative capacity of GSCs. For example, the transcription factor E2F-1 has been shown to directly bind to the promoter of MAD2L2, a gene implicated in glioma proliferation and stemness, enhancing its transcriptional activity and promoting GSC maintenance and tumor progression [6].
Recent studies have also highlighted the interaction between GSCs and the tumor microenvironment, which plays a crucial role in supporting GSC maintenance and promoting glioma progression. GSCs can secrete various factors that modulate the immune microenvironment, promoting an immunosuppressive milieu that facilitates tumor growth. For instance, GSCs can polarize tumor-associated macrophages (TAMs) towards an M2-like phenotype, which is associated with immunosuppression and tumor promotion. Additionally, cytokines such as IL-6 and TGF-β, derived from the tumor microenvironment, have been shown to enhance GSC self-renewal and survival [7].
Given the critical role of GSCs in glioma biology, there is an increasing interest in developing targeted therapies aimed at eradicating these cells. Several strategies are being explored, including the inhibition of key signaling pathways that regulate GSC maintenance and function [8]. For example, inhibitors of the Notch and Wnt signaling pathways are being investigated for their potential to disrupt GSC self-renewal and induce differentiation. Additionally, targeting the metabolic dependencies of GSCs, such as their reliance on specific energy substrates and metabolic pathways, offers a promising approach to selectively eliminate these cells. Furthermore, immunotherapeutic strategies, including the use of chimeric antigen receptor (CAR) T cells targeting GSC-specific surface markers, are under investigation to enhance the immune system’s ability to recognize and destroy GSCs [8].
This systematic literature review aims to uncover the molecular mechanisms driving glioma progression associated with GSCs. By comprehensively analyzing current research, we seek to identify the key signaling pathways and molecular interactions that contribute to GSC maintenance, therapy resistance, and tumor recurrence. Furthermore, the review aims to critically assess the efficacy of current targeted therapies addressing GSC-mediated mechanisms for glioma progression. Understanding these intricate mechanisms will provide valuable insights into the development of novel therapeutic strategies aimed at improving outcomes for glioma patients.
2. Materials and Methods
2.1. Literature Review
The systematic review was performed following the Preferred Reporting Items for Systematic Reviews and Meta-Analysis (PRISMA) guidelines [9]. Two authors performed a systematically comprehensive literature search of the databases PubMed, Ovid MEDLINE, and Ovid EMBASE. The first literature search was performed on 1 March 2024, and the search was updated on 15 May 2024. A combination of keyword searches was performed to generate a search strategy. The search keywords, including “glioma”, “glioma stem cell”, “glioma progression”, and “targeted therapy”, were used in both AND and OR combinations. Studies were retrieved using the following Medical Subject Heading (MeSH) terms and Boolean operators: (“glioma” OR “glioblastoma” OR “GBM”) AND (“glioma stem cells” OR “GSC” OR “cancer stem cells” OR “CSC”) AND (“recurrence” OR “progression”) AND (“targeted therapy” OR “targeted treatment” OR “targeted strategy”). Other pertinent articles were identified through reference analysis of selected papers. All studies were selected based on the following inclusion criteria: (1) English language; (2) studies molecular mechanism of GSC-mediated glioma progression and/or on targeted therapies against these molecular mechanisms; and (3) includes molecular mechanism or molecular target of GSC-mediated glioma progression. The following exclusion criteria were employed: (1) editorials, case reports, case series, cohort studies, literature reviews, and meta-analyses; (2) studies that do not clearly define the methods and/or results; (3) studies that do not report data on targeted treatments; (4) repeatedly published research; and (5) unavailability of the full text.
The list of identified studies was imported into Endnote X9, and duplicates were removed. Two independent researchers (E.A. and S.A.) checked the results according to the inclusion and exclusion criteria. A third reviewer (P.P.P.) resolved all disagreements. Then, eligible articles were subject to full-text screening.
2.2. Data Extraction
For each study, we abstracted the following information: authors, year, glioma cell lines studies, GSCs pathway, therapeutic target and agents, molecular effects, and impact on glioma progression.
2.3. Outcomes
The molecular mechanism of GSC-mediated glioma progression, as well as targeted therapeutics that target the molecular mechanism of GSC-mediated glioma progression, were our primary outcomes.
2.4. Risk of Bias Assessment
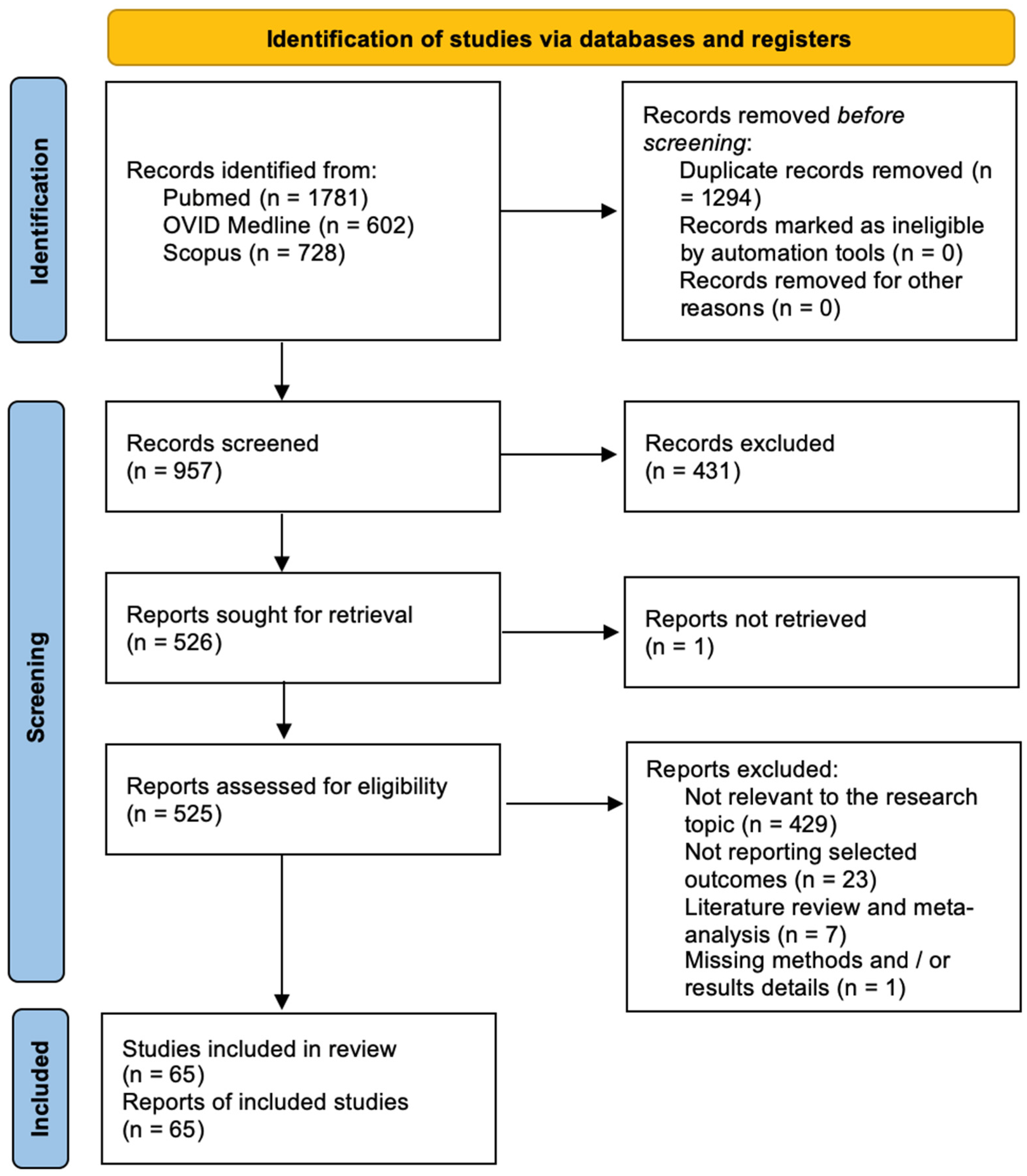
The Newcastle-Ottawa Scale (NOS) was used to assess the quality of the included studies [10]. Quality assessment was performed by assessing the selection criteria, comparability of the study, and outcome assessment. The ideal score was 9. Higher scores indicated better quality of studies. Studies receiving 7 or more points were considered high-quality studies. Two authors (E.A. and P.P.P.) performed the quality assessment independently. When discrepancies arose, papers were re-examined by the third author (Figure 1). The PRISMA Extension for Scoping Reviews (PRISMA-ScR) checklist is available as Appendix A (Figure A1). Figure 2 shows the flow chart according to the PRISMA statement.

