
Identification of Ribonuclease Inhibitors for the Control of Pathogenic Bacteria
 , , and
, , and
Abstract
:1. Introduction
2. Results
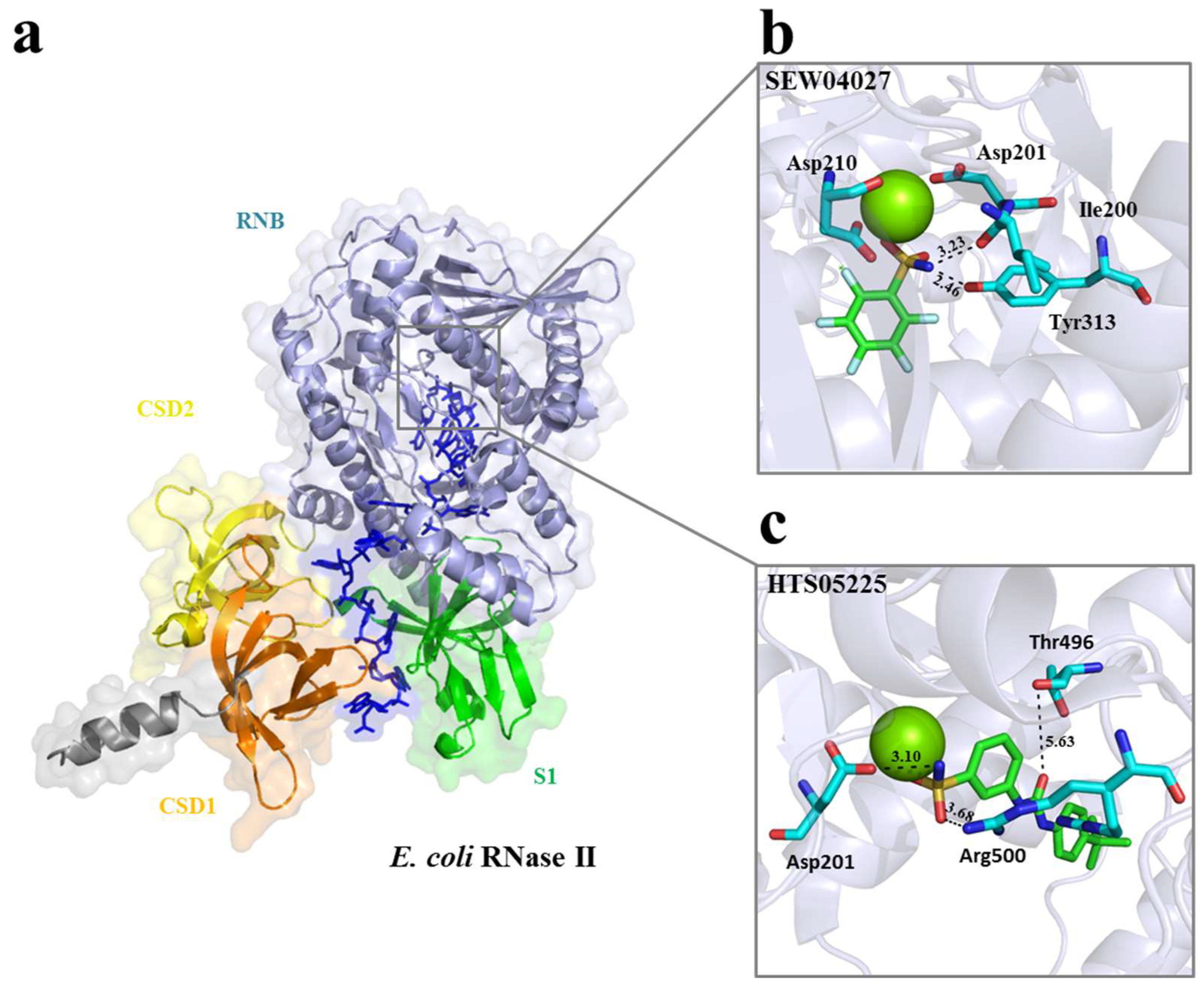
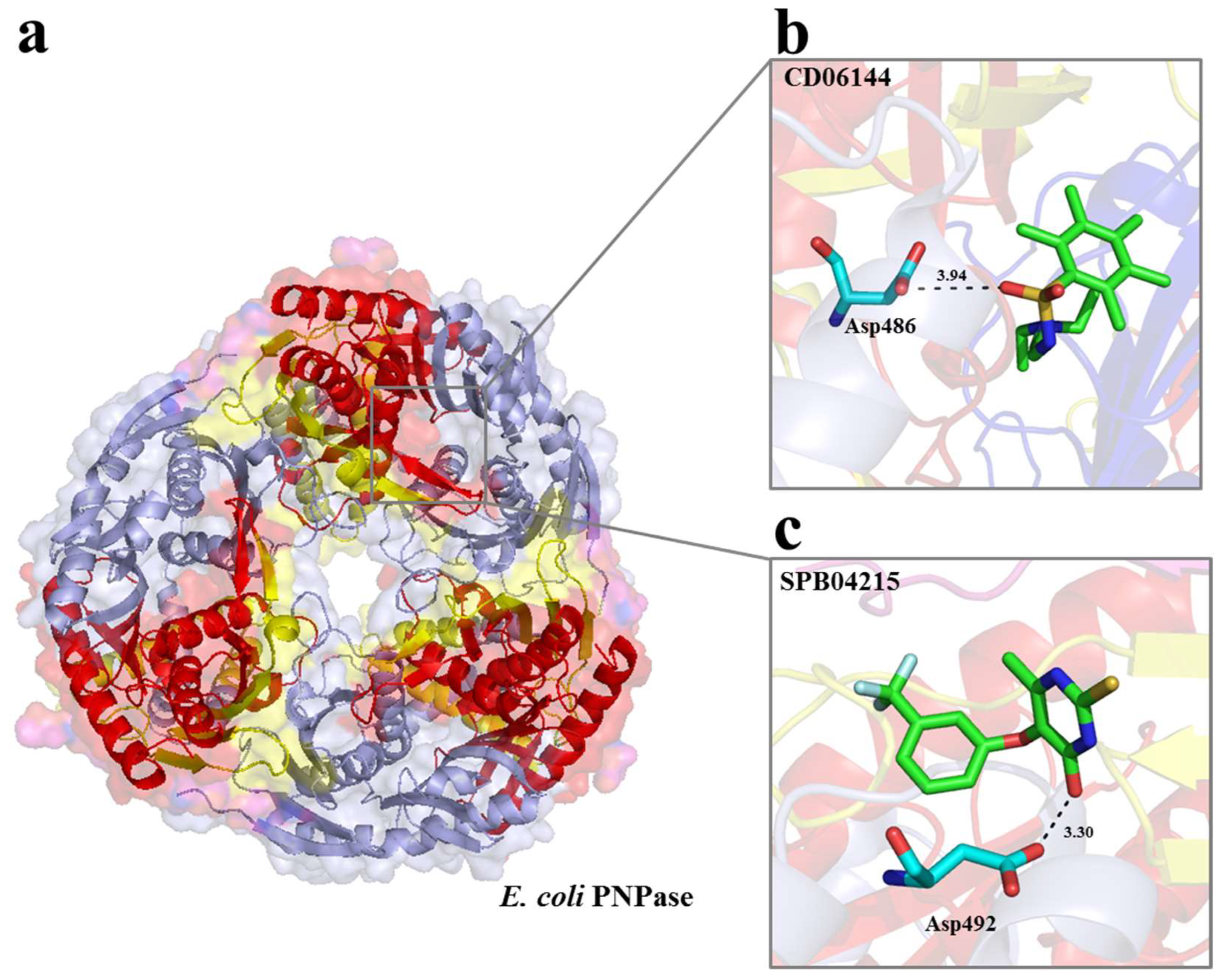
2.1. Selection of Small Molecules Targeting E. coli RNase II and PNPase by vHTS
2.2. RNase II Activity Is Affected In Vitro by Some Chemical Compounds
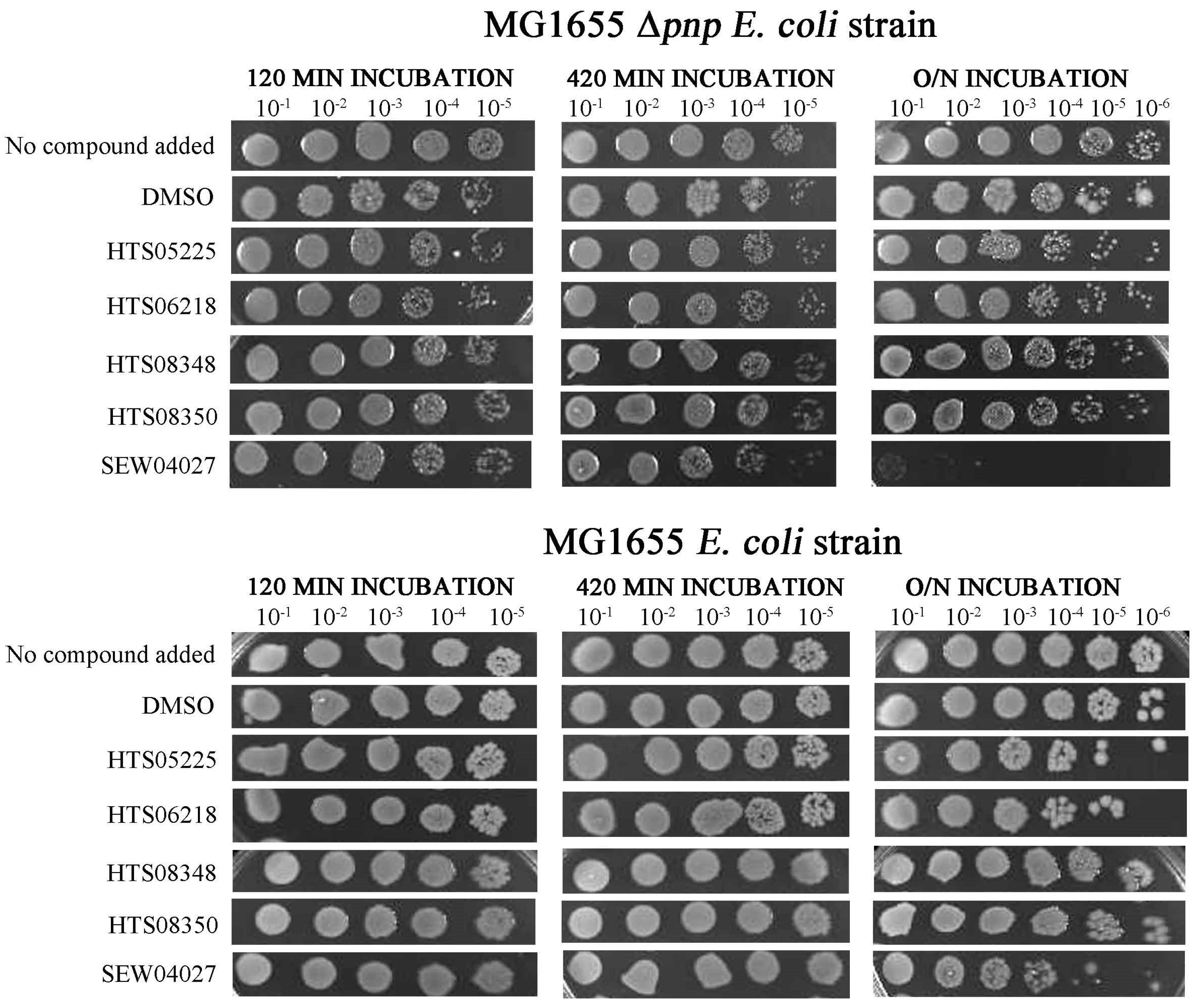
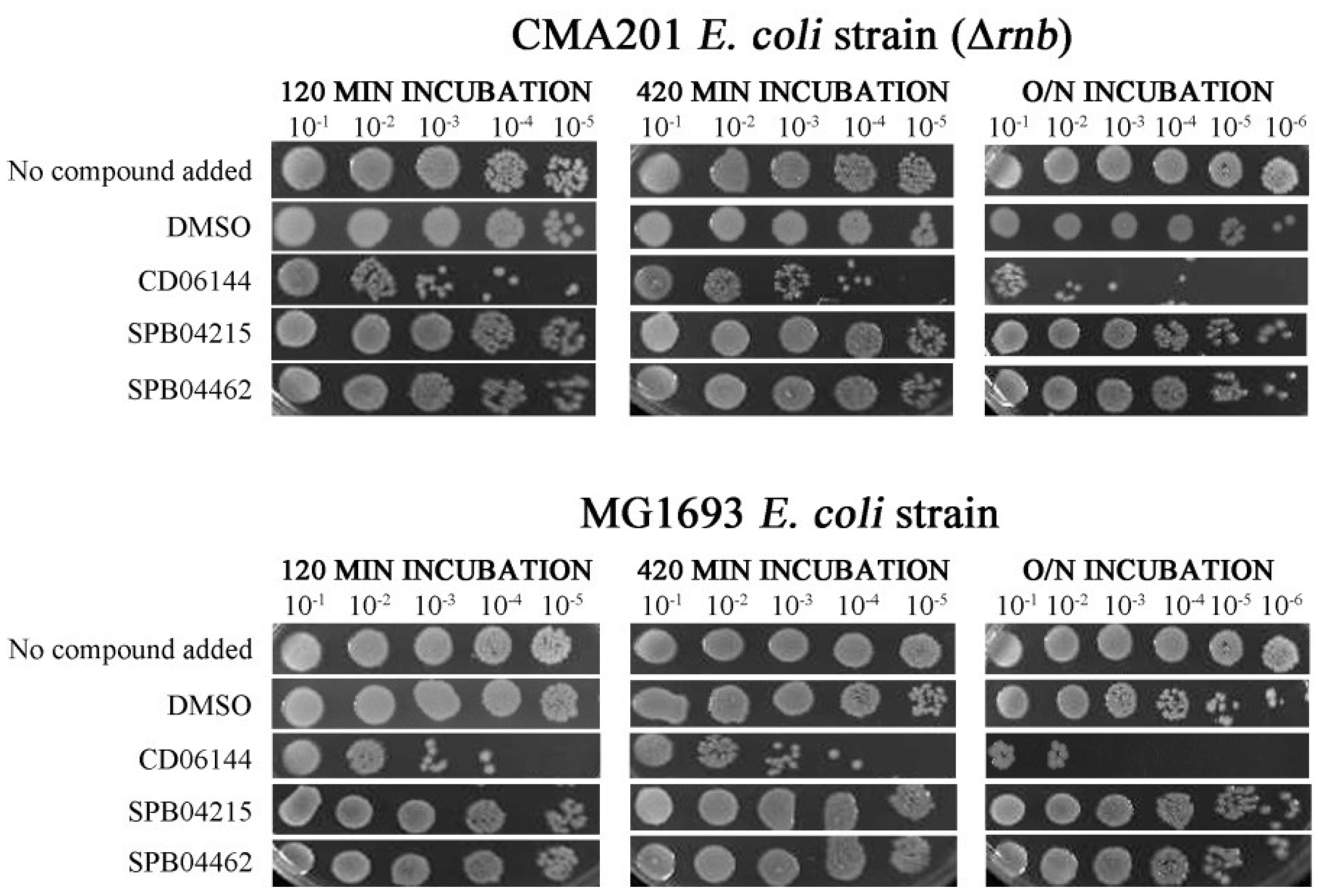
2.3. One Chemical Compound Inhibits RNase II Activity In Vivo
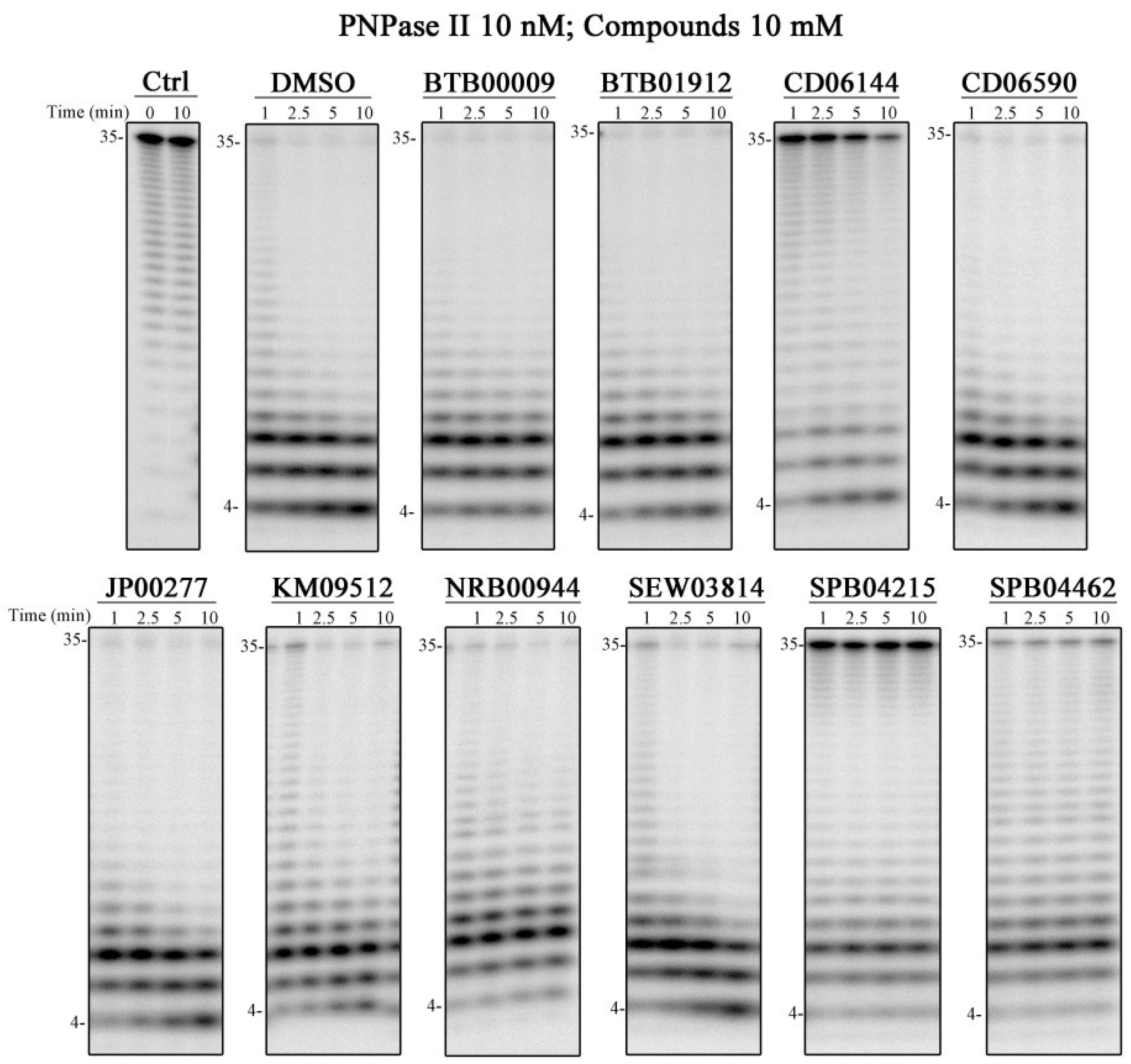
2.4. Compounds That Affect PNPase Activity In Vitro
2.5. Compounds That Inhibit PNPase Reduce Viability of Wild-Type Cells
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Molecular Modelling
4.3. Overexpression and Purification of E. coli RNase II, RNase R, and PNPase
4.4. Activity Assays
4.5. In Vivo Assays
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fischbach, M.A.; Walsh, C.T. Antibiotics for emerging pathogens. Science 2009, 325, 1089–1093. [Google Scholar] [CrossRef] [PubMed]
- Eidem, T.M.; Roux, C.M.; Dunman, P.M. Rna decay: A novel therapeutic target in bacteria. Wiley Interdiscip. Rev. RNA 2012, 3, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Matos, R.G.; Barria, C.; Pobre, V.; Andrade, J.M.; Arraiano, C.M. Exoribonucleases as modulators of virulence in pathogenic bacteria. Front. Cell Infect. Microbiol. 2012, 2, 65. [Google Scholar] [CrossRef] [PubMed]
- Kime, L.; Vincent, H.A.; Gendoo, D.M.; Jourdan, S.S.; Fishwick, C.W.; Callaghan, A.J.; McDowall, K.J. The first small-molecule inhibitors of members of the ribonuclease e family. Sci. Rep. 2015, 5, 8028. [Google Scholar] [CrossRef] [PubMed]
