
Potential Transcriptional Enhancers in Coronaviruses: From Infectious Bronchitis Virus to SARS-CoV-2



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
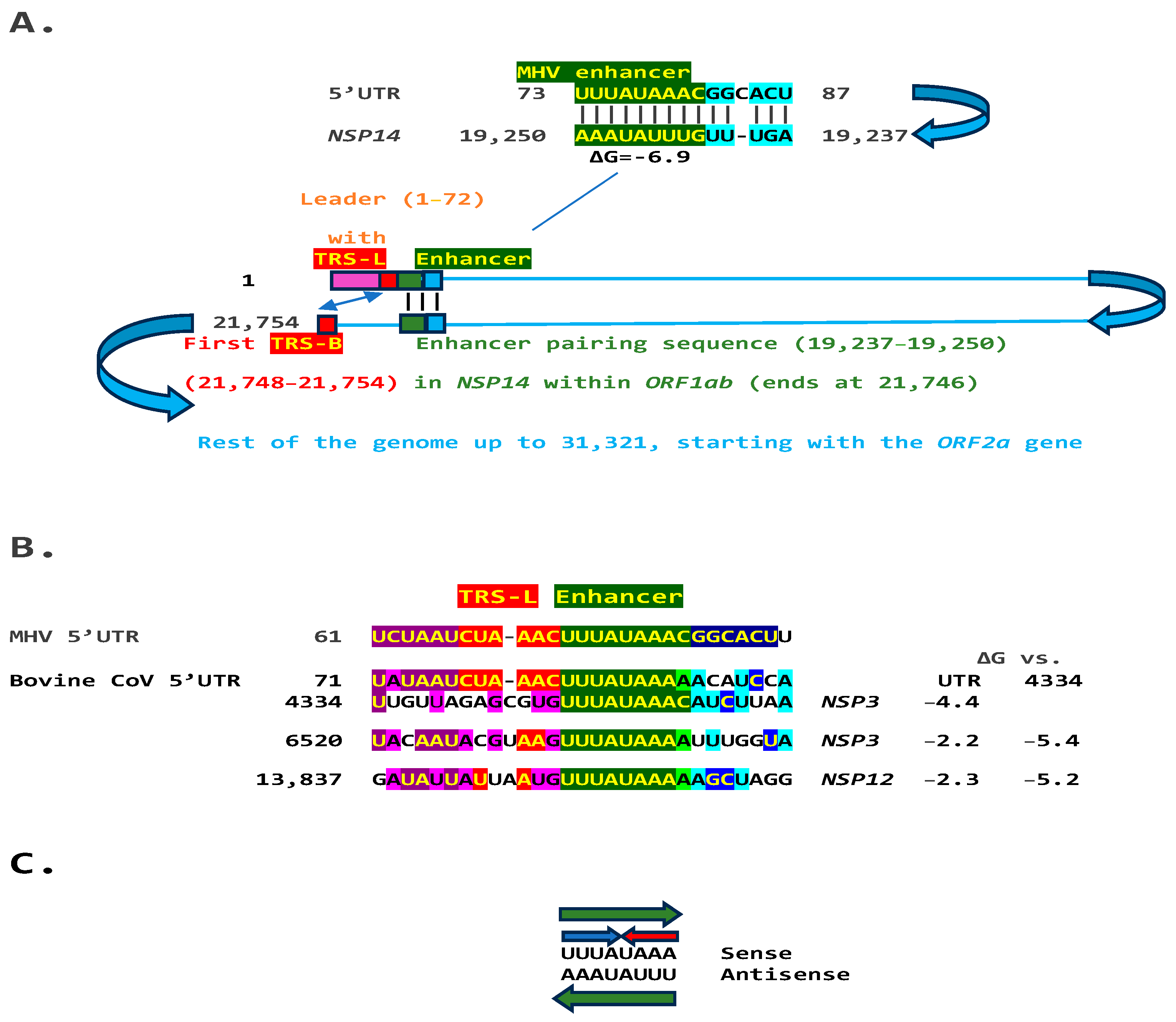
2.1. TGEV Enhancer-Based Model for Murine Hepatitis Virus (MHV) Enhancer and Potentially Bovine Coronavirus
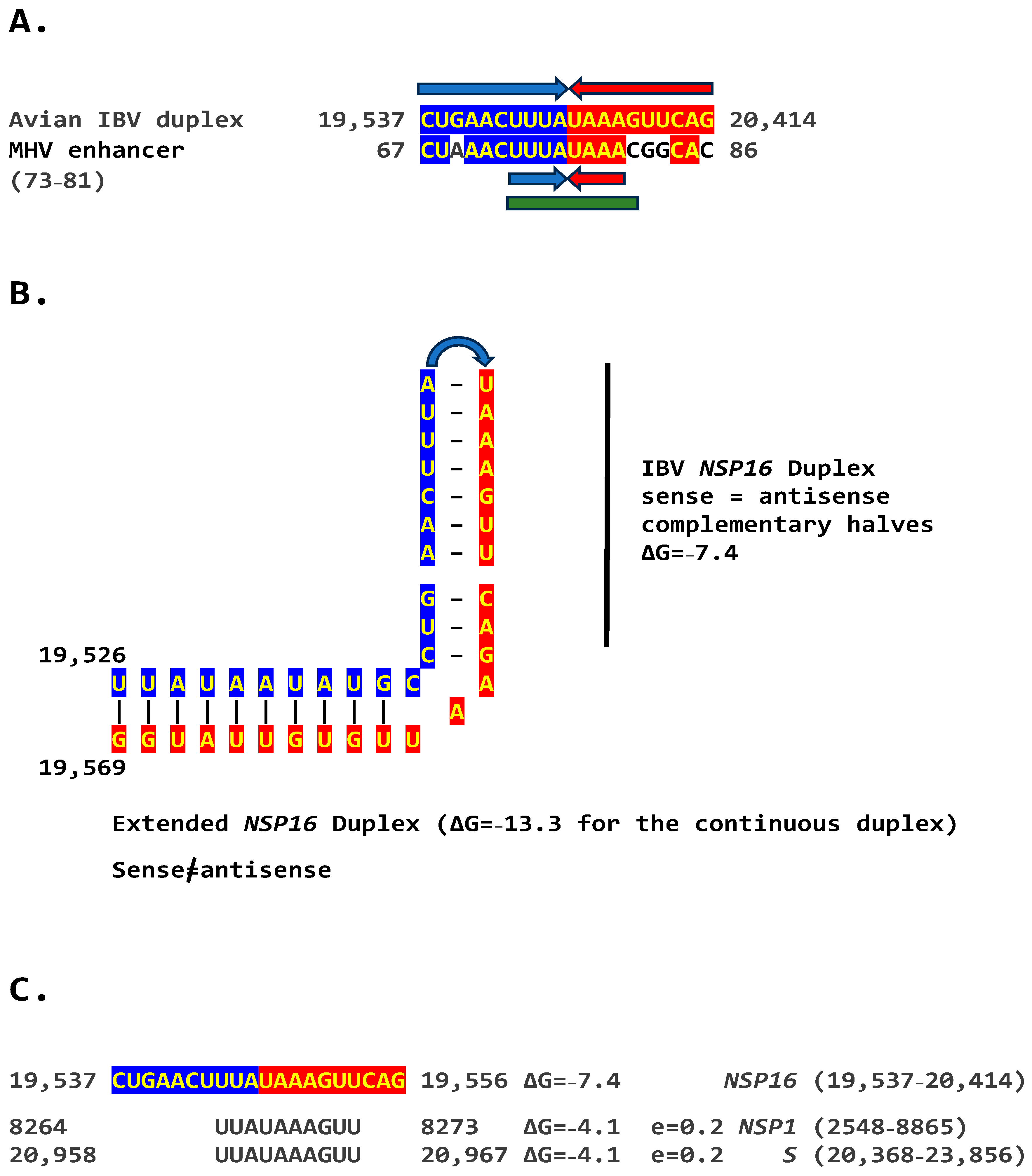
2.2. Potential Enhancer in Infectious Bronchitis Virus (IBV)
2.2.1. Variation in NSP16 Duplex, Its Distal Binding Sequence in S, or Both, Consistent with Reported Cases of Viral Attenuation or Its Reversal
2.2.2. Further Analysis of NSP16 Duplex Variation and Its Possible Association with Viral Attenuation or Its Reversal
2.2.3. Sequences Similar to the IBV NSP16 Extended Duplex Are Present in the Rousettus Bat Betacoronaviruses (Nobecoviruses), Related to SARS-CoV-1/-2
2.3. Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2) and Other Betacoronaviruses Infecting Humans
3. Discussion
4. Materials and Methods
4.1. Rationale for Primary and Secondary Structure Analyses and Use of TGEV Enhancer Model
4.2. Detection of Duplex-Forming Genomic Sequences Reading Similarly in the Sense and Antisense Directions with Complementary Halves as Potential Core Enhancer Elements
4.3. Detection of Coronaviral Genomic Sequences That Could Pair to a Core Enhancer Element
4.4. Characterization of Extended Duplexes and Visualization of RNA Secondary Structures and Estimation of Minimum Free Energies of Duplexes
4.5. Determination of Similarities Between Coronaviral Extended Duplexes with Viridae Sequences in GenBank and Analysis of Possible Nucleotide and Amino Acid Sequence Variations Being Consistent with Attenuation
4.6. Assessment of Sequence Similarities in Multiple Alignments of Potential Coronaviral Extended-Duplex Core Enhancer Elements
4.7. Assessment of Attenuation Status of IBV Strains
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Gorbalenya, A.E.; Enjuanes, L.; Ziebuhr, J.; Snijder, E.J. Nidovirales: Evolving the largest RNA virus genome. Virus Res. 2006, 117, 17–37. [Google Scholar] [CrossRef]
- Lauber, C.; Goeman, J.J.; Parquet Mdel, C.; Nga, P.T.; Snijder, E.J.; Morita, K.; Gorbalenya, A.E. The footprint of genome architecture in the largest genome expansion in RNA viruses. PLoS Pathog. 2013, 9, e1003500. [Google Scholar] [CrossRef]
- Masters, P.S. The molecular biology of coronaviruses. Adv. Virus Res. 2006, 66, 193–292. [Google Scholar] [CrossRef]
- Forni, D.; Cagliani, R.; Clerici, M.; Sironi, M. Molecular Evolution of Human Coronavirus Genomes. Trends Microbiol. 2017, 25, 35–48. [Google Scholar] [CrossRef]
- Ziv, O.; Gabryelska, M.M.; Lun, A.T.L.; Gebert, L.F.R.; Sheu-Gruttadauria, J.; Meredith, L.W.; Liu, Z.Y.; Kwok, C.K.; Qin, C.F.; MacRae, I.J.; et al. COMRADES determines in vivo RNA structures and interactions. Nat. Methods 2018, 15, 785–788. [Google Scholar] [CrossRef]
- Chen, S.C.; Olsthoorn, R.C.L. Group-specific structural features of the 5′-proximal sequences of coronavirus genomic RNAs. Virology 2010, 401, 29–41. [Google Scholar] [CrossRef]
- Sawicki, S.G.; Sawicki, D.L. Coronavirus transcription: A perspective. Curr. Top. Microbiol. Immunol. 2005, 287, 31–55. [Google Scholar] [CrossRef]
- Wang, D.; Jiang, A.; Feng, J.; Li, G.; Guo, D.; Sajid, M.; Wu, K.; Zhang, Q.; Ponty, Y.; Will, S.; et al. The SARS-CoV-2 subgenome landscape and its novel regulatory features. Mol. Cell 2021, 81, 2135–2147.e5. [Google Scholar] [CrossRef]
- Zúñiga, S.; Cruz, J.L.; Sola, I.; Mateos-Gómez, P.A.; Palacio, L.; Enjuanes, L. Coronavirus nucleocapsid protein facilitates template switching and is required for efficient transcription. J. Virol. 2010, 84, 2169–2175. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Kang, H.; Liu, P.; Makkinje, N.; Williamson, S.T.; Leibowitz, J.L.; Giedroc, D.P. Structural lability in stem-loop 1 drives a 5’ UTR-3’ UTR interaction in coronavirus replication. J. Mol. Biol. 2008, 377, 790–803. [Google Scholar] [CrossRef]
- Miao, Z.; Tidu, A.; Eriani, G.; Martin, F. Secondary structure of the SARS-CoV-2 5’-UTR. RNA Biol. 2021, 18, 447–456. [Google Scholar] [CrossRef]
- Hahn, C.S.; Hahn, Y.S.; Rice, C.M.; Lee, E.; Dalgarno, L.; Strauss, E.G.; Strauss, J.H. Conserved elements in the 3’ untranslated region of flavivirus RNAs and potential cyclization sequences. J. Mol. Biol. 1987, 198, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.Y.; Tsai, T.L.; Lin, C.N.; Lin, C.H.; Wu, H.Y. Interaction of coronavirus nucleocapsid protein with the 5’- and 3’-ends of the coronavirus genome is involved in genome circularization and negative-strand RNA synthesis. FEBS J. 2019, 286, 3222–3239. [Google Scholar] [CrossRef]
- Goebel, S.J.; Hsue, B.; Dombrowski, T.F.; Masters, P.S. Characterization of the RNA components of a putative molecular switch in the 3’ untranslated region of the murine coronavirus genome. J. Virol. 2004, 78, 669–682. [Google Scholar] [CrossRef]
- Xue, X.; Yang, H.; Shen, W.; Zhao, Q.; Li, J.; Yang, K.; Chen, C.; Jin, Y.; Bartlam, M.; Rao, Z. Production of authentic SARS-CoV M(pro) with enhanced activity: Application as a novel tag-cleavage endopeptidase for protein overproduction. J. Mol. Biol. 2007, 366, 965–975. [Google Scholar] [CrossRef] [PubMed]
- Jonassen, C.M.; Jonassen, T.O.; Grinde, B. A common RNA motif in the 3’ end of the genomes of astroviruses, avian infectious bronchitis virus and an equine rhinovirus. J. Gen. Virol. 1998, 79, 715–718. [Google Scholar] [CrossRef]