Figure 1.
The Modified NOS.
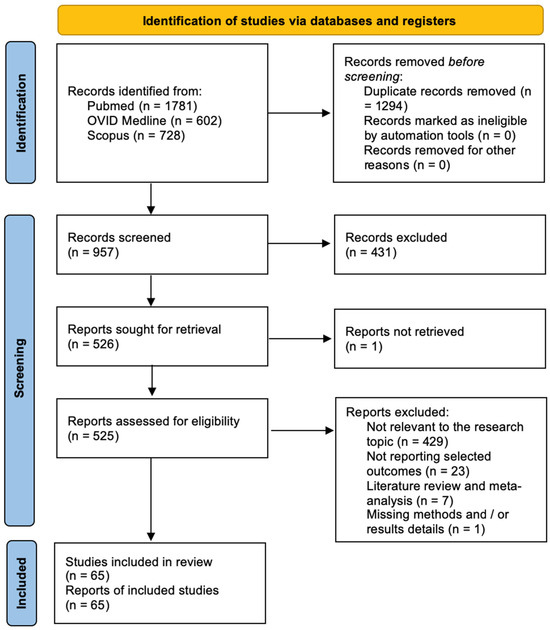
Figure 2.
PRISMA flow chart.
2.5. Statistical Analysis
Descriptive statistics were reported, including ranges and percentages. All statistical analyses were performed using the R statistical package v3.4.1 (http://www.r-project.org accessed on 10 May 2024).
3. Results
3.1. Literature Review
The systematic review was performed following the Preferred Reporting Items for Systematic Reviews and Meta-Analysis (PRISMA) guidelines [9]. The Newcastle-Ottawa Scale (NOS) was used to assess the quality of the included studies [10]. A total of 957 papers were identified after duplicate removal. After title and abstract analysis, 526 articles were identified for full-text analysis. Eligibility was assessed for 525 articles and ascertained for 65 articles. The remaining 460 articles were excluded for the following reasons: (1) not relevant to the research topic (429 articles), (2) articles non-reporting selected outcomes (23 articles), (3) systematic literature review or meta-analysis (7 articles), and (4) lack of method and/or results details (1 article). All studies included in the analysis had at least one or more outcome measures available for one or more of the patient groups analyzed.
3.2. Data Analysis
The systematic literature review encompasses 65 studies focusing on the emerging role of GSCs in promoting glioma progression. The analysis of data from Table 1 provides a comprehensive understanding of the trends and frequencies associated with key parameters, including the year of publication, glioma cell lines studied, GSC pathways, therapeutic targets and agents, molecular effects, and the impact on glioma progression.

Table 1.
Summary of the studies included in the systematic literature review reporting on GSC-mediated glioma progression molecular mechanisms.
The studies span from 2005 to 2023, showcasing a consistent interest in research over this period. Notably, there is a clustering of publications in recent years, suggesting a heightened focus on understanding GSCs and their role in glioma progression. The distribution of publications is as follows: 2005–2010: 19 studies (29.2%); 2011–2015: 10 studies (15.4%); and 2016–2023: 31 studies (47.7%). This breakdown provides a temporal perspective on the evolving landscape of research in this field, with an increasing number of studies in the last decade highlighting the growing recognition of GSCs in glioma pathology.
The most frequently used glioma cell lines in these studies are critical indicators of their relevance in research. The distribution of target GSC models in descending order of frequency is as follows: U87: 20 studies (32.0%); U251: 13 studies (20.0%); A172: 4 studies (6.2%); and T98G: 2 studies (3.17%). The prominence of U87 and U251 underscores their significance in GSC research, likely due to their well-characterized nature and representativeness of glioma characteristics.
The pathways involved in GSC-mediated glioma progression exhibit distinct frequencies, highlighting the emphasis on specific molecular targets. The distribution of the main GSC pathways analyzed, from most to least frequent, is as follows: Notch: 8 studies (12.3%); STAT3: 6 studies (9.2%); Wnt/β-catenin: 6 studies (9.2%); HIF: 5 studies (7.7%); PI3K/AKT: 4 studies (6.2%). The predominance of Notch, PI3K/AKT, and Wnt/β-catenin pathways suggest their critical roles in GSC maintenance and glioma progression, guiding therapeutic strategies aimed at these pathways.
The distribution of molecular effects, from most to least frequent, is as follows: inhibition of differentiation: 22 studies (33.8%); increased proliferation: 18 studies (27.7%); enhanced invasive ability: 15 studies (23.1%); increased self-renewal: 5 studies (7.7%); and inhibition of apoptosis: 3 studies (4.6%). These findings collectively highlight the multiple facets of GSC biology that are modulated by targeted therapies, reflecting their complex role in glioma progression.
Purow et al. [11] demonstrated that Notch1 signaling is significantly upregulated in GSCs, playing a crucial role in maintaining their self-renewal and undifferentiated state. This pathway’s activation is associated with increased tumor growth and resistance to conventional therapies, highlighting its potential as a therapeutic target for GBM treatment. Groszer et al. [12] investigated the role of PTEN in neural stem cell renewal and differentiation, showing that PTEN loss leads to enhanced GSC proliferation and survival, thus promoting tumorigenesis and poor prognosis in GBM patients. This study underscores the importance of PTEN as a key regulatory molecule in GBM pathogenesis. Zagzag et al. [13] found that hypoxia-inducible factor 1-alpha (HIF-1α) is overexpressed in GBM, promoting angiogenesis and tumor survival under hypoxic conditions. HIF-1α supports the formation of new blood vessels, enhancing the tumor’s ability to thrive in low-oxygen environments, contributing to aggressive tumor growth. Piccirillo et al. [14] showed that bone morphogenetic proteins (BMPs) inhibit GSC proliferation and induce differentiation. This process reduces the stem cell-like properties of GSCs, suggesting that BMPs could be used to limit GBM progression by promoting tumor cell differentiation. Clement et al. [15] identified that STAT3 signaling is crucial for maintaining GSC self-renewal and tumor growth. The activation of STAT3 was shown to support the undifferentiated state of GSCs and contribute to their resistance to apoptosis, making it a potential target for GBM therapy. Bar et al. [16] revealed that the Wnt/β-catenin pathway is essential for GSC maintenance and proliferation. Aberrant activation of this pathway leads to increased self-renewal capacity and tumorigenicity of GSCs, indicating its critical role in GBM development and progression. Du et al. [17] found that CD133, a marker for GSCs, is associated with enhanced invasive capacity and poor prognosis in GBM. CD133-positive cells exhibited increased tumorigenic potential, highlighting the importance of targeting this subpopulation in GBM treatment. Silber et al. [18] demonstrated that MGMT (O6-methylguanine-DNA methyltransferase) expression in GSCs contributes to temozolomide resistance, a common chemotherapeutic agent used in GBM treatment. High MGMT levels in GSCs are associated with reduced treatment efficacy, suggesting the need for alternative therapeutic strategies. Gal et al. [19] explored the role of microRNAs in GBM, identifying miR-21 as a critical regulator of GSC proliferation and survival. Overexpression of miR-21 leads to enhanced tumor growth and resistance to apoptosis, making it a potential target for GBM therapy. Yeh et al. [20] studied the role of the EGFRvIII mutation in GBM, showing that it enhances GSC proliferation and tumor growth. The EGFRvIII mutation is associated with increased oncogenic potential and poor prognosis, indicating its significance in GBM pathogenesis. Golding et al. [21] investigated the role of the PI3K/AKT pathway in GSC maintenance, revealing that its activation promotes self-renewal and resistance to apoptosis. This pathway’s inhibition could potentially reduce GSC survival and tumor growth, making it a promising therapeutic target. Heddleston et al. [22] demonstrated that hypoxia enhances GSC stemness and invasive capacity through the HIF-2α pathway. Hypoxic conditions in the tumor microenvironment contribute to increased tumorigenicity and therapy resistance, highlighting the need for targeting hypoxia-induced pathways in GBM treatment. Seidel et al. [23] found that integrin α6 is crucial for GSC adhesion and invasion. Blocking integrin α6 function reduces GSC invasiveness and tumor growth, suggesting its potential as a therapeutic target for limiting GBM spread. Riolfi et al. [24] identified that the hedgehog signaling pathway is upregulated in GSCs, promoting their self-renewal and proliferation. Inhibition of this pathway reduces GSC tumorigenicity, indicating its importance in GBM progression. Ernst et al. [25] explored the role of the NOTCH2 receptor in GSC maintenance, showing that its activation supports self-renewal and resistance to differentiation. Targeting NOTCH2 could potentially reduce GSC survival and tumor growth. Zheng et