- Mardle, C.E.; Goddard, L.R.; Spelman, B.C.; Atkins, H.S.; Butt, L.E.; Cox, P.A.; Gowers, D.M.; Vincent, H.A.; Callaghan, A.J. Identification and analysis of novel small molecule inhibitors of RNase E: Implications for antibacterial targeting and regulation of RNase E. Biochem. Biophys. Rep. 2020, 23, 100773. [Google Scholar] [CrossRef] [PubMed]
- Grunberg-Manago, M. Polynucleotide phosphorylase. Progress. Nucl. Acid. Res. Mol. Biol. 1963, 1, 93–133. [Google Scholar]
- Carpousis, A.J.; Van Houwe, G.; Ehretsmann, C.; Krisch, H.M. Copurification of E. coli RNase E and PNPase: Evidence for a specific association between two enzymes important in RNA processing and degradation. Cell 1994, 76, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Donovan, W.P.; Kushner, S.R. Polynucleotide phosphorylase and ribonuclease II are required for cell viability and mRNA turnover in Escherichia coli K-12. Proc. Natl. Acad. Sci. USA 1986, 83, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Haddad, N.; Tresse, O.; Rivoal, K.; Chevret, D.; Nonglaton, Q.; Burns, C.M.; Prévost, H.; Cappelier, J.M. Polynucleotide phosphorylase has an impact on cell biology of Campylobacter jejuni. Front. Cell. Inf. Microbiol. 2012, 2, 20298. [Google Scholar] [CrossRef]
- Clements, M.O.; Eriksson, S.; Thompson, A.; Lucchini, S.; Hinton, J.C.; Normark, S.; Rhen, M. Polynucleotide phosphorylase is a global regulator of virulence and persistency in Salmonella enterica. Proc. Natl. Acad. Sci. USA 2002, 99, 8784–8789. [Google Scholar] [CrossRef]