- Robertson, M.P.; Igel, H.; Baertsch, R.; Haussler, D.; Ares, M., Jr.; Scott, W.G. The structure of a rigorously conserved RNA element within the SARS virus genome. PLoS Biol. 2005, 3, e5. [Google Scholar] [CrossRef] [PubMed]
- Spagnolo, J.F.; Hogue, B.G. Host protein interactions with the 3’ end of bovine coronavirus RNA and the requirement of the poly(A) tail for coronavirus defective genome replication. J. Virol. 2000, 74, 5053–5065. [Google Scholar] [CrossRef]
- Tarun, S.Z., Jr.; Wells, S.E.; Deardorff, J.A.; Sachs, A.B. Translation initiation factor eIF4G mediates in vitro poly(A) tail-dependent translation. Proc. Natl. Acad. Sci. USA 1997, 94, 9046–9051. [Google Scholar] [CrossRef]
- Viswanathan, T.; Arya, S.; Chan, S.H.; Qi, S.; Dai, N.; Misra, A.; Park, J.G.; Oladunni, F.; Kovalskyy, D.; Hromas, R.A.; et al. Structural basis of RNA cap modification by SARS-CoV-2. Nat. Commun. 2020, 11, 3718. [Google Scholar] [CrossRef]
- Thiel, V.; Ivanov, K.A.; Putics, Á.; Hertzig, T.; Schelle, B.; Bayer, S.; Weißbrich, B.; Snijder, E.J.; Rabenau, H.; Doerr, H.W.; et al. Mechanisms and enzymes involved in SARS coronavirus genome expression. J. Gen. Virol. 2003, 84, 2305–2315. [Google Scholar] [CrossRef]
- Lai, M.M. Cellular factors in the transcription and replication of viral RNA genomes: A parallel to DNA-dependent RNA transcription. Virology 1998, 244, 1–12. [Google Scholar] [CrossRef]
- Bentley, K.; Keep, S.M.; Armesto, M.; Britton, P. Identification of a noncanonically transcribed subgenomic mRNA of infectious bronchitis virus and other gammacoronaviruses. J. Virol. 2013, 87, 2128–2136. [Google Scholar] [CrossRef]
- Moreno, J.L.; Zúñiga, S.; Enjuanes, L.; Sola, I. Identification of a coronavirus transcription enhancer. J. Virol. 2008, 82, 3882–3893. [Google Scholar] [CrossRef]
- Mateos-Gómez, P.A.; Morales, L.; Zuñiga, S.; Enjuanes, L.; Sola, I. Long-distance RNA-RNA interactions in the coronavirus genome form high-order structures promoting discontinuous RNA synthesis during transcription. J. Virol. 2013, 87, 177–186. [Google Scholar] [CrossRef]
- Patarca, R.; Haseltine, W.A. Bioinformatics Insights on Viral Gene Expression Transactivation: From HIV-1 to SARS-CoV-2. Int. J. Mol. Sci. 2024, 25, 3378. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, X. The leader RNA of coronavirus mouse hepatitis virus contains an enhancer-like element for subgenomic mRNA transcription. J. Virol. 2000, 74, 10571–10580. [Google Scholar] [CrossRef]
- Lindenbach, B.D.; Sgro, J.Y.; Ahlquist, P. Long-distance base pairing in flock house virus RNA1 regulates subgenomic RNA3 synthesis and RNA2 replication. J. Virol. 2002, 76, 3905–3919. [Google Scholar] [CrossRef]
- Choi, I.R.; White, K.A. An RNA activator of subgenomic mRNA1 transcription in tomato bushy stunt virus. J. Biol. Chem. 2002, 277, 3760–3766. [Google Scholar] [CrossRef]
- Lin, H.X.; White, K.A. A complex network of RNA-RNA interactions controls subgenomic mRNA transcription in a tombusvirus. EMBO J. 2004, 23, 3365–3374. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Grigull, J.; Ore, M.O.; Morin, S.; White, K.A. Global organization of a positive-strand RNA virus genome. PLoS Pathog. 2013, 9, e1003363. [Google Scholar] [CrossRef]
- Gong, T.; Ju, F.; Bu, D. Accurate prediction of RNA secondary structure including pseudoknots through solving minimum-cost flow with learned potentials. Commun. Biol. 2024, 7, 297. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wimmer, E.; Paul, A.V. Cis-acting RNA elements in human and animal plus-strand RNA viruses. Biochim. Biophys. Acta 2009, 1789, 495–517. [Google Scholar] [CrossRef]
- Pathak, K.B.; Pogany, J.; Nagy, P.D. Non-template functions of the viral RNA in plant RNA virus replication. Curr. Opin. Virol. 2011, 1, 332–338. [Google Scholar] [CrossRef]
- Sztuba-Solińska, J.; Stollar, V.; Bujarski, J.J. Subgenomic messenger RNAs: Mastering regulation of (+)-strand RNA virus life cycle. Virology 2011, 412, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, S.A.; Thornbrough, J.M.; Zhang, R.; Jha, B.K.; Li, Y.; Elliott, R.; Quiroz-Figueroa, K.; Chen, A.I.; Silverman, R.H.; Weiss, S.R. Lineage A Betacoronavirus NS2 Proteins and the Homologous Torovirus Berne pp1a Carboxy-Terminal Domain Are Phosphodiesterases That Antagonize Activation of RNase L. J. Virol. 2017, 91, e02201-16. [Google Scholar] [CrossRef]
- Goldstein, S.A.; Elde, N.C. Recurrent viral capture of cellular phosphodiesterases that antagonize OAS-RNase L. Proc. Natl. Acad. Sci. USA 2024, 121, e2312691121. [Google Scholar] [CrossRef]
- Zhao, L.; Jha, B.K.; Wu, A.; Elliott, R.; Ziebuhr, J.; Gorbalenya, A.E.; Silverman, R.H.; Weiss, S.R. Antagonism of the interferon-induced OAS-RNase L pathway by murine coronavirus ns2 protein is required for virus replication and liver pathology. Cell Host Microbe 2012, 11, 607–616. [Google Scholar] [CrossRef]
- Wertheim, J.O.; Chu, D.K.; Peiris, J.S.; Kosakovsky Pond, S.L.; Poon, L.L. A case for the ancient origin of coronaviruses. J. Virol. 2013, 87, 7039–7045. [Google Scholar] [CrossRef]
- Zhang, G.; Slowinski, V.; White, K.A. Subgenomic mRNA regulation by a distal RNA element in a (+)-strand RNA virus. RNA 1999, 5, 550–561. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Wang, Y.; Zhang, Y.; Song, X.; Zou, Y.; Li, L.; Zhao, X.; Yin, Z. Current Knowledge on Infectious Bronchitis Virus Non-structural Proteins: The Bearer for Achieving Immune Evasion Function. Front. Vet. Sci. 2022, 9, 820625. [Google Scholar] [CrossRef]
