
Targeting the Tumor Vascular Supply to Enhance Radiation Therapy Administered in Single or Clinically Relevant Fractionated Schedules


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
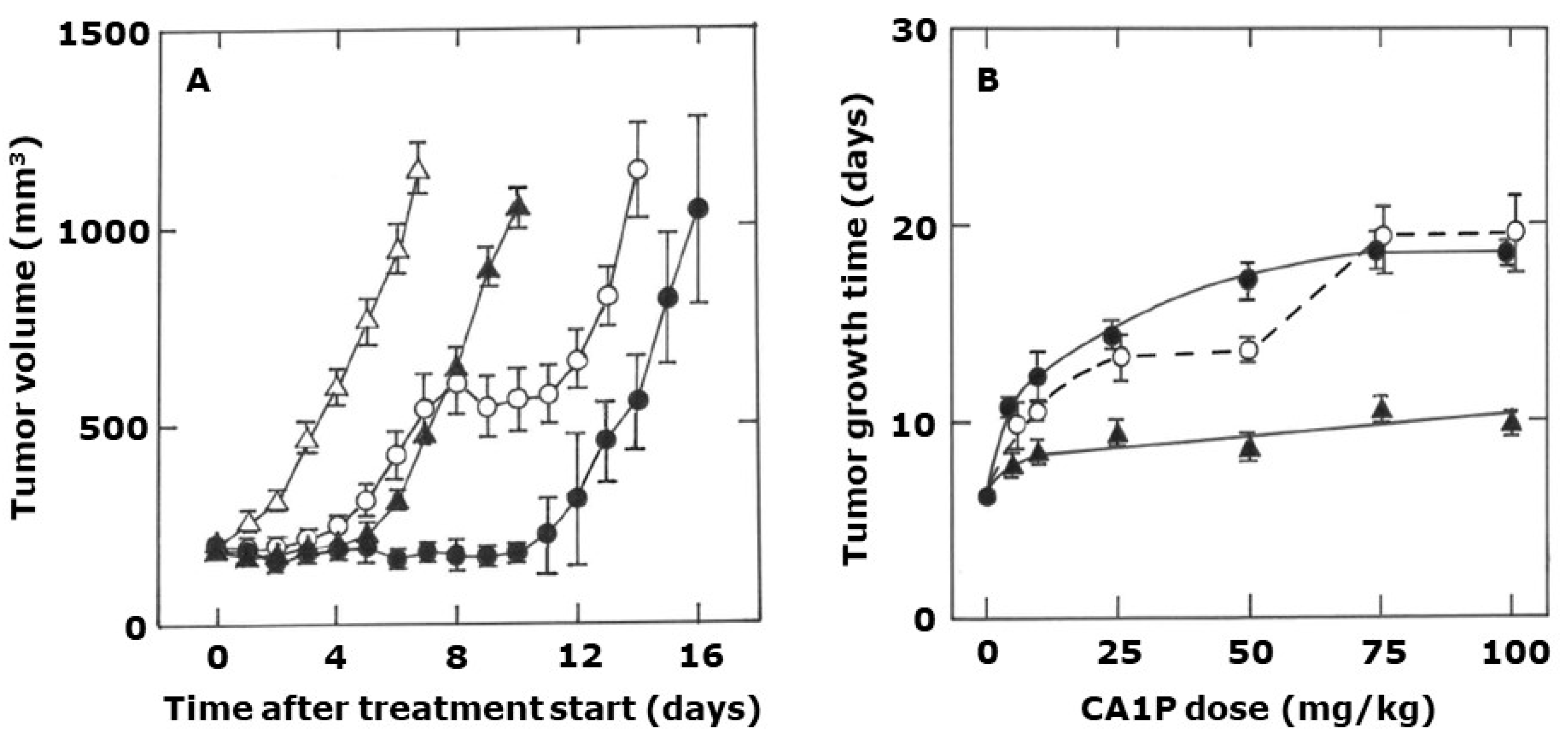
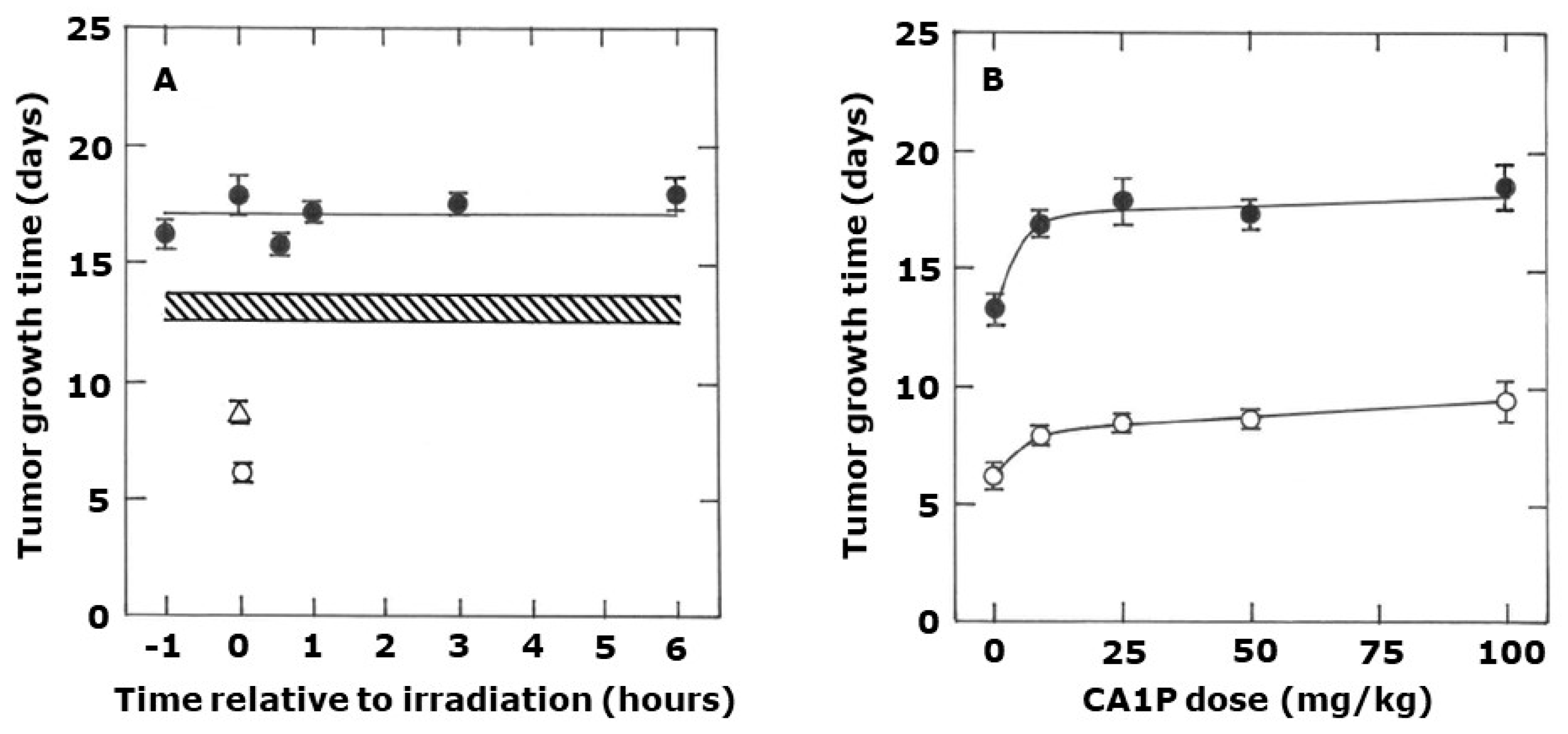
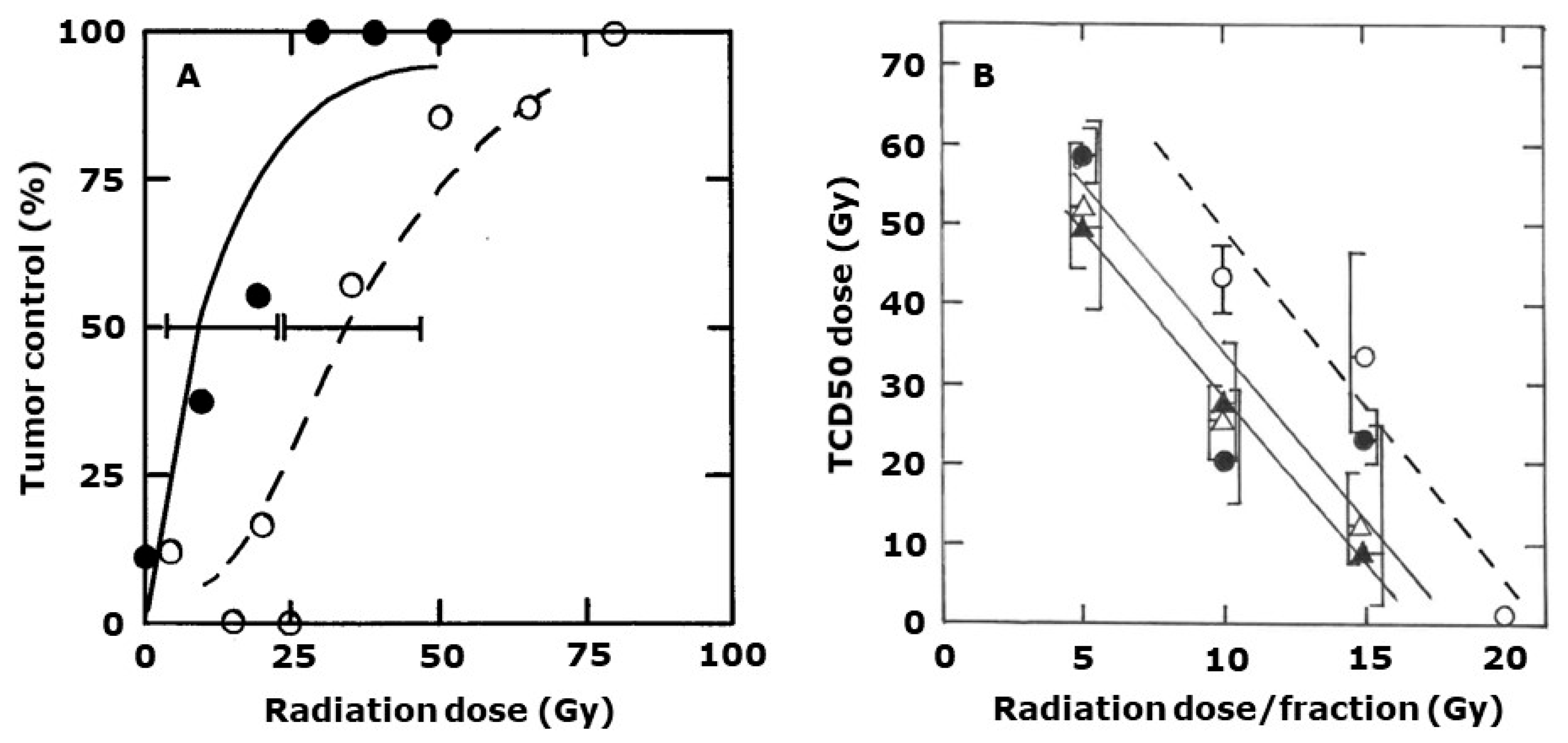