- Rosenzweig, J.A.; Chromy, B.; Echeverry, A.; Yang, J.; Adkins, B.; Plano, G.V.; McCutchen-Maloney, S.; Schesser, K. Polynucleotide phosphorylase independently controls virulence factor expression levels and export in Yersinia spp. FEMS Microbiol. Lett. 2007, 270, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Zhu, M.J. Defects in polynucleotide phosphorylase impairs virulence in Escherichia coli O157:H7. Front. Microbiol. 2015, 6, 806. [Google Scholar] [CrossRef] [PubMed]
- Symmons, M.F.; Jones, G.H.; Luisi, B.F. A duplicated fold is the structural basis for polynucleotide phosphorylase catalytic activity, processivity, and regulation. Structure 2000, 8, 1215–1226. [Google Scholar] [CrossRef]
- Nurmohamed, S.; Vaidialingam, B.; Callaghan, A.J.; Luisi, B.F. Crystal structure of Escherichia coli polynucleotide phosphorylase core bound to RNase E, RNA and manganese: Implications for catalytic mechanism and RNA degradosome assembly. J. Mol. Biol. 2009, 389, 17–33. [Google Scholar] [CrossRef]
- Shi, Z.; Yang, W.Z.; Lin-Chao, S.; Chak, K.F.; Yuan, H.S. Crystal structure of Escherichia coli PNPase: Central channel residues are involved in processive RNA degradation. RNA 2008, 14, 2361–2371. [Google Scholar] [CrossRef]
- Arraiano, C.M.; Andrade, J.M.; Domingues, S.; Guinote, I.B.; Malecki, M.; Matos, R.G.; Moreira, R.N.; Pobre, V.; Reis, F.P.; Saramago, M.; et al. The critical role of RNA processing and degradation in the control of gene expression. FEMS Microbiol. Rev. 2010, 34, 883–923. [Google Scholar] [CrossRef]