- Cavanagh, D.; Casais, R.; Armesto, M.; Hodgson, T.; Izadkhasti, S.; Davies, M.; Lin, F.; Tarpey, I.; Britton, P. Manipulation of the infectious bronchitis coronavirus genome for vaccine development and analysis of the accessory proteins. Vaccine 2007, 25, 5558–5562. [Google Scholar] [CrossRef]
- Kint, J.; Dickhout, A.; Kutter, J.; Maier, H.J.; Britton, P.; Koumans, J.; Pijlman, G.P.; Fros, J.J.; Wiegertjes, G.F.; Forlenza, M. Infectious Bronchitis Coronavirus Inhibits STAT1 Signaling and Requires Accessory Proteins for Resistance to Type I Interferon Activity. J. Virol. 2015, 89, 12047–12057. [Google Scholar] [CrossRef]
- Jindal, G.A.; Bantle, A.T.; Solvason, J.J.; Grudzien, J.L.; D’Antonio-Chronowska, A.; Lim, F.; Le, S.H.; Song, B.P.; Ragsac, M.F.; Klie, A.; et al. Single-nucleotide variants within heart enhancers increase binding affinity and disrupt heart development. Dev. Cell 2023, 58, 2206–2216.e5. [Google Scholar] [CrossRef]
- Phillips, J.E.; Jackwood, M.W.; McKinley, E.T.; Thor, S.W.; Hilt, D.A.; Acevedol, N.D.; Williams, S.M.; Kissinger, J.C.; Paterson, A.H.; Robertson, J.S.; et al. Changes in nonstructural protein 3 are associated with attenuation in avian coronavirus infectious bronchitis virus. Virus Genes 2012, 44, 63–74. [Google Scholar] [CrossRef]
- Lin, S.Y.; Li, Y.T.; Chen, Y.T.; Chen, T.C.; Hu, C.J.; Chen, H.W. Identification of an infectious bronchitis coronavirus strain exhibiting a classical genotype but altered antigenicity, pathogenicity, and innate immunity profile. Sci. Rep. 2016, 6, 37725. [Google Scholar] [CrossRef]
- Zhao, Y.; Liang, R.; Cheng, J.; Zhao, J.; Xue, J.; Zhang, G. Attenuated Viral Replication of Avian Infectious Bronchitis Virus with a Novel 82-Nucleotide Deletion in the 5a Gene Indicates a Critical Role for 5a in Virus-Host Interactions. Microbiol. Spectr. 2022, 10, e0140522. [Google Scholar] [CrossRef]
- Zhao, Y.; Cheng, J.; Yan, S.; Jia, W.; Zhang, K.; Zhang, G. S gene and 5a accessory gene are responsible for the attenuation of virulent infectious bronchitis coronavirus. Virology 2019, 533, 12–20. [Google Scholar] [CrossRef]
- Tsai, C.T.; Wu, H.Y.; Wang, C.H. Genetic sequence changes related to the attenuation of avian infectious bronchitis virus strain TW2575/98. Virus Genes 2020, 56, 369–379. [Google Scholar] [CrossRef]
- Ammayappan, A.; Upadhyay, C.; Gelb, J., Jr.; Vakharia, V.N. Identification of sequence changes responsible for the attenuation of avian infectious bronchitis virus strain Arkansas DPI. Arch. Virol. 2009, 154, 495–499. [Google Scholar] [CrossRef]
- Amarasinghe, A.; De Silva Senapathi, U.; Abdul-Cader, M.S.; Popowich, S.; Marshall, F.; Cork, S.C.; van der Meer, F.; Gomis, S.; Abdul-Careem, M.F. Comparative features of infections of two Massachusetts (Mass) infectious bronchitis virus (IBV) variants isolated from Western Canadian layer flocks. BMC Vet. Res. 2018, 14, 391. [Google Scholar] [CrossRef]
- Zhao, F.; Han, Z.; Zhang, T.; Shao, Y.; Kong, X.; Ma, H.; Liu, S. Genomic characteristics and changes of avian infectious bronchitis virus strain CK/CH/LDL/971 after serial passages in chicken embryos. Intervirology 2014, 57, 319–330. [Google Scholar] [CrossRef]
- Liu, X.L.; Su, J.L.; Zhao, J.X.; Zhang, G.Z. Complete genome sequence analysis of a predominant infectious bronchitis virus (IBV) strain in China. Virus Genes 2009, 38, 56–65. [Google Scholar] [CrossRef]
- Han, Z.; Sun, C.; Yan, B.; Zhang, X.; Wang, Y.; Li, C.; Zhang, Q.; Ma, Y.; Shao, Y.; Liu, Q.; et al. A 15-year analysis of molecular epidemiology of avian infectious bronchitis coronavirus in China. Infect. Genet. Evol. 2011, 11, 190–200. [Google Scholar] [CrossRef]
- McKinley, E.T.; Jackwood, M.W.; Hilt, D.A.; Kissinger, J.C.; Robertson, J.S.; Lemke, C.; Paterson, A.H. Attenuated live vaccine usage affects accurate measures of virus diversity and mutation rates in avian coronavirus infectious bronchitis virus. Virus Res. 2011, 158, 225–234. [Google Scholar] [CrossRef]
- Thor, S.W.; Hilt, D.A.; Kissinger, J.C.; Paterson, A.H.; Jackwood, M.W. Recombination in avian gamma-coronavirus infectious bronchitis virus. Viruses 2011, 3, 1777–1799. [Google Scholar] [CrossRef]
- Jackwood, M.W.; Hall, D.; Handel, A. Molecular evolution and emergence of avian gammacoronaviruses. Infect. Genet. Evol. 2012, 12, 1305–1311. [Google Scholar] [CrossRef]
- Reddy, V.R.; Theuns, S.; Roukaerts, I.D.; Zeller, M.; Matthijnssens, J.; Nauwynck, H.J. Genetic Characterization of the Belgian Nephropathogenic Infectious Bronchitis Virus (NIBV) Reference Strain B1648. Viruses 2015, 7, 4488–4506. [Google Scholar] [CrossRef]
- Lin, S.Y.; Chen, H.W. Infectious Bronchitis Virus Variants: Molecular Analysis and Pathogenicity Investigation. Int. J. Mol. Sci. 2017, 18, 2030. [Google Scholar] [CrossRef]
- Bande, F.; Arshad, S.S.; Omar, A.R.; Hair-Bejo, M.; Mahmuda, A.; Nair, V. Global distributions and strain diversity of avian infectious bronchitis virus: A review. Anim. Health Res. Rev. 2017, 18, 70–83. [Google Scholar] [CrossRef]
- Liu, X.; Shao, Y.; Ma, H.; Sun, C.; Zhang, X.; Li, C.; Han, Z.; Yan, B.; Kong, X.; Liu, S. Comparative analysis of four Massachusetts type infectious bronchitis coronavirus genomes reveals a novel Massachusetts type strain and evidence of natural recombination in the genome. Infect. Genet. Evol. 2013, 14, 29–38. [Google Scholar] [CrossRef]