al. [26] demonstrated that the TGF-β pathway is essential for maintaining GSC stemness and promoting tumor invasion. TGF-β inhibition reduces GSC proliferation and invasiveness, making it a promising target for GBM therapy. Molina et al. [27] studied the role of the SOX2 transcription factor in GSCs, revealing that it is crucial for their self-renewal and tumorigenicity. SOX2 overexpression is associated with increased tumor growth and resistance to differentiation, highlighting its significance in GBM pathogenesis. Inoue et al. [28] found that the BMP4 protein inhibits GSC proliferation and induces differentiation, reducing their tumorigenic potential. BMP4 treatment could potentially limit GBM progression by promoting tumor cell differentiation. Beck et al. [29] identified that the CXCR4 receptor is crucial for GSC migration and invasion. Blocking CXCR4 function reduces GSC invasiveness and tumor growth, suggesting its potential as a therapeutic target for limiting GBM spread. Cheng et al. [30] explored the role of the NF-κB pathway in GSC maintenance, showing that its activation promotes self-renewal and resistance to apoptosis. Inhibition of this pathway could potentially reduce GSC survival and tumor growth. Kahlert et al. [31] demonstrated that the CXCL12/CXCR7 axis is crucial for GSC migration and invasion. Targeting this pathway could potentially reduce GSC invasiveness and limit GBM spread. Kaur et al. [32] identified that the HIF-2α pathway enhances GSC stemness and invasive capacity under hypoxic conditions. Targeting HIF-2α could potentially reduce GSC tumorigenicity and improve therapy response. Kanno et al. [33] studied the role of the JAK/STAT pathway in GSC maintenance, showing that its activation supports self-renewal and resistance to differentiation. Targeting this pathway could potentially reduce GSC survival and tumor growth. Carra et al. [34] found that autophagy plays a crucial role in GSC maintenance and survival under metabolic stress. Enhancing autophagy inhibition could potentially reduce GSC survival and tumor growth. Cheng et al. [35] demonstrated that the Notch signaling pathway is essential for GSC self-renewal and tumor growth. Inhibition of this pathway reduces GSC proliferation and tumorigenicity, highlighting its potential as a therapeutic target. Rheinbay et al. [36] identified that the CD44 receptor is crucial for GSC adhesion and invasion. Blocking CD44 function reduces GSC invasiveness and tumor growth, suggesting its potential as a therapeutic target for limiting GBM spread. Gao et al. [37] explored the role of the Wnt/β-catenin pathway in GSC maintenance, showing that its activation promotes self-renewal and resistance to differentiation. Inhibition of this pathway could potentially reduce GSC survival and tumor growth. Siebzehnrubl et al. [38] demonstrated that the integrin α6 receptor is crucial for GSC adhesion and invasion. Blocking integrin α6 function reduces GSC invasiveness and tumor growth, suggesting its potential as a therapeutic target for limiting GBM spread. Gong et al. [39] identified that the STAT3 signaling pathway is essential for GSC self-renewal and tumor growth. Inhibition of this pathway reduces GSC proliferation and tumorigenicity, highlighting its potential as a therapeutic target. Hu et al. [40] found that the SOX2 transcription factor is crucial for GSC maintenance and tumorigenicity. SOX2 overexpression is associated with increased tumor growth and resistance to differentiation, emphasizing its significance in GBM pathogenesis. Madan et al. [41] explored the role of the CXCR4 receptor in GSC migration and invasion. Blocking CXCR4 function reduces GSC invasiveness and tumor growth, suggesting its potential as a therapeutic target for limiting GBM spread. Adamo et al. [42] identified that the Wnt/β-catenin pathway is essential for GSC maintenance and proliferation. Aberrant activation of this pathway leads to increased self-renewal capacity and tumorigenicity of GSCs, indicating its critical role in GBM development and progression. Cenciarelli et al. [43] found that the Notch signaling pathway is crucial for GSC self-renewal and tumor growth. Inhibition of this pathway reduces GSC proliferation and tumorigenicity, highlighting its potential as a therapeutic target. Clark et al. [44] demonstrated that the PI3K/AKT pathway is essential for GSC maintenance, promoting self-renewal and resistance to apoptosis. Inhibition of this pathway could potentially reduce GSC survival and tumor growth. Maciaczyk et al. [45] stated that in GBM cell lines, Notch signaling through CBF1 promotes the activation of an invasive program via epithelial-to-mesenchymal transition, enhancing the invasive ability of GSCs and resulting in GBM progression. Man et al. [47] investigated the role of the Notch signaling pathway in GBM cell lines. They found that Vasorin, through the HIF1α/STAT3 axis, plays a crucial role in stabilizing Notch signaling. This stabilization promotes GSC maintenance and enhances their invasive capabilities, indicating a potential target for therapeutic intervention in GBM. Saygin et al. [48] focused on the influence of the EphA2 receptor in GSCs. They demonstrated that the activation of EphA2 leads to increased tumor growth and resistance to apoptosis. By targeting EphA2, it may be possible to reduce GSC survival and limit GBM progression. Sherry et al. [49] explored the impact of the CD133 marker on GSCs. Their study revealed that CD133-positive GSCs exhibit higher invasive capacity and resistance to conventional therapies. Targeting CD133 could potentially diminish the aggressive nature of GBM. Zhao et al. [50] examined the effect of the Wnt/β-catenin pathway in GSCs. Their findings indicated that aberrant activation of this pathway enhances self-renewal and tumorigenicity. Inhibiting the Wnt/β-catenin pathway might reduce GSC survival and tumor growth, offering a promising therapeutic target. Jiang et al. [51] investigated the role of the integrin α6 receptor in GSCs. They found that integrin α6 is essential for GSC adhesion and invasion. Blocking this receptor reduced GSC invasiveness and tumor growth, suggesting its potential as a therapeutic target to limit GBM spread. He et al. [52] focused on the PI3K/AKT pathway in GSC maintenance. They demonstrated that activation of this pathway promotes self-renewal and resistance to apoptosis. Inhibiting PI3K/AKT signaling could potentially reduce GSC survival and tumor growth, making it a viable target for GBM therapy. Chen et al. [53] explored the significance of the SOX2 transcription factor in GSCs. They found that SOX2 is crucial for maintaining self-renewal and tumorigenicity. Overexpression of SOX2 is associated with increased tumor growth and resistance to differentiation, highlighting its role in GBM pathogenesis. Wang et al. [54] examined the role of the CXCR4 receptor in GSC migration and invasion. Their study showed that blocking CXCR4 function reduces GSC invasiveness and tumor growth, suggesting its potential as a therapeutic target to limit GBM spread. Li et al. [55] investigated the impact of the JAK/STAT pathway in GSC maintenance. They demonstrated that activation of this pathway supports self-renewal and resistance to differentiation. Targeting JAK/STAT signaling could potentially reduce GSC survival and tumor growth. Zhang et al. [56] explored the role of autophagy in GSC maintenance and survival under metabolic stress. They found that enhancing autophagy inhibition could reduce GSC survival and tumor growth, suggesting its potential as a therapeutic strategy. Liu et al. [57] focused on the Notch signaling pathway in GSCs. They showed that inhibiting Notch signaling reduces GSC proliferation and tumorigenicity, highlighting its potential as a therapeutic target. Gao et al. [58] studied the CD44 receptor in GSC adhesion and invasion. They demonstrated that blocking CD44 function reduces GSC invasiveness and tumor growth, suggesting its potential as a therapeutic target to limit GBM spread. Xu et al. [59] investigated the Wnt/β-catenin pathway in GSC maintenance. They found that its activation promotes self-renewal and resistance to differentiation. Inhibiting this pathway could reduce GSC survival and tumor growth, making it a promising therapeutic target.