- Frazão, C.; McVey, C.E.; Amblar, M.; Barbas, A.; Vonrhein, C.; Arraiano, C.M.; Carrondo, M.A. Unravelling the dynamics of RNA degradation by ribonuclease II and its RNA-bound complex. Nature 2006, 443, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Matos, R.G.; Barbas, A.; Gomez-Puertas, P.; Arraiano, C.M. Swapping the domains of exoribonucleases RNase II and RNase R: Conferring upon RNase II the ability to degrade dsRNA. Proteins 2011, 79, 1853–1867. [Google Scholar] [CrossRef]
- Vincent, H.A.; Deutscher, M.P. The roles of individual domains of RNase R in substrate binding and exoribonuclease activity. The nuclease domain is sufficient for digestion of structured RNA. J. Biol. Chem. 2009, 284, 486–494. [Google Scholar] [CrossRef]
- Chu, L.Y.; Hsieh, T.J.; Golzarroshan, B.; Chen, Y.P.; Agrawal, S.; Yuan, H.S. Structural insights into RNA unwinding and degradation by RNase R. Nucleic Acids Res. 2017, 45, 12015–12024. [Google Scholar] [CrossRef]
- Dehbanipour, R.; Ghalavand, Z. Anti-virulence therapeutic strategies against bacterial infections: Recent advances. Germs 2022, 12, 262–275. [Google Scholar] [CrossRef]
- Rasko, D.A.; Sperandio, V. Anti-virulence strategies to combat bacteria-mediated disease. Nat. Rev. Drug Discov. 2010, 9, 117–128. [Google Scholar] [CrossRef]
- Seifert, M.H.; Kraus, J.; Kramer, B. Virtual high-throughput screening of molecular databases. Curr. Opin. Drug Discov. Devel 2007, 10, 298–307. [Google Scholar]
- Shoichet, B.K. Virtual screening of chemical libraries. Nature 2004, 432, 862–865. [Google Scholar] [CrossRef]
- DeLano, W.L. The Pymol Molecular Graphics System, 0.83 ed.; DeLano Scientific: San Carlos, CA, USA, 2002. [Google Scholar]