- Zhou, S.; Tang, M.; Jiang, Y.; Chen, X.; Shen, X.; Li, J.; Dai, Y.; Zou, J. Complete genome sequence of a novel infectious bronchitis virus strain circulating in China with a distinct S gene. Virus Genes 2014, 49, 152–156. [Google Scholar] [CrossRef]
- Zhang, T.; Han, Z.; Xu, Q.; Wang, Q.; Gao, M.; Wu, W.; Shao, Y.; Li, H.; Kong, X.; Liu, S. Serotype shift of a 793/B genotype infectious bronchitis coronavirus by natural recombination. Infect. Genet. Evol. 2015, 32, 377–387. [Google Scholar] [CrossRef]
- Jakhesara, S.J.; Nath, B.; Pal, J.K.; Joshi, C.G.; Kumar, S. Emergence of a genotype I variant of avian infectious bronchitis virus from Northern part of India. Acta Trop. 2018, 183, 57–60. [Google Scholar] [CrossRef]
- Jiang, L.; Han, Z.; Chen, Y.; Zhao, W.; Sun, J.; Zhao, Y.; Liu, S. Characterization of the complete genome, antigenicity, pathogenicity, tissue tropism, and shedding of a recombinant avian infectious bronchitis virus with a ck/CH/LJL/140901-like backbone and an S2 fragment from a 4/91-like virus. Virus Res. 2018, 244, 99–109. [Google Scholar] [CrossRef]
- Al-Jallad, T.; Kassouha, M.; Salhab, M.; Alomar, A.; Al-Masalma, M.; Abdelaziz, F. Molecular characterization of isolated infectious bronchitis viruses from affected vaccinated broiler flocks in Syria. BMC Vet. Res. 2020, 16, 449. [Google Scholar] [CrossRef]
- Dinan, A.M.; Keep, S.; Bickerton, E.; Britton, P.; Firth, A.E.; Brierley, I. Comparative Analysis of Gene Expression in Virulent and Attenuated Strains of Infectious Bronchitis Virus at Subcodon Resolution. J. Virol. 2019, 93, e00714-19. [Google Scholar] [CrossRef]
- Hassan, M.S.H.; Ojkic, D.; Coffin, C.S.; Cork, S.C.; van der Meer, F.; Abdul-Careem, M.F. Delmarva (DMV/1639) Infectious Bronchitis Virus (IBV) Variants Isolated in Eastern Canada Show Evidence of Recombination. Viruses 2019, 11, 1054. [Google Scholar] [CrossRef]
- Goraichuk, I.V.; Davis, J.F.; Kulkarni, A.B.; Afonso, C.L.; Suarez, D.L. A 25-Year-Old Sample Contributes the Complete Genome Sequence of Avian Coronavirus Vaccine Strain ArkDPI, Reisolated from Commercial Broilers in the United States. Microbiol. Resour. Announc. 2020, 9, e00067-20. [Google Scholar] [CrossRef]
- Guzmán, M.; Hidalgo, H. Live Attenuated Infectious Bronchitis Virus Vaccines in Poultry: Modifying Local Viral Populations Dynamics. Animal 2020, 10, 2058. [Google Scholar] [CrossRef]
- Legnardi, M.; Tucciarone, C.M.; Franzo, G.; Cecchinato, M. Infectious Bronchitis Virus Evolution, Diagnosis and Control. Vet. Sci. 2020, 7, 79. [Google Scholar] [CrossRef] [PubMed]
- Bali, K.; Bálint, Á.; Farsang, A.; Marton, S.; Nagy, B.; Kaszab, E.; Belák, S.; Palya, V.; Bányai, K. Recombination Events Shape the Genomic Evolution of Infectious Bronchitis Virus in Europe. Viruses 2021, 13, 535. [Google Scholar] [CrossRef] [PubMed]
- Quinteros, J.A.; Ignjatovic, J.; Chousalkar, K.K.; Noormohammadi, A.H.; Browning, G.F. Infectious bronchitis virus in Australia: A model of coronavirus evolution—A review. Avian Pathol. 2021, 50, 295–310. [Google Scholar] [CrossRef] [PubMed]
- Quinteros, J.A.; Noormohammadi, A.H.; Lee, S.W.; Browning, G.F.; Diaz-Méndez, A. Genomics and pathogenesis of the avian coronavirus infectious bronchitis virus. Aust. Vet. J. 2022, 100, 496–512. [Google Scholar] [CrossRef] [PubMed]
- Lisowska, A.; Pikuła, A.; Opolska, J.; Jasik, A.; Kycko, A.; Domańska-Blicharz, K. Virulence Properties of GI-23 Infectious Bronchitis Virus Isolated in Poland and Efficacy of Different Vaccination strategies. Pathogens 2021, 10, 522. [Google Scholar] [CrossRef] [PubMed]
- Houta, M.H.; Hassan, K.E.; El-Sawah, A.A.; Elkady, M.F.; Kilany, W.H.; Ali, A.; Abdel-Moneim, A.S. The emergence, evolution and spread of infectious bronchitis virus genotype GI-23. Arch. Virol. 2021, 166, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Lv, D.; Dong, Z.H.; Fan, W.S.; Tang, N.; Wang, L.; Wei, L.P.; Ji, Z.H.; Tang, J.W.; Lin, L.T.; Wei, T.C.; et al. Identification of a novel avian coronavirus infectious bronchitis virus variant with three-nucleotide-deletion in nucleocapsid gene in China. J. Vet. Med. Sci. 2021, 83, 1608–1619. [Google Scholar] [CrossRef] [PubMed]
- da Silva, A.P.; Jude, R.; Gallardo, R.A. Infectious Bronchitis Virus: A Comprehensive Multilocus Genomic Analysis to Compare DMV/1639 and QX Strains. Viruses 2022, 14, 1998. [Google Scholar] [CrossRef] [PubMed]
- Marandino, A.; Mendoza-González, L.; Panzera, Y.; Tomás, G.; Williman, J.; Techera, C.; Gayosso-Vázquez, A.; Ramírez-Andoney, V.; Alonso-Morales, R.; Realpe-Quintero, M.; et al. Genome Variability of Infectious Bronchitis Virus in Mexico: High Lineage Diversity and Recurrent Recombination. Viruses 2023, 15, 1581. [Google Scholar] [CrossRef]
- Rafique, S.; Jabeen, Z.; Pervaiz, T.; Rashid, F.; Luo, S.; Xie, L.; Xie, Z. Avian infectious bronchitis virus (AIBV) review by continent. Front. Cell Infect. Microbiol. 2024, 14, 1325346. [Google Scholar] [CrossRef]
- Rohaim, M.A.; El Naggar, R.F.; Abdelsabour, M.A.; Mohamed, M.H.A.; El-Sabagh, I.M.; Munir, M. Evolutionary Analysis of Infectious Bronchitis Virus Reveals Marked Genetic Diversity and Recombination Events. Genes 2020, 11, 605. [Google Scholar] [CrossRef]