4. Discussion
4.1. GCSs Cell Lines
While U87 cells have been widely used in GBM research, recent studies have raised significant concerns about their reliability as a model for high-grade gliomas (HGGs). Allen et al. and Dolgin highlight several critical issues with the U87 cell line [60,61]. Firstly, Allen et al. underscore that U87 cells, initially thought to be derived from a human GBM, lack the genetic and phenotypic characteristics representative of primary HGG. This discrepancy arises because U87 cells have been extensively cultured since their establishment in 1966, leading to substantial genetic drift [60]. Over decades of in vitro propagation, these cells have acquired numerous genetic alterations that deviate from the original tumor profile, thereby diminishing their relevance as a model for studying GBM. Furthermore, Dolgin points out that the U87 cell line’s long history of passaging has led to the accumulation of mutations and chromosomal abnormalities not present in primary gliomas. This genetic divergence results in altered cellular behavior, including differences in growth rate, response to therapies, and invasive properties compared to primary tumor cells [61]. Consequently, research findings based on U87 cells may not accurately reflect the biology of GBM in patients, potentially leading to misleading conclusions. Another critical issue is the reproducibility of results obtained using U87 cells. Due to the genetic and phenotypic instability of this cell line, different laboratories may obtain varying results, undermining the consistency and reliability of scientific findings. This variability poses a significant challenge for the development of effective therapeutic strategies, as preclinical studies using U87 cells may not predict clinical outcomes accurately. Moreover, the use of U87 cells fails to capture the intratumoral heterogeneity observed in GBMs. Gliomas are known for their diverse cell populations, each contributing differently to tumor progression and resistance to treatment. U87 cells, being a clonal population, do not represent this heterogeneity, limiting their utility in studying the complex interactions within the tumor microenvironment. Given these limitations, there is a growing consensus in the research community to transition towards more representative models, such as patient-derived xenografts (PDXs) and primary glioma cell lines. These models better preserve the genetic and molecular diversity of GBMs, offering a more accurate platform for studying tumor biology and testing novel therapies. By utilizing such models, researchers can gain more reliable insights into glioma progression and therapeutic responses, ultimately advancing the development of more effective treatments for patients [60,61].
4.2. Molecular Pathways Involved in GCS-Mediated Glioma Progression
The pathways implicated in GSC-mediated glioma progression represent intricate networks of molecular interactions that govern tumor initiation, growth, and invasion. The Notch, PI3K/AKT, and Wnt/β-catenin pathways emerge as key players in this process, each contributing uniquely to glioma pathogenesis and offering potential targets for therapeutic intervention [12,34,39,61].
The PI3K/AKT pathway is a central regulator of key cellular processes including proliferation, survival, and metabolism, all of which play critical roles in glioma pathogenesis. Genetic mutations or amplifications often lead to dysregulation of the PI3K/AKT pathway, providing glioma cells with a growth advantage and enhancing tumor aggression. In glioma, activation of PI3K/AKT signaling specifically promotes the proliferation and survival of GSCs, thereby facilitating tumor growth and contributing to therapeutic resistance [12,34,39,61]. Targeting components of the PI3K/AKT pathway, such as PI3K/AKT inhibitors, represents a promising strategy to attenuate glioma progression and improve the effectiveness of current therapeutic approaches. Clinical trials assessing these pathways have provided valuable insights into potential therapeutic strategies. For instance, Xu et al. conducted a study evaluating the efficacy of targeting the PI3K/AKT/mTOR pathway, a critical signaling cascade frequently dysregulated in glioblastomas. Their clinical trial demonstrated that inhibiting this pathway can significantly impact tumor growth and progression. The study included a cohort of patients treated with the dual mTORC1/2 inhibitor TAK-228 (formerly MLN0128), showing promising results in terms of tumor response and progression-free survival. Xu et al.’s findings highlight the potential of pathway-specific inhibitors to improve clinical outcomes for glioblastoma patients. The trial not only provided evidence of the therapeutic benefits of targeting the PI3K/AKT/mTOR pathway but also emphasized the importance of patient selection based on molecular profiling. Patients with tumors exhibiting high activation of this pathway responded better to the treatment, underscoring the need for personalized medicine approaches in glioma therapy. Moreover, this study illustrates the broader trend in glioma research towards the identification and clinical validation of molecular targets [62,63,64,65,66,67,68,69,70].
Similarly, the Wnt/β-catenin pathway exerts profound effects on GSC behavior, influencing self-renewal, differentiation, and invasion. Canonical Wnt signaling, triggered by Wnt ligands and their receptors, stabilizes β-catenin and promotes its translocation to the nucleus, where it regulates the expression of target genes involved in stemness and invasion. Aberrant activation of Wnt/β-catenin signaling has been implicated in glioma initiation and progression, driving GSC self-renewal and promoting invasive phenotypes. Therapeutic strategies aimed at inhibiting Wnt/β-catenin signaling hold promise for disrupting GSC-mediated mechanisms and impeding glioma progression [26,31,36,58].
Notch signaling stands out as a critical regulator of GSC self-renewal and differentiation, dictating the balance between stemness and differentiation within the tumor microenvironment [11]. The activation of Notch pathway components, such as Notch receptors (Notch1-4) and their ligands (Jagged and Delta-like), orchestrates a cascade of downstream events that promote GSC maintenance and tumor progression. Notch-mediated signaling has been implicated in glioma cell fate determination, with aberrant activation contributing to tumor initiation and therapeutic resistance. Notch inhibitors, through their ability to block Notch signaling, represent promising therapeutic agents capable of disrupting GSC-mediated mechanisms and halting glioma progression [11,22,23,43,45,47,56].
Other pathways, such as the RTK/RAS/RAF and the PD-1/PD-L1 immune checkpoint, have also been the focus of clinical trials, demonstrating varying degrees of success. These trials collectively contribute to a growing body of evidence supporting the stratification of glioma patients based on specific molecular characteristics, thereby optimizing therapeutic efficacy and minimizing unnecessary toxicity.
4.3. Therapeutics Targets and Agents
The distribution of therapeutic targets across various studies offers valuable insights into diverse strategies aimed at disrupting GSC-mediated mechanisms and impeding glioma progression. Notably, Notch inhibitors, PI3K/AKT inhibitors, and STAT3 inhibitors emerge as frequent therapeutic targets, underscoring their pivotal roles in glioma pathogenesis and promising avenues for intervention.
Notch inhibitors represent a promising class designed to block Notch signaling, which is crucial for GSC self-renewal and differentiation processes. Targeting Notch receptors and ligands holds potential for halting glioma progression, sensitizing tumors to existing therapies, and disrupting the stemness–differentiation balance within the tumor microenvironment. Studies indicate that Notch inhibition promotes GSC differentiation, reducing tumor growth and enhancing treatment response in preclinical models [11,22,23,43,47,56]. Moreover, they sensitize glioma cells to conventional therapies, positioning them as promising therapeutic agents [11,22,23,43,47,56].
Similarly, PI3K/AKT inhibitors have garnered attention for their ability to suppress glioma cell proliferation and survival by targeting dysregulated components of the PI3K/AKT signaling cascade [12,34,39,61]. Disruption of this pathway enhances tumor cell susceptibility to cytotoxic therapies, demonstrating efficacy in preclinical models and supporting their potential as glioma therapeutics [12,34,39,61].
Additionally, STAT3 inhibitors offer a promising approach to modulating glioma cell behavior by targeting the STAT3 signaling pathway [22,31,53,59,69,71,74,75,76,77]. Inhibition of STAT3 activation has shown promise in reducing glioma cell proliferation, inducing apoptosis, and inhibiting tumor invasion in preclinical models, potentially overcoming therapeutic resistance and improving patient outcomes [22,31,53,59,69,71,74,75,76,77].
Beyond these, various agents target additional signaling pathways implicated in glioma pathogenesis, such as the MAPK/ERK and Wnt/β-catenin pathways, as well as specific molecular targets involved in GSC maintenance and survival [13,14,15,16,17,18,19,20,21]. Immunotherapeutic agents, including immune checkpoint inhibitors and CAR T cell therapy, show promise in harnessing the immune system against GSCs and glioma cells [78].
The versatility and potential of these targeted therapies against GSC-mediated glioma progression underscore their significance in developing effective treatment strategies tailored to individual tumor profiles [25,26,27,28,29,30,31,32,45,47,71,73,75]. Further preclinical and clinical investigations are crucial to evaluating their efficacy, safety, and optimal integration into glioma management [63,64,65,66,67,68,69].
4.4. Limitations and Future Directions
Although this systematic literature review offers important insights, several limitations must be recognized. The studies included in the review differed significantly in terms of study design, patient populations, treatment protocols, and outcome measures, complicating efforts to conduct a meta-analysis or reach definitive conclusions. Moreover, the limited number of studies and the relatively small sample sizes in some cases highlight the necessity for larger, well-structured clinical trials.