- Piedade, J.; Zilhão, R.; Arraiano, C.M. Construction and characterization of an absolute deletion mutant of Escherichia coli ribonuclease II. FEMS Microbiol. Lett. 1995, 127, 187–193. [Google Scholar] [CrossRef]
- Lalonde, M.S.; Zuo, Y.; Zhang, J.; Gong, X.; Wu, S.; Malhotra, A.; Li, Z. Exoribonuclease R in Mycoplasma genitalium can carry out both RNA processing and degradative functions and is sensitive to RNA ribose methylation. RNA 2007, 13, 1957–1968. [Google Scholar] [CrossRef]
- Chen, Y.C. Beware of docking! Trends Pharmacol. Sci. 2015, 36, 78–95. [Google Scholar] [CrossRef]
- Saikia, S.; Bordoloi, M. Molecular docking: Challenges, advances and its use in drug discovery perspective. Curr. Drug Targets 2019, 20, 501–521. [Google Scholar] [CrossRef]
- Reygaert, W.C. An overview of the antimicrobial resistance mechanisms of bacteria. Aims Microbiol. 2018, 4, 482–501. [Google Scholar] [CrossRef]
- Chowdhury, S.; Zielinski, D.C.; Dalldorf, C.; Rodrigues, J.V.; Palsson, B.O.; Shakhnovich, E.I. Empowering drug off-target discovery with metabolic and structural analysis. Nat. Commun. 2023, 14, 3390. [Google Scholar] [CrossRef]
- Feng, J.; Zheng, Y.L.; Ma, W.Q.; Ihsan, A.; Hao, H.H.; Cheng, G.Y.; Wang, X. Multitarget antibacterial drugs: An effective strategy to combat bacterial resistance. Pharmacol. Ther. 2023, 252, 108550. [Google Scholar] [CrossRef]
- Moffat, J.G.; Vincent, F.; Lee, J.A.; Eder, J.; Prunotto, M. Opportunities and challenges in phenotypic drug discovery: An industry perspective. Nat. Rev. Drug Discov. 2017, 16, 531–543. [Google Scholar] [CrossRef]