- Wang, C.; Hou, B. A pathogenic and recombinant infectious bronchitis virus variant (CK/CH/GX/202109) with multiorgan tropism. Vet. Res. 2023, 54, 54. [Google Scholar] [CrossRef]
- Li, H.; Liu, G.; Zhou, Q.; Yang, H.; Zhou, C.; Kong, W.; Su, J.; Li, G.; Si, H.; Ou, C. Which strain of the avian coronavirus vaccine will become the prevalent one in China next? Front. Vet. Sci. 2023, 10, 1139089. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.G.; Shen, S.; Tay, F.P.; Liu, D.X. Selection of and recombination between minor variants lead to the adaptation of an avian coronavirus to primate cells. Biochem. Biophys. Res. Commun. 2005, 336, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Lim, X.F.; Lee, C.B.; Pascoe, S.M.; How, C.B.; Chan, S.; Tan, J.H.; Yang, X.; Zhou, P.; Shi, Z.; Sessions, O.M.; et al. Detection and characterization of a novel bat-borne coronavirus in Singapore using multiple molecular approaches. J. Gen. Virol. 2019, 100, 1363–1374. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Pan, Y.F.; Yang, L.F.; Yang, W.H.; Lv, K.; Luo, C.M.; Wang, J.; Kuang, G.P.; Wu, W.C.; Gou, Q.Y.; et al. Individual bat virome analysis reveals co-infection and spillover among bats and virus zoonotic potential. Nat. Commun. 2023, 14, 4079. [Google Scholar] [CrossRef]
- Cao, Y.; Yang, R.; Lee, I.; Zhang, W.; Sun, J.; Wang, W.; Meng, X. Characterization of the SARS-CoV-2 E Protein: Sequence, Structure, Viroporin, and Inhibitors. Protein Sci. 2021, 30, 1114–1130. [Google Scholar] [CrossRef] [PubMed]
- Khare, S.; Gurry, C.; Freitas, L.; Schultz, M.B.; Bach, G.; Diallo, A.; Akite, N.; Ho, J.; Lee, R.T.; Yeo, W.; et al. GISAID’s Role in Pandemic Response. China CDC Wkly. 2021, 3, 1049–1051. [Google Scholar] [CrossRef] [PubMed]
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef]
- Shu, Y.; McCauley, J. GISAID: From vision to reality. EuroSurveillance 2017, 22, 30494. [Google Scholar] [CrossRef]
- Konno, Y.; Kimura, I.; Uriu, K.; Fukushi, M.; Irie, T.; Koyanagi, Y.; Sauter, D.; Gifford, R.J.; USFQ-COVID19 Consortium; Nakagawa, S.; et al. SARS-CoV-2 ORF3b Is a Potent Interferon Antagonist Whose Activity Is Increased by a Naturally Occurring Elongation Variant. Cell Rep. 2020, 32, 108185. [Google Scholar] [CrossRef]
- Lim, F.; Solvason, J.J.; Ryan, G.E.; Le, S.H.; Jindal, G.A.; Steffen, P.; Jandu, S.K.; Farley, E.K. Affinity-optimizing enhancer variants disrupt development. Nature 2024, 626, 151–159. [Google Scholar] [CrossRef]
- de Wit, J.J.S.; Cook, J.K.A. Spotlight on avian pathology: Infectious bronchitis virus. Avian Pathol. 2019, 48, 393–395. [Google Scholar] [CrossRef] [PubMed]
- Cavanagh, D. Coronavirus avian infectious bronchitis virus. Vet. Res. 2007, 38, 281–297. [Google Scholar] [CrossRef]
- Keep, S.; StevensonLeggett, P.; Dowgier, G.; Foldes, K.; Webb, I.; Fones, A.; Littolff, K.; Everest, H.; Britton, P.; Bickerton, E. A Temperature-Sensitive Recombinant of Avian Coronavirus Infectious Bronchitis Virus Provides Complete Protection against Homologous Challenge. J. Virol. 2002, 96, e01100-22. [Google Scholar] [CrossRef] [PubMed]
- Sawicki, S.G.; Sawicki, D.L.; Younker, D.; Meyer, Y.; Thiel, V.; Stokes, H.; Siddell, S.G. Functional and genetic analysis of coronavirus replicase-transcriptase proteins. PLoS Pathog. 2005, 1, e39. [Google Scholar] [CrossRef] [PubMed]
- Haijema, B.J.; Volders, H.; Rottier, P.J. Live, attenuated coronavirus vaccines through the directed deletion of group-specific genes provide protection against feline infectious peritonitis. J. Virol. 2004, 78, 3863–3871, Erratum in PLoS Pathog. 2006, 2, e17. [Google Scholar] [CrossRef]
- de Haan, C.A.; Masters, P.S.; Shen, X.; Weiss, S.; Rottier, P.J. The group-specific murine coronavirus genes are not essential, but their deletion, by reverse genetics, is attenuating in the natural host. Virology 2002, 296, 177–189. [Google Scholar] [CrossRef]
- Laconi, A.; van Beurden, S.J.; Berends, A.J.; Krämer-Kühl, A.; Jansen, C.A.; Spekreijse, D.; Chénard, G.; Philipp, H.C.; Mundt, E.; Rottier, P.J.M.; et al. Deletion of accessory genes 3a, 3b, 5a or 5b from avian coronavirus infectious bronchitis virus induces an attenuated phenotype both in vitro and in vivo. J. Gen. Virol. 2018, 99, 1381–1390. [Google Scholar] [CrossRef]
- Huang, Y.P.; Wang, C.H. Sequence changes of infectious bronchitis virus isolates in the 3’ 7.3 kb of the genome after attenuating passage in embryonated eggs. Avian Pathol. 2007, 36, 59–67. [Google Scholar] [CrossRef]
- Liu, S.; Han, Z.; Chen, J.; Liu, X.; Shao, Y.; Kong, X.; Tong, G.; Rong, J. S1 gene sequence heterogeneity of a pathogenic infectious bronchitis virus strain and its embryo-passaged, attenuated derivatives. Avian Pathol. 2007, 36, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, T.; Casais, R.; Dove, B.; Britton, P.; Cavanagh, D. Recombinant infectious bronchitis coronavirus Beaudette with the spike protein gene of the pathogenic M41 strain remains attenuated but induces protective immunity. J. Virol. 2004, 78, 13804–13811. [Google Scholar] [CrossRef]
- Armesto, M.; Cavanagh, D.; Britton, P. The replicase gene of avian coronavirus infectious bronchitis virus is a determinant of pathogenicity. PLoS ONE 2009, 4, e7384. [Google Scholar] [CrossRef]
- Vo Ngoc, L.; Wang, Y.L.; Kassavetis, G.A.; Kadonaga, J.T. The punctilious RNA polymerase II core promoter. Genes Dev. 2017, 31, 1289–1301. [Google Scholar] [CrossRef]