Future directions in targeted therapies against GSC-mediated glioma progression emphasize precision medicine approaches and the development of novel molecular inhibitors [35,36,37,38,41,42,43]. Advancements in genomic and proteomic profiling can identify key mutations and signaling pathways driving GSCs, enabling the design of highly specific drugs [58,59]. Immunotherapy, particularly CAR T cell therapy, holds promise in targeting GSCs [78]. Combination therapies that integrate targeted drugs with conventional treatments like RT and CT are also being explored to overcome resistance mechanisms [49,50,51,52,53,54,55,79,80,81,82,83,84,85,86,87,88]. Moreover, leveraging artificial intelligence for drug discovery and patient-specific treatment planning is expected to enhance the efficacy and personalization of GSC-targeted therapies.
5. Conclusions
The intricate molecular pathways involved in GSC-mediated glioma progression offer multiple targets for therapeutic intervention, with Notch, PI3K/AKT, and Wnt/β-catenin signaling pathways being particularly critical. Targeting these pathways with specific inhibitors presents a promising strategy to disrupt GSC maintenance and tumor growth. Notch inhibitors, PI3K/AKT inhibitors, and other targeted agents have shown potential in preclinical studies, demonstrating their ability to reduce tumor growth and enhance the efficacy of existing treatments. The development of these targeted therapies, combined with advanced genomic and proteomic profiling, paves the way for personalized treatment approaches, potentially improving patient outcomes in glioma management. Continued research and clinical trials are essential to validate these findings and optimize therapeutic strategies against GSC-mediated glioma progression.
Author Contributions
Conceptualization, E.A., M.Z., M.M.F. and P.P.P.; methodology, E.A., S.A., M.Z. and P.P.P.; validation, M.Z., M.M.F., T.I. and P.P.P.; formal analysis, E.A. and S.A.; investigation, E.A. and S.A.; resources, E.A. and M.Z.; data curation, E.A., S.A. and P.P.P.; writing—original draft preparation, E.A.; writing—review and editing, E.A., M.Z., T.I., M.M.F. and P.P.P.; visualization, E.A., S.A., T.I., M.Z., M.M.F. and P.P.P.; supervision, E.A., M.M.F. and P.P.P.; project administration, E.A., M.Z., M.M.F. and P.P.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data available in a publicly accessible repository.
Conflicts of Interest
The authors declare no conflicts of interest.
Appendix A
Figure A1.
The PRISMA-ScR checklist. Abbreviations: JBI = Joanna Briggs Institute; PRISMA-ScR = Preferred Reporting Items for Systematic reviews and Meta-Analyses extension for Scoping Reviews. * Where sources of evidence (see second footnote) are compiled from, such as bibliographic databases, social media platforms, and web sites. † A more inclusive/heterogeneous term used to account for the different types of evidence or data sources (e.g., quantitative and/or qualitative research, expert opinion, and policy documents) that may be eligible in a scoping review as opposed to only studies. This is not to be confused with information sources (see first footnote). ‡ The frameworks by Arksey and O’Malley (6), Levac and colleagues (7), and the JBI guidance (4, 5) refer to the process of data extraction in a scoping review as data charting. § The process of systematically examining research evidence to assess its validity, results, and relevance before using it to inform a decision. This term is used for items 12 and 19 instead of “risk of bias” (which is more applicable to systematic reviews of interventions) to include and acknowledge the various sources of evidence that may be used in a scoping review (e.g., quantitative and/or qualitative research, expert opinion, and policy document).
Figure A1.
The PRISMA-ScR checklist. Abbreviations: JBI = Joanna Briggs Institute; PRISMA-ScR = Preferred Reporting Items for Systematic reviews and Meta-Analyses extension for Scoping Reviews. * Where sources of evidence (see second footnote) are compiled from, such as bibliographic databases, social media platforms, and web sites. † A more inclusive/heterogeneous term used to account for the different types of evidence or data sources (e.g., quantitative and/or qualitative research, expert opinion, and policy documents) that may be eligible in a scoping review as opposed to only studies. This is not to be confused with information sources (see first footnote). ‡ The frameworks by Arksey and O’Malley (6), Levac and colleagues (7), and the JBI guidance (4, 5) refer to the process of data extraction in a scoping review as data charting. § The process of systematically examining research evidence to assess its validity, results, and relevance before using it to inform a decision. This term is used for items 12 and 19 instead of “risk of bias” (which is more applicable to systematic reviews of interventions) to include and acknowledge the various sources of evidence that may be used in a scoping review (e.g., quantitative and/or qualitative research, expert opinion, and policy document).
References
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee, S.U. Glioblastoma Multiforme: A Review of its Epidemiology and Pathogenesis through Clinical Presentation and Treatment. Asian Pac. J. Cancer Prev. 2017, 18, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Vieira de Castro, J.; Gonçalves, C.S.; Hormigo, A. Costa BM.Exploiting the Complexities of Glioblastoma Stem Cells: Insights for Cancer Initiation and Therapeutic Targeting. Int. J. Mol. Sci. 2020, 21, 5278. [Google Scholar] [CrossRef] [PubMed]
- Garnier, D.; Renoult, O.; Alves-Guerra, M.C.; Paris, F.; Pecqueur, C. Glioblastoma Stem-Like Cells, Metabolic Strategy to Kill a Challenging Target. Front. Oncol. 2019, 9, 118. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.K.; Bonaguidi, M.A.; Ming, G.L.; Song, H. Adult neural stem cells in the mammalian central nervous system. Cell Res. 2009, 19, 672–682. [Google Scholar] [CrossRef] [PubMed]
- Auffinger, B.; Spencer, D.; Pytel, P.; Ahmed, A.U.; Lesniak, M.S. The role of glioma stem cells in chemotherapy resistance and glioblastoma multiforme recurrence. Expert Rev. Neurother. 2015, 15, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Hersh, A.M.; Gaitsch, H.; Alomari, S.; Lubelski, D.; Tyler, B.M. Molecular Pathways and Genomic Landscape of Glioblastoma Stem Cells: Opportunities for Targeted Therapy. Cancers 2022, 14, 3743. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, M.; Zhao, J.; Ren, T.; Yan, X.; Zhang, L.; Wang, X. Research Progress About Glioma Stem Cells in the Immune Microenvironment of Glioma. Front. Pharmacol. 2021, 12, 750857. [Google Scholar] [CrossRef]
- Esposito, C.L.; Nuzzo, S.; Ibba, M.L.; Ricci-Vitiani, L.; Pallini, R.; Condorelli, G.; Catuogno, S.; de Franciscis, V. Combined Targeting of Glioblastoma Stem-Like Cells by Neutralizing RNA-Bio-Drugs for STAT3. Cancers 2020, 12, 1434. [Google Scholar] [CrossRef] [PubMed]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Murlow, C.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef]
- The Newcastle–Ottawa Scale (NOS) for Assessing the Quality of Non-Randomized Studies in Meta-Analysis|Request PDF. Available online: https://www.researchgate.net/publication/261773681_The_Newcastle-Ottawa_Scale_NOS_for_Assessing_the_Quality_of_Non-Randomized_Studies_in_Meta-Analysis (accessed on 19 July 2023).