- Studier, F.W.; Moffatt, B.A. Use of bacteriophage t7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 1986, 189, 113–130. [Google Scholar] [CrossRef]
- Bachmann, B.J.; Low, K.B. Linkage map of Escherichia coli K-12, edition 6. Microbiol. Rev. 1980, 44, 1–56. [Google Scholar] [CrossRef]
- Bachmann, B.J. Pedigrees of some mutant strains of Escherichia coli K-12. Bacteriol. Rev. 1972, 36, 525–557. [Google Scholar] [CrossRef]
- Dressaire, C.; Pobre, V.; Laguerre, S.; Girbal, L.; Arraiano, C.M.; Cocaign-Bousquet, M. PNPase is involved in the coordination of mRNA degradation and expression in stationary phase cells of Escherichia coli. BMC Genom. 2018, 19, 848. [Google Scholar] [CrossRef]
- Zsoldos, Z.; Reid, D.; Simon, A.; Sadjad, S.B.; Johnson, A.P. Ehits: A new fast, exhaustive flexible ligand docking system. J. Mol. Graph. Model. 2007, 26, 198–212. [Google Scholar] [CrossRef]
- Gillet, V.J.; Newell, W.; Mata, P.; Myatt, G.; Sike, S.; Zsoldos, Z.; Johnson, A.P. Sprout: Recent developments in the de novo design of molecules. J. Chem. Inf. Comput. Sci. 1994, 34, 207–217. [Google Scholar] [CrossRef]
- Amblar, M.; Barbas, A.; Goméz-Puertas, P.; Arraiano, C.M. The role of the S1 domain in exoribonucleolytic activity: Substrate specificity and multimerization. RNA 2007, 13, 317–327. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Cairrão, F.; Chora, A.; Zilhão, R.; Carpousis, A.J.; Arraiano, C.M. RNase II levels change according to the growth conditions: Characterization of gmr, a new Escherichia coli gene involved in the modulation of RNase II. Mol. Microbiol. 2001, 39, 1550–1561. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Maybridge Code | eHiTS Score | Target | Name |