- FANTOM Consortium and the RIKEN PMI and CLST (DGT). A promoter-level mammalian expression atlas. Nature 2014, 507, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Xi, H.; Yu, Y.; Fu, Y.; Foley, J.; Halees, A.; Weng, Z. Analysis of overrepresented motifs in human core promoters reveals dual regulatory roles of YY1. Genome Res. 2007, 17, 798–806. [Google Scholar] [CrossRef] [PubMed]
- Dudnyk, K.; Cai, D.; Shi, C.; Xu, J.; Zhou, J. Sequence basis of transcription initiation in the human genome. Science 2024, 384, eadj0116. [Google Scholar] [CrossRef]
- Kawasaki, K.; Fukaya, T. Regulatory landscape of enhancer-mediated transcriptional activation. Trends Cell Biol. 2024. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Li, W. Enhancer RNA: What we know and what we can achieve. Cell Prolif. 2022, 55, e13202. [Google Scholar] [CrossRef]
- Liang, L.; Cao, C.; Ji, L.; Cai, Z.; Wang, D.; Ye, R.; Chen, J.; Yu, X.; Zhou, J.; Bai, Z.; et al. Complementary Alu sequences mediate enhancer-promoter selectivity. Nature 2023, 619, 868–875. [Google Scholar] [CrossRef]
- Schoenfelder, S.; Fraser, P. Long-range enhancer-promoter contacts in gene expression control. Nat. Rev. Genet. 2019, 20, 437–455. [Google Scholar] [CrossRef] [PubMed]
- Pombo, A.; Dillon, N. Three-dimensional genome architecture: Players and mechanisms. Nat. Rev. Mol. Cell Biol. 2015, 16, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Long, H.K.; Prescott, S.L.; Wysocka, J. Ever-Changing Landscapes: Transcriptional Enhancers in Development and Evolution. Cell 2016, 167, 1170–1187. [Google Scholar] [CrossRef]
- Furlong, E.E.M.; Levine, M. Developmental enhancers and chromosome topology. Science 2018, 361, 1341–1345. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Liu, Y.; Liu, S.; Zhu, X.; Wu, L.; Zhu, Y.; Zhao, D.; Xu, X.; Chemparathy, A.; Wang, H.; et al. Nested epistasis enhancer networks for robust genome regulation. Science 2022, 377, 1077–1085. [Google Scholar] [CrossRef] [PubMed]
- Mönttinen, H.A.M.; Frilander, M.J.; Löytynoja, A. Generation of de novo miRNAs from template switching during DNA replication. Proc. Natl. Acad. Sci. USA 2023, 120, e2310752120. [Google Scholar] [CrossRef]
- Lau, B.; Kerr, K.; Camiolo, S.; Nightingale, K.; Gu, Q.; Antrobus, R.; Suárez, N.M.; Loney, C.; Stanton, R.J.; Weekes, M.P.; et al. Human Cytomegalovirus RNA2.7 Is Required for Upregulating Multiple Cellular Genes To Promote Cell Motility and Viral Spread Late in Lytic Infection. J. Virol. 2021, 95, e0069821. [Google Scholar] [CrossRef]
- Miller, R.H.; Zimmer, A.; Moutot, G.; Mesnard, J.M.; Chazal, N. Retroviral Antisense Transcripts and Genes: 33 Years after First Predicted, a Silent Retroviral Revolution? Viruses 2021, 13, 2221. [Google Scholar] [CrossRef]
- Toyoda, K.; Matsuoka, M. Functional and Pathogenic Roles of Retroviral Antisense Transcripts. Front. Immunol. 2022, 13, 875211. [Google Scholar] [CrossRef]
- Lin, E.; Panfil, A.R.; Sandel, G.; Jain, P. Novel perspectives on antisense transcription in HIV-1, HTLV-1, and HTLV-2. Front. Microbiol. 2022, 13, 1042761. [Google Scholar] [CrossRef]
- Georg, J.; Hess, W.R. Widespread Antisense Transcription in Prokaryotes. Microbiol. Spectr. 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- Werner, A.; Kanhere, A.; Wahlestedt, C.; Mattick, J.S. Natural antisense transcripts as versatile regulators of gene expression. Nat. Rev. Genet. 2024, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Patarca, R.; Haseltine, W.A. Forty years of HIV research inspires the development of SARS-CoV-2 therapy. J. Mol. Cell Biol. 2024, 15, mjad065. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Caico, I.; Xu, Z.; Iqbal, M.S.; Romerio, F. Epigenetic Regulation of HIV-1 Sense and Antisense Transcription in Response to Latency-Reversing Agents. Noncoding RNA 2023, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Ward, S.; Childs, A.; Staley, C.; Waugh, C.; Watts, J.A.; Kotowska, A.M.; Bhosale, R.; Borkar, A.N. Integrating cryo-OrbiSIMS with computational modelling and metadynamics simulations enhances RNA structure prediction at atomic resolution. Nat. Commun. 2024, 15, 4367. [Google Scholar] [CrossRef] [PubMed]
- Enjuanes, L.; Zuñiga, S.; Castaño-Rodriguez, C.; Gutierrez-Alvarez, J.; Canton, J.; Sola, I. Molecular Basis of Coronavirus Virulence and Vaccine Development. Adv. Virus Res. 2016, 96, 245–286. [Google Scholar] [CrossRef]
- Menachery, V.D.; Gralinski, L.E.; Mitchell, H.D.; Dinnon, K.H., 3rd; Leist, S.R.; Yount, B.L., Jr.; McAnarney, E.T.; Graham, R.L.; Waters, K.M.; Baric, R.S. Combination Attenuation Offers Strategy for Live Attenuated Coronavirus Vaccines. J. Virol. 2018, 92, e00710-18. [Google Scholar] [CrossRef] [PubMed]
- van Beurden, S.J.; Berends, A.J.; Krämer-Kühl, A.; Spekreijse, D.; Chénard, G.; Philipp, H.C.; Mundt, E.; Rottier, P.J.M.; Verheije, M.H. A reverse genetics system for avian coronavirus infectious bronchitis virus based on targeted RNA recombination. Virol. J. 2017, 14, 109. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36, W5–W9. [Google Scholar] [CrossRef]
- ICTV Coronaviridae Study Group. International Committee on Taxonomy of Viruses (ICTV). 2021. Available online: https://talk.ictvonline.org/ictv-reports/ictv_9th_report/positive-sense-rna-viruses-2011/w/posrna_viruses/223/coronaviridae-figures (accessed on 12 January 2024).