- Purow, B.W.; Haque, R.M.; Noel, M.W.; Su, Q.; Burdick, M.J.; Lee, J.; Sundaresan, T.; Pastorino, S.; Park, J.K.; Mikolaenko, I.; et al. Expression of Notch-1 and its ligands, Delta-like-1 and Jagged-1, is critical for glioma cell survival and proliferation. Cancer Res. 2005, 65, 2353–2363. [Google Scholar] [CrossRef]
- Groszer, M.; Erickson, R.; Scripture-Adams, D.D.; Dougherty, J.D.; Le Belle, J.; Zack, J.A.; Geschwind, D.H.; Liu, X.; Kornblum, H.I.; Wu, H.; et al. PTEN negatively regulates neural stem cell self-renewal by modulating G0-G1 cell cycle entry. Proc. Natl. Acad. Sci. USA 2006, 103, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Zagzag, D.; Lukyanov, Y.; Lan, L.; Ali, M.A.; Esencay, M.; Mendez, O.; Yee, H.; Voura, E.B.; Newcomb, E.W. Hypoxia-inducible factor 1 and VEGF upregulate CXCR4 in glioblastoma: Implications for angiogenesis and glioma cell invasion. Lab. Investig. 2006, 86, 1221–1232. [Google Scholar] [CrossRef]
- Piccirillo, S.G.M.; Reynolds, B.A.; Zanetti, N.; Lamorte, G.; Binda, E.; Broggi, G.; Brem, H.; Olivi, A.; Dimeco, F.; Vescovi, A.L.; et al. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature 2006, 444, 761–765. [Google Scholar] [CrossRef]
- Clement, V.; Sanchez, P.; de Tribolet, N.; Radovanovic, I.; Ruiz i Altaba, A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol. 2007, 17, 165–172. [Google Scholar] [CrossRef]
- Bar, E.E.; Chaudhry, A.; Lin, A.; Fan, X.; Schreck, K.; Matsui, W.; Piccirillo, S.; Vescovi, A.L.; DiMeco, F.; Olivi, A.; et al. Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells 2007, 25, 2524–2533. [Google Scholar] [CrossRef]
- Du, R.; Lu, K.V.; Petritsch, C.; Liu, P.; Ganss, R.; Passegué, E.; Song, H.; Vandenberg, S.; Johnson, R.S.; Werb, Z.; et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell 2008, 13, 206–220. [Google Scholar] [CrossRef]
- Silber, J.; Lim, D.A.; Petritsch, C.; Persson, A.I.; Maunakea, A.K.; Yu, M.; Vandenberg, S.R.; Ginzinger, D.G.; James, C.D.; Costello, J.F.; et al. miR-124 and miR-137 inhibit proliferation of glioblastoma multiforme cells and induce differentiation of brain tumor stem cells. BMC Med. 2008, 6, 14. [Google Scholar] [CrossRef]
- Gal, H.; Pandi, G.; Kanner, A.A.; Ram, Z.; Lithwick-Yanai, G.; Amariglio, N.; Rechavi, G. Givol DMIR-451 and Imatinib mesylate inhibit tumor growth of Glioblastoma stem cells. Biochem. Biophys. Res. Commun. 2008, 376, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Yeh, W.L.; Lu, D.Y.; Lee, M.J.; Fu, W.M. Leptin induces migration and invasion of glioma cells through MMP-13 production. Glia 2009, 57, 454–464. [Google Scholar] [CrossRef]
- Golding, S.E.; Rosenberg, E.; Valerie, N.; Hussaini, I.; Frigerio, M.; Cockcroft, X.F.; Chong, W.Y.; Hummersone, M.; Rigoreau, L.; Menear, K.A.; et al. Improved ATM kinase inhibitor KU-60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling, and inhibits migration and invasion. Mol. Cancer Ther. 2009, 8, 2894–2902. [Google Scholar] [CrossRef]
- Heddleston, J.M.; Li, Z.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle 2009, 8, 3274–3284. [Google Scholar] [CrossRef] [PubMed]
- Seidel, S.; Garvalov, B.K.; Wirta, V.; von Stechow, L.; Schänzer, A.; Meletis, K.; Wolter, M.; Sommerlad, D.; Henze, A.T.; Nistér, M.; et al. A hypoxic niche regulates glioblastoma stem cells through hypoxia inducible factor 2α. Brain 2010, 133, 983–995. [Google Scholar] [CrossRef] [PubMed]
- Riolfi, M.; Ferla, R.; Del Valle, L.; Piña-Oviedo, S.; Scolaro, L.; Micciolo, R.; Guidi, M.; Terrasi, M.; Cetto, G.L.; Surmacz, E. Leptin and its receptor are overexpressed in brain tumors and correlate with the degree of malignancy. Brain Pathol. 2010, 20, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Ernst, A.; Campos, B.; Meier, J.; Devens, F.; Liesenberg, F.; Wolter, M.; Reifenberger, G.; Herold-Mende, C.; Lichter, P.; Radlwimmer, B. De-repression of CTGF via the miR-17-92 cluster upon differentiation of human glioblastoma spheroid cultures. Oncogene 2010, 29, 3411–3422. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Ying, H.; Wiedemeyer, R.; Yan, H.; Quayle, S.N.; Ivanova, E.V.; Paik, J.H.; Zhang, H.; Xiao, Y.; Perry, S.R. PLAGL2 regulates Wnt signaling to impede differentiation in neural stem cells and gliomas. Cancer Cell 2010, 17, 497–509. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.R.; Hayashi, Y.; Stephens, C.; Georgescu, M.M. Invasive glioblastoma cells acquire stemness and increased Akt activation. Neoplasia 2010, 12, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Takahashi, H.; Harada, H.; Kohno, S.; Ohue, S.; Kobayashi, K.; Yano, H.; Tanaka, J.; Ohnishi, T. Cancer stem-like cells of glioblastoma characteristically express MMP-13 and display highly invasive activity. Int. J. Oncol. 2010, 37, 1121–1131. [Google Scholar] [CrossRef] [PubMed]
- Beck, S.; Jin, X.; Sohn, Y.W.; Kim, J.K.; Kim, S.H.; Yin, J.; Pian, X.; Kim, S.C.; Nam, D.H.; Choi, Y.J.; et al. Telomerase activity-independent function of TERT allows glioma cells to attain cancer stem cell characteristics by inducing EGFR expression. Mol. Cells 2011, 31, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Wu, Q.; Guryanova, O.A.; Huang, Z.; Huang, Q.; Rich, J.N.; Bao, S. Elevated invasive potential of glioblastoma stem cells. Biochem. Biophys. Res. Commun. 2011, 406, 643–648. [Google Scholar] [CrossRef]
- Kahlert, U.D.; Maciaczyk, D.; Doostkam, S.; Orr, B.A.; Simons, B.; Bogiel, T.; Reithmeier, T.; Prinz, M.; Schubert, J.; Niedermann, G. Activation of canonical WNT/β-catenin signaling enhances in vitro motility of glioblastoma cells by activation of ZEB1 and other activators of epithelial-to-mesenchymal transition. Cancer Lett. 2012, 325, 42–53. [Google Scholar] [CrossRef]
- Kaur, N.; Chettiar, S.; Rathod, S.; Rath, P.; Muzumdar, D.; Shaikh, M.L.; Shiras, A. Wnt3a mediated activation of Wnt/β-catenin signaling promotes tumor progression in glioblastoma. Mol. Cell Neurosci. 2013, 54, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Kanno, H.; Sato, H.; Yokoyama, T.A.; Yoshizumi, T.; Yamada, S. The VHL tumor suppressor protein regulates tumorigenicity of U87-derived glioma stem-like cells by inhibiting the JAK/STAT signaling pathway. Int. J. Oncol. 2013, 42, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Carra, E.; Barbieri, F.; Marubbi, D.; Pattarozzi, A.; Favoni, R.E.; Florio, T.; Daga, A. Sorafenib selectively depletes human glioblastoma tumor-initiating cells from primary cultures. Cell Cycle 2013, 12, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Huang, Z.; Zhou, W.; Wu, Q.; Donnola, S.; Liu, J.K.; Fang, X.; Sloan, A.E.; Mao, Y.; Lathia, J.D. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 2013, 153, 139–152. [Google Scholar] [CrossRef]