|---|---|---|---|
| BTB00009 | −6.7 | PNP | ethyl 4-(4-methylphenyl)-2-oxo-6-phenylcyclohex-3-ene-1-carboxylate |
| BTB01912 | −6.8 | PNP | 2-[2-nitro-4-(trifluoromethyl)phenyl]-1,2,3,4-tetrahydroisoquinoline |
| CD06144 | −7.4 | PNP | N1-[3-(diethylamino)propyl]-2,3,4,5,6-pentamethylbenzene-1-sulfonamide |
| CD06590 | −6.9 | PNP | 2,6-dimethyl-4-[(2,3,4,5,6-pentamethylphenyl)sulfonyl]morpholine |
| HTS05225 | −7.1 | RNB | 3-({[4-(tert-butyl)anilino]carbonyl}amino)benzenesulfonamide |
| HTS05245 | −6.9 | RNB | N-[3-(aminosulfonyl)phenyl]-2,4-difluorobenzamide |
| HTS06218 | −6.9 | RNB | N-[3-(aminosulfonyl)phenyl]cyclohexanecarboxamide |
| HTS07176 | −7.4 | RNB | 4-[(anilinocarbonyl)amino]benzenesulfonamide |
| HTS08348 | −7 | RNB | N-[4-(aminosulfonyl)phenyl]-4-ethylbenzamide |
| HTS08350 | −6.9 | RNB | N-[4-(aminosulfonyl)phenyl]-2,3-dimethylbenzamide |
| JFD01016 | −7.0 | RNB | ethyl 2-(3,4-dimethylanilino)-3,3,3-trifluoro-2-(propionylamino)propanoate |
| JP00277 | −6.6 | PNP | 2,6-dimethyl-4-{[3-(trifluoromethyl)phenyl]sulfonyl}morpholine |
| KM09512 | −7.0 | PNP | 2,8-di(trifluoromethyl)quinolin-4-ol |
| NRB00944 | −7.4 | PNP | 2-[(4,5-diphenyl-1H-imidazol-2-yl)thio]acetic acid |
| RJC02746 | −6.8 | RNB | N1-[3-(2,3,4,5,6-pentafluorophenoxy)phenyl]acetamide |
| SEW03814 | −7.1 | PNP | 4-oxo-4-[1-(trifluoromethyl)-2,3,4,9-tetrahydro-1H-beta-carbolin-2-yl]butanoic a |
| SEW04027 | −6.9 | RNB | 2,3,4,5,6-pentafluorobenzenesulfonamide |
| SPB04215 | −7.0 | PNP | 2-mercapto-6-methyl-5-[3-(trifluoromethyl)phenoxy]pyrimidin-4-ol |
| SPB04462 | −7.3 | PNP | N1-{4-hydroxy-6-methyl-5-[3-(trifluoromethyl)phenoxy]pyrimidin-2-yl}acetamide |