- Patarca, R.; Haseltine, W.A. Intragenomic rearrangements involving 5’-untranslated region segments in SARS-CoV-2, other betacoronaviruses, and alphacoronaviruses. Virol. J. 2023, 20, 36. [Google Scholar] [CrossRef]
- Kerpedjiev, P.; Hammer, S.; Hofacker, I.L. Forna (force-directed RNA): Simple and effective online RNA secondary structure diagrams. Bioinformatics 2015, 31, 3377–3379. [Google Scholar] [CrossRef] [PubMed]
- Gruber, A.R.; Lorenz, R.; Bernhart, S.H.; Neuböck, R.; Hofacker, I.L. The Vienna RNA Websuite. Nucleic Acids Res. 2008, 36, W70–W74. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, R.; Bernhart, S.H.; Höner Zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Bikandi, J.; San Millán, R.; Rementeria, A.; Garaizar, J. In silico analysis of complete bacterial genomes: PCR, AFLP-PCR and endonuclease restriction. Bioinformatics 2004, 20, 798–799. [Google Scholar] [CrossRef] [PubMed]
- Duvaud, S.; Gabella, C.; Lisacek, F.; Stockinger, H.; Ioannidis, V.; Durinx, C. Expasy, the Swiss Bioinformatics Resource Portal, as designed by its users. Nucleic Acids Res. 2021, 49, W216–W227. [Google Scholar] [CrossRef]