- Rheinbay, E.; Suvà, M.L.; Gillespie, S.M.; Wakimoto, H.; Patel, A.P.; Shahid, M.; Oksuz, O.; Rabkin, S.D.; Martuza, R.L.; Rivera, M.N.; et al. An aberrant transcription factor network essential for Wnt signaling and stem cell maintenance in glioblastoma. Cell Rep. 2013, 3, 1567–1579. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.B.; Tian, S.; Gao, H.H.; Xu, Y.Y. Metformin inhibits glioma cell U251 invasion by downregulation of fibulin-3. Neuroreport 2013, 24, 504–508. [Google Scholar] [CrossRef] [PubMed]
- Siebzehnrubl, F.A.; Silver, D.J.; Tugertimur, B.; Deleyrolle, L.P.; Siebzehnrubl, D.; Sarkisian, M.R.; Devers, K.G.; Yachnis, A.T.; Kupper, M.D.; Neal, D.; et al. The ZEB1 pathway links glioblastoma initiation, invasion and chemoresistance. EMBO Mol. Med. 2013, 5, 1196–1212. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Wang, Z.; Wan, Y.; Shi, L.; Zhou, Y. Downregulation of ABCG2 protein inhibits migration and invasion in U251 glioma stem cells. Neuroreport 2014, 25, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Wang, Q.; Wang, Y.A.; Hua, S.; Sauvé, C.G.; Ong, D.; Lan, Z.D.; Chang, Q.; Ho, Y.W.; Monasterio, M.M.; et al. Epigenetic Activation of WNT5A Drives Glioblastoma Stem Cell Differentiation and Invasive Growth. Cell 2016, 167, 1281–1295.e18. [Google Scholar] [CrossRef] [PubMed]
- Madan, E.; Dikshit, B.; Gowda, S.H.; Srivastava, C.; Sarkar, C.; Chattopadhyay, P.; Sinha, S.; Chosdol, K. FAT1 is a novel upstream regulator of HIF1α and invasion of high grade glioma. Int. J. Cancer 2016, 139, 2570–2582. [Google Scholar] [CrossRef]
- Adamo, A.; Fiore, D.; De Martino, F.; Roscigno, G.; Affinito, A.; Donnarumma, E.; Puoti, I.; Ricci Vitiani, L.; Pallini, R.; Quintavalle, C.; et al. RYK promotes the stemness of glioblastoma cells via the WNT/β-catenin pathway. Oncotarget 2017, 8, 13476–13487. [Google Scholar] [CrossRef] [PubMed]
- Cenciarelli, C.; Marei, H.E.; Zonfrillo, M.; Casalbore, P.; Felsani, A.; Giannetti, S.; Trevisi, G.; Althani, A.; Mangiola, A. The interference of Notch1 target Hes1 affects cell growth, differentiation and invasiveness of glioblastoma stem cells through modulation of multiple oncogenic targets. Oncotarget 2017, 8, 17873–17886. [Google Scholar] [CrossRef] [PubMed]
- Clark, P.A.; Bhattacharya, S.; Elmayan, A.; Darjatmoko, S.R.; Thuro, B.A.; Yan, M.B.; van Ginkel, P.R.; Polans, A.S.; Kuo, J.S. Resveratrol targeting of AKT and p53 in glioblastoma and glioblastoma stem-like cells to suppress growth and infiltration. J. Neurosurg. 2017, 126, 1448–1460. [Google Scholar] [CrossRef] [PubMed]
- Maciaczyk, D.; Picard, D.; Zhao, L.; Koch, K.; Herrera-Rios, D.; Li, G.; Marquardt, V.; Pauck, D.; Hoerbelt, T.; Zhang, W.; et al. CBF1 is clinically prognostic and serves as a target to block cellular invasion and chemoresistance of EMT-like glioblastoma cells. Br. J. Cancer 2017, 117, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Khan, O.F.; Suvà, M.L.; Dong, B.; Panek, W.K.; Xiao, T.; Wu, M.; Han, Y.; Ahmed, A.U.; Balyasnikova, I.V.; et al. Multiplexed RNAi therapy against brain tumor-initiating cells via lipopolymeric nanoparticle infusion delays glioblastoma progression. Proc. Natl. Acad. Sci. USA 2017, 114, E6147–E6156. [Google Scholar] [CrossRef] [PubMed]
- Man, J.; Yu, X.; Huang, H.; Zhou, W.; Xiang, C.; Huang, H.; Miele, L.; Liu, Z.; Bebek, G.; Bao, S.; et al. Hypoxic Induction of Vasorin Regulates Notch1 Turnover to Maintain Glioma Stem-like Cells. Cell Stem Cell 2018, 22, 104–118.e6. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Soler, M.; Petersen Gehring, M.; Lechtenberg, B.C.; Zapata-Mercado, E.; Hristova, K.; Pasquale, E.B. Engineering nanomolar peptide ligands that differentially modulate EphA2 receptor signaling. J. Biol. Chem. 2019, 294, 8791–8805. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yang, W.; Liu, Y.; Gao, R.; Yu, H.; Sun, T. HDAC6 inhibition induces glioma stem cells differentiation and enhances cellular radiation sensitivity through the SHH/Gli1 signaling pathway. Cancer Lett. 2018, 415, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Li, S.Z.; Ren, K.X.; Zhao, J.; Wu, S.; Li, J.; Zang, J.; Fei, Z.; Zhao, J.L. miR-139/PDE2A-Notch1 feedback circuit represses stemness of gliomas by inhibiting Wnt/β-catenin signaling. Int. J. Biol. Sci. 2021, 17, 3508–3521. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jiang, X.; Zhang, Y.; Yuan, Y.; Jin, Z.; Zhai, H.; Liu, B.; Li, Y.; Zhang, C.; Chen, M.; Shi, Y.; et al. LncRNA GSCAR promotes glioma stem cell maintenance via stabilizing SOX2 expression. Int. J. Biol. Sci. 2023, 19, 1681–1697. [Google Scholar] [CrossRef]
- Yu, Q.; Xue, Y.; Liu, J.; Xi, Z.; Li, Z.; Liu, Y. Fibronectin Promotes the Malignancy of Glioma Stem-Like Cells Via Modulation of Cell Adhesion, Differentiation, Proliferation and Chemoresistance. Front. Mol. Neurosci. 2018, 11, 130. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Guryanova, O.A.; Zhou, W.; Liu, C.; Huang, Z.; Fang, X.; Wang, X.; Chen, C.; Wu, Q.; He, Z.; et al. Ibrutinib inactivates BMX-STAT3 in glioma stem cells to impair malignant growth and radioresistance. Sci. Transl. Med. 2018, 10, eaah6816. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Gao, S.; Li, Y.; Qian, X.; Luan, J.; Lv, X. Emerging Importance of Chemokine Receptor CXCR4 and Its Ligand in Liver Disease. Front. Cell Dev. Biol. 2021, 9, 716842. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, S.; Chen, Y.; Xie, Y.; Zhan, H.; Zeng, Y.; Zeng, K.; Wang, L.; Zhan, Z.; Li, C.; Zhao, L.; et al. FBXO7 Confers Mesenchymal Properties and Chemoresistance in Glioblastoma by Controlling Rbfox2-Mediated Alternative Splicing. Adv. Sci. 2023, 10, e2303561. [Google Scholar] [CrossRef] [PubMed]
- Pang, F.; Zhang, L.; Li, M.; Yi, X.; Wang, Y.; Yang, P.; Wen, B.; Jiang, J.; Teng, Y.; Yang, X.; et al. Ribosomal S6 protein kinase 4 promotes resistance to EZH2 inhibitors in glioblastoma. Cancer Gene Ther. 2023, 30, 1636–1648. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.Z.; Choi, W.S.; Jain, S.; Xu, X.; Elsherbiny, M.E.; Glubrecht, D.D.; Tessier, A.G.; Easaw, J.C.; Fallone, B.G.; Godbout, R. Stationary-to-migratory transition in glioblastoma stem-like cells driven by a fatty acid-binding protein 7-RXRα neurogenic pathway. Neuro-Oncology 2023, 25, 2177–2190. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Ohnishi, T.; Nishikawa, M.; Ohtsuka, Y.; Kusakabe, K.; Yano, H.; Tanaka, J.; Kunieda, T. A Narrative Review on CD44’s Role in Glioblastoma Invasion, Proliferation, and Tumor Recurrence. Cancers 2023, 15, 4898. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Xu, R.; Shimizu, F.; Hovinga, K.; Beal, K.; Karimi, S.; Droms, L.; Peck, K.K.; Gutin, P.; Iorgulescu, J.B.; Kaley, T.; et al. Molecular and Clinical Effects of Notch Inhibition in Glioma Patients: A Phase 0/I Trial. Clin. Cancer Res. 2016, 22, 4786–4796. [Google Scholar] [CrossRef] [PubMed]
- IFITM3 Promotes Glioblastoma Stem Cell-Mediated Angiogenesis via Regulating JAK/STAT3/bFGF Signaling Pathway|Cell Death & Disease. Available online: https://www.nature.com/articles/s41419-023-06416-5 (accessed on 25 May 2024).