| SPB05935 | −6.8 | RNB | N1-[4-(trifluoromethyl)phenyl]-4-(trifluoromethyl)piperidine-1-carboxamide |
| E. coli Strain | Relevant Properties | Reference |
|---|---|---|
| Bl21(DE3) | F− rB− mB− gal ompT [int::PlacUV5 T7 gen1imm21 nin5] | [34] |
| MG1693 | thyA715 F− λ−rph-1 | [35] |
| CMA201 | MG1693 Δrnb-201::tet | [26] |
| MG1655 | F− λ−rph-1 | [36] |
| MG1655_Δpnp | MG1655 Δpnp::sr | [37] |
| Plasmid | Characteristics | Reference |
|---|---|---|
| pFCT6.1 | pET15b derivative expressing E. coli RNase II with a N-terminal Histidine tag | [42] |
| pABA–RNR | pET15b derivative expressing E. coli RNase R with a N-terminal Histidine tag | [40] |
| pABA–PNP | pET15b derivative expressing E. coli PNPase with a N-terminal Histidine tag | [40] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matos, R.G.; Simmons, K.J.; Fishwick, C.W.G.; McDowall, K.J.; Arraiano, C.M. Identification of Ribonuclease Inhibitors for the Control of Pathogenic Bacteria. Int. J. Mol. Sci. 2024, 25, 8048. https://doi.org/10.3390/ijms25158048
Matos RG, Simmons KJ, Fishwick CWG, McDowall KJ, Arraiano CM. Identification of Ribonuclease Inhibitors for the Control of Pathogenic Bacteria. International Journal of Molecular Sciences. 2024; 25(15):8048. https://doi.org/10.3390/ijms25158048
Chicago/Turabian StyleMatos, Rute G., Katie J. Simmons, Colin W. G. Fishwick, Kenneth J. McDowall, and Cecília M. Arraiano. 2024. "Identification of Ribonuclease Inhibitors for the Control of Pathogenic Bacteria" International Journal of Molecular Sciences 25, no. 15: 8048. https://doi.org/10.3390/ijms25158048