- Madeira, F.; Pearce, M.; Tivey, A.R.N.; Basutkar, P.; Lee, J.; Edbali, O.; Madhusoodanan, N.; Kolesnikov, A.; Lopez, R. Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic Acids Res. 2022, 50, W276–W279. [Google Scholar] [CrossRef]


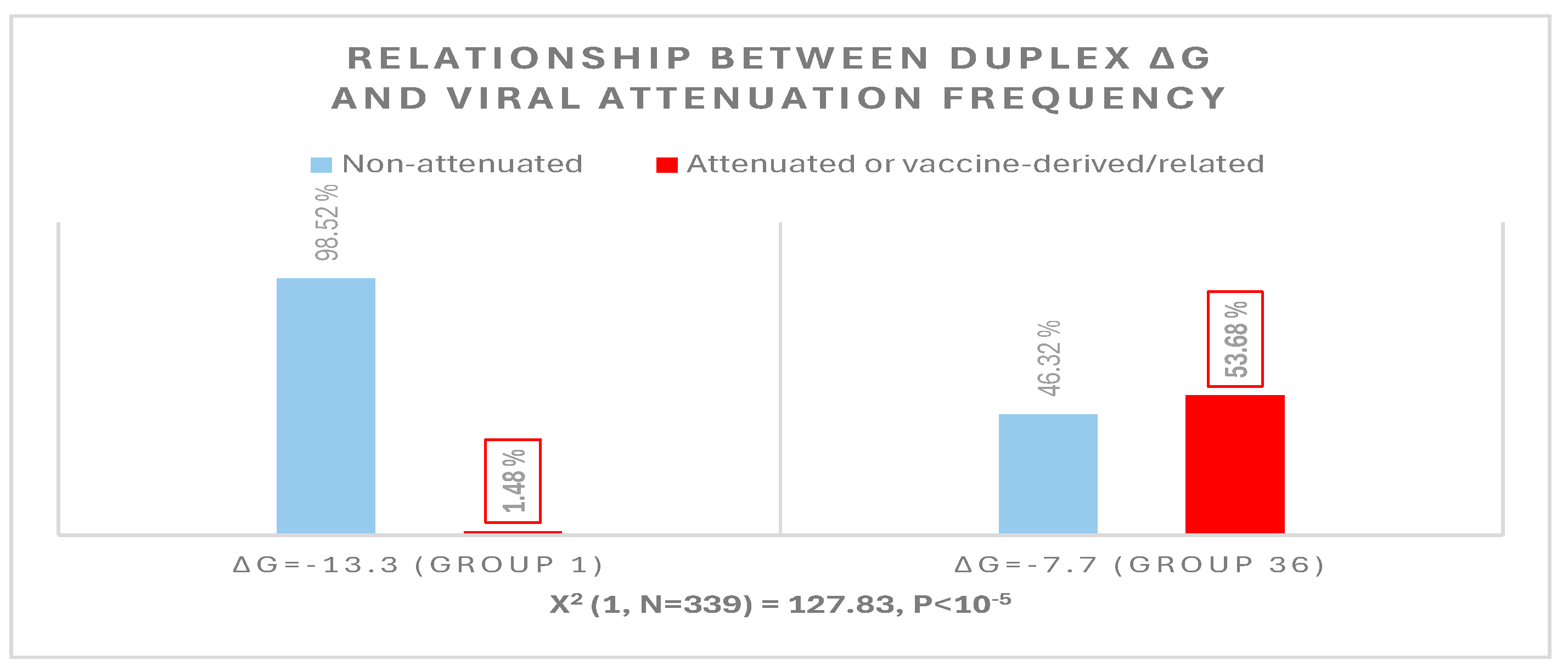

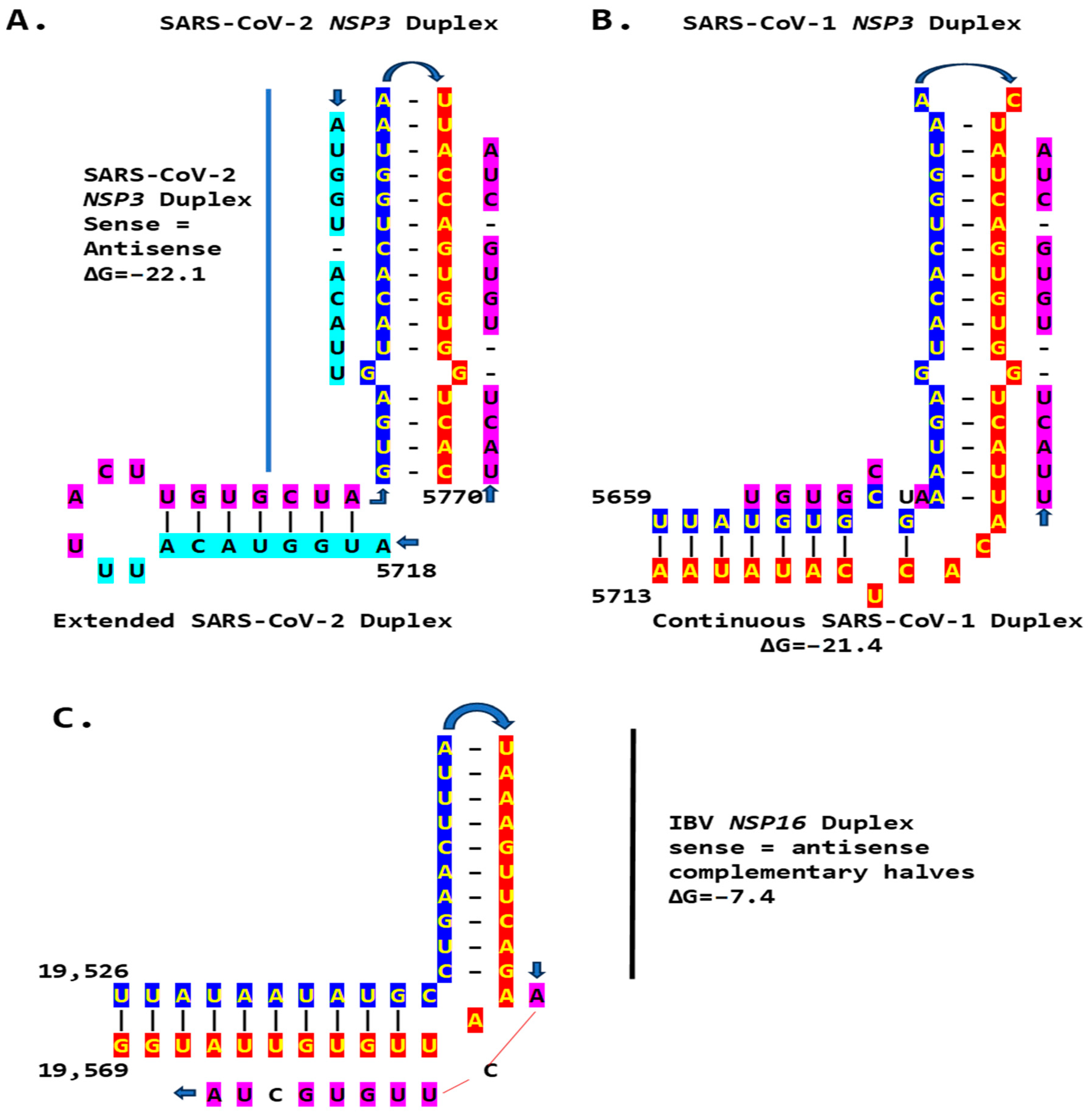
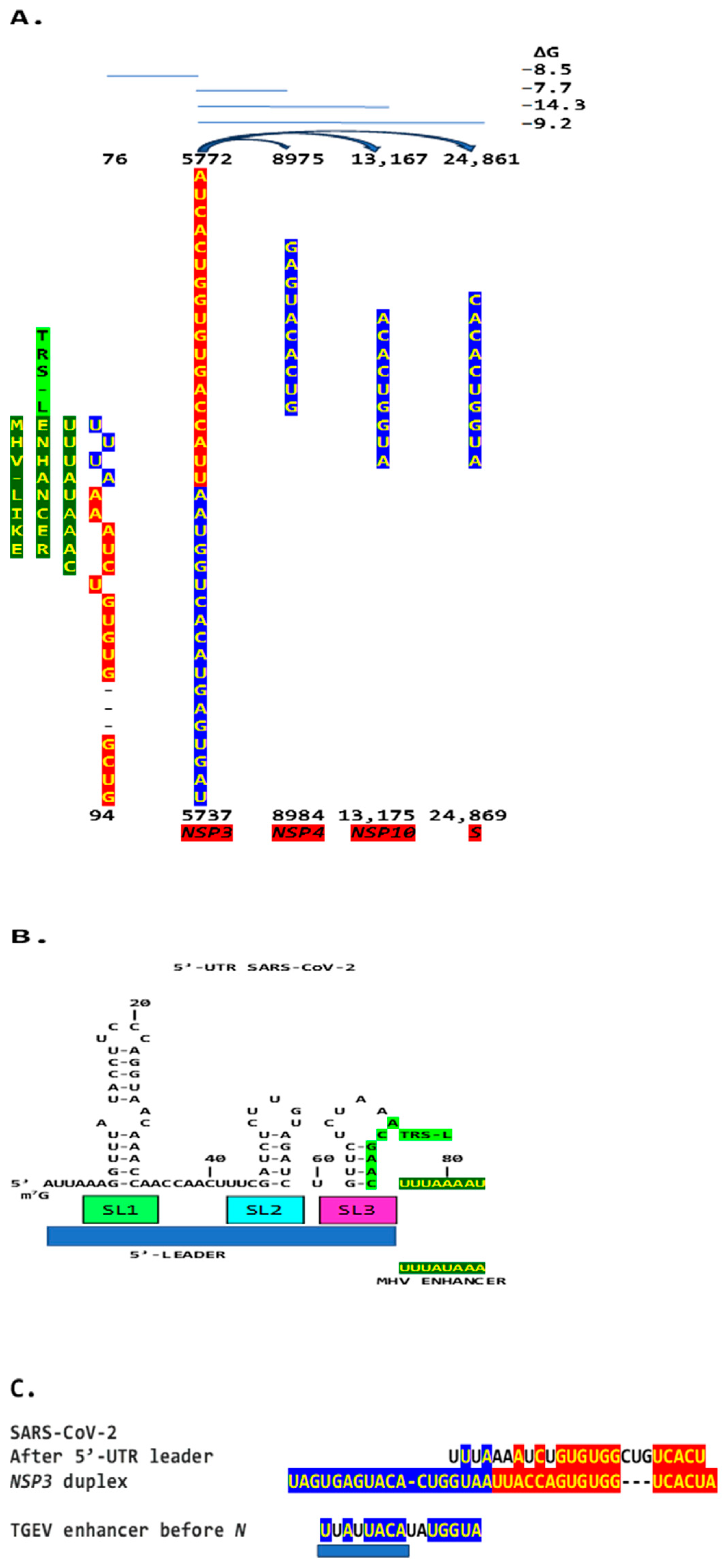




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patarca, R.; Haseltine, W.A. Potential Transcriptional Enhancers in Coronaviruses: From Infectious Bronchitis Virus to SARS-CoV-2. Int. J. Mol. Sci. 2024, 25, 8012. https://doi.org/10.3390/ijms25158012
Patarca R, Haseltine WA. Potential Transcriptional Enhancers in Coronaviruses: From Infectious Bronchitis Virus to SARS-CoV-2. International Journal of Molecular Sciences. 2024; 25(15):8012. https://doi.org/10.3390/ijms25158012
Chicago/Turabian StylePatarca, Roberto, and William A. Haseltine. 2024. "Potential Transcriptional Enhancers in Coronaviruses: From Infectious Bronchitis Virus to SARS-CoV-2" International Journal of Molecular Sciences 25, no. 15: 8012. https://doi.org/10.3390/ijms25158012
APA StylePatarca, R., & Haseltine, W. A. (2024). Potential Transcriptional Enhancers in Coronaviruses: From Infectious Bronchitis Virus to SARS-CoV-2. International Journal of Molecular Sciences, 25(15), 8012. https://doi.org/10.3390/ijms25158012