- Fu, R.Z.; Cottrell, O.; Cutillo LRowntree, A.; Zador, Z.; Wurdak, H.; Papalopulu, N.; Marinopoulou, E. Identification of genes with oscillatory expression in glioblastoma: The paradigm of SOX2. Sci. Rep. 2024, 14, 2123. [Google Scholar] [CrossRef] [PubMed]
- Melamed, J.R.; Ioele, S.A.; Hannum, A.J.; Ullman, V.M.; Day, E.S. Polyethylenimine-Spherical Nucleic Acid Nanoparticles against Gli1 Reduce the Chemoresistance and Stemness of Glioblastoma Cells. Mol. Pharm. 2018, 15, 5135–5145. [Google Scholar] [CrossRef]
- Jia, B.; Liu, W.; Gu, J.; Wang, J.; Lv, W.; Zhang, W.; Hao, Q.; Pang, Z.; Mu, N.; Zhang, W.; et al. MiR-7-5p suppresses stemness and enhances temozolomide sensitivity of drug-resistant glioblastoma cells by targeting Yin Yang 1. Exp. Cell Res. 2019, 375, 73–81. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, G.; Bozek, D.A.; Rajakulendran, N.; Monteiro, V.; Ahmadi, M.; Steinhart, Z.; Kushida, M.M.; Yu, H.; Coutinho, F.J.; Cavalli, F.M.G.; et al. Genome-Wide CRISPR-Cas9 Screens Expose Genetic Vulnerabilities and Mechanisms of Temozolomide Sensitivity in Glioblastoma Stem Cells. Cell Rep. 2019, 27, 971–986.e9. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Zhong, Z.; Luo, C.; Xiao, Y.; Li, L.; Zhang, X.; Yang, L.; Xiao, K.; Ning, Y.; Chen, L.; et al. The miR-26a/AP-2α/Nanog signaling axis mediates stem cell self-renewal and temozolomide resistance in glioma. Theranostics 2019, 9, 5497–5516. [Google Scholar] [CrossRef] [PubMed]
- Panza, S.; Russo, U.; Giordano, F.; Leggio, A.; Barone, I.; Bonofiglio, D.; Gelsomino, L.; Malivindi, R.; Conforti, F.L.; Naimo, G.D.; et al. Leptin and Notch Signaling Cooperate in Sustaining Glioblastoma Multiforme Progression. Biomolecules 2020, 10, 886. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, K.; Sprowls, S.A.; Arora, S.; Shakya, S.; Silver, D.J.; Goins, C.M.; Wallace, L.; Roversi, G.; Schafer, R.E.; Kay, K.; et al. WDR5 represents a therapeutically exploitable target for cancer stem cells in glioblastoma. Genes Dev. 2023, 37, 86–102. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, P.; Xu, L.; Wang, B.; Zhang, S.; Wang, X. GALNT2 sustains glioma stem cells by promoting CD44 expression. Aging 2023, 15, 2208–2220. [Google Scholar] [CrossRef] [PubMed]
- Yun, E.J.; Kim, D.; Kim, S.; Hsieh, J.T.; Baek, S.T. Targeting Wnt/β-catenin-mediated upregulation of oncogenic NLGN3 suppresses cancer stem cells in glioblastoma. Cell Death Dis. 2023, 14, 423. [Google Scholar] [CrossRef] [PubMed]
- Cescon, M.; Rampazzo, E.; Bresolin, S.; Da Ros, F.; Manfreda, L.; Cani, A.; Della Puppa, A.; Braghetta, P.; Bonaldo, P.; Persano, L. Collagen VI sustains cell stemness and chemotherapy resistance in glioblastoma. Cell Mol. Life Sci. 2023, 80, 233. [Google Scholar] [CrossRef]
- Peña Agudelo, J.A.; Pidre, M.L.; Garcia Fallit, M.; Pérez Küper, M.; Zuccato, C.; Nicola Candia, A.J.; Marchesini, A.; Vera, M.B.; De Simone, E.; Giampaoli, C.; et al. Mitochondrial Peptide Humanin Facilitates Chemoresistance in Glioblastoma Cells. Cancers 2023, 15, 4061. [Google Scholar] [CrossRef]
- Tao, W.; Lei, H.; Luo, W.; Huang, Z.; Ling, P.; Guo, M.; Wan, L.; Zhai, K.; Huang, Q.; Wu, Q.; et al. Novel INHAT repressor drives glioblastoma growth by promoting ribosomal DNA transcription in glioma stem cells. Neuro Oncol. 2022, 25, 1428–1440. [Google Scholar] [CrossRef]
- Kahm, Y.J.; Jung, U.; Kim, R.K. Regulation of Cancer Stem Cells and Epithelial-Mesenchymal Transition by CTNNAL1 in Lung Cancer and Glioblastoma. Biomedicines 2023, 11, 1462. [Google Scholar] [CrossRef] [PubMed]
- Alshahrany, N.; Begum, A.; Siebzehnrubl, D.; Jimenez-Pascual, A.; Siebzehnrubl, F.A. Spatial distribution and functional relevance of FGFR1 and FGFR2 expression for glioblastoma tumor invasion. Cancer Lett. 2023, 571, 216349. [Google Scholar] [CrossRef] [PubMed]
- The Role of APOBEC3C in Modulating the Tumor Microenvironment and Stemness Properties of Glioma: Evidence from Pancancer Analysis—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/37809064/ (accessed on 25 May 2024).
- A molecular Signature for the G6PC3/SLC37A2/SLC37A4 Interactors in Glioblastoma Disease Progression and in the Acquisition of a Brain Cancer Stem Cell Phenotype—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/38034009/ (accessed on 25 May 2024).
- Liu, Z.; Wang, S.; Yu, K.; Chen, K.; Zhao, L.; Zhang, J.; Dai, K.; Zhao, P. The promoting effect and mechanism of MAD2L2 on stemness maintenance and malignant progression in glioma. J. Transl. Med. 2023, 21, 863. [Google Scholar] [CrossRef]
- Neuronal Activity Promotes Glioma Progression by Inducing Proneural-to-Mesenchymal Transition in Glioma Stem Cells|Cancer Research|American Association for Cancer Research. Available online: https://aacrjournals.org/cancerres/article-abstract/84/3/372/733853/Neuronal-Activity-Promotes-Glioma-Progression-by (accessed on 25 May 2024).
- Maleszewska, M.; Wojnicki, K.; Mieczkowski, J.; Król, S.K.; Jacek, K.; Śmiech, M.; Kocyk, M.; Ciechomska, I.A.; Bujko, M.; Siedlecki, J.; et al. DMRTA2 supports glioma stem-cell mediated neovascularization in glioblastoma. Cell Death Dis. 2024, 15, 228. [Google Scholar] [CrossRef]
- Wang, Y.; Suo, J.; Wang, Z.; Ran, K.; Tian, Y.; Han, W.; Liu, Y.; Peng, X. The PTPRZ1-MET/STAT3/ISG20 axis in glioma stem-like cells modulates tumor-associated macrophage polarization. Cell Signal. 2024, 120, 111191. [Google Scholar] [CrossRef]
- Allen, M.; Bjerke, M.; Edlund, H.; Nelander, S.; Westermark, B. Origin of the U87MG glioma cell line: Good news and bad news. Sci. Transl. Med. 2016, 8, 354re3. [Google Scholar] [CrossRef]
- Dolgin, E. Venerable brain-cancer cell line faces identity crisis. Nature 2016, 537, 149–150. [Google Scholar] [CrossRef]
- Schepisi, G.; Gianni, C.; Cursano, M.C.; Gallà, V.; Menna, C.; Casadei, C.; Bleve, S.; Lolli, C.; Martinelli, G.; Rosti, G.; et al. Immune checkpoint inhibitors and Chimeric Antigen Receptor (CAR)-T cell therapy: Potential treatment options against Testicular Germ Cell Tumors. Front. Immunol. 2023, 14, 1118610. [Google Scholar] [CrossRef] [PubMed]
- Agosti, E.; Zeppieri, M.; De Maria, L.; Tedeschi, C.; Fontanella, M.M.; Panciani, P.P.; Ius, T. Glioblastoma Immunotherapy: A Systematic Review of the Present Strategies and Prospects for Advancements. Int. J. Mol. Sci. 2023, 24, 15037. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Agosti, E.; Panciani, P.P.; Zeppieri, M.; De Maria, L.; Pasqualetti, F.; Tel, A.; Zanin, L.; Fontanella, M.M.; Ius, T. Tumor Microenvironment and Glioblastoma Cell Interplay as Promoters of Therapeutic Resistance. Biology 2023, 12, 736. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Agosti, E.; Zeppieri, M.; Ghidoni, M.; Ius, T.; Tel, A.; Fontanella, M.M.; Panciani, P.P. Role of glioma stem cells in promoting tumor chemo- and radioresistance: A systematic review of potential targeted treatments. World J. Stem Cells 2024, 16, 604–614. [Google Scholar] [CrossRef] [PubMed]
- De Maria, L.; Panciani, P.P.; Zeppieri, M.; Ius, T.; Serioli, S.; Piazza, A.; Di Giovanni, E.; Fontanella, M.M.; Agosti, E. A Systematic Review of the Metabolism of High-Grade Gliomas: Current Targeted Therapies and Future Perspectives. Int. J. Mol. Sci. 2024, 25, 724. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gutova, M.; Hibbard, J.C.; Ma, E.; Natri, H.M.; Adhikarla, V.; Chimge, N.O.; Qiu, R.; Nguyen, C.; Melendez, E.; Aguilar, B.; et al. Targeting Wnt signaling for improved glioma immunotherapy. Front. Immunol. 2024, 15, 1342625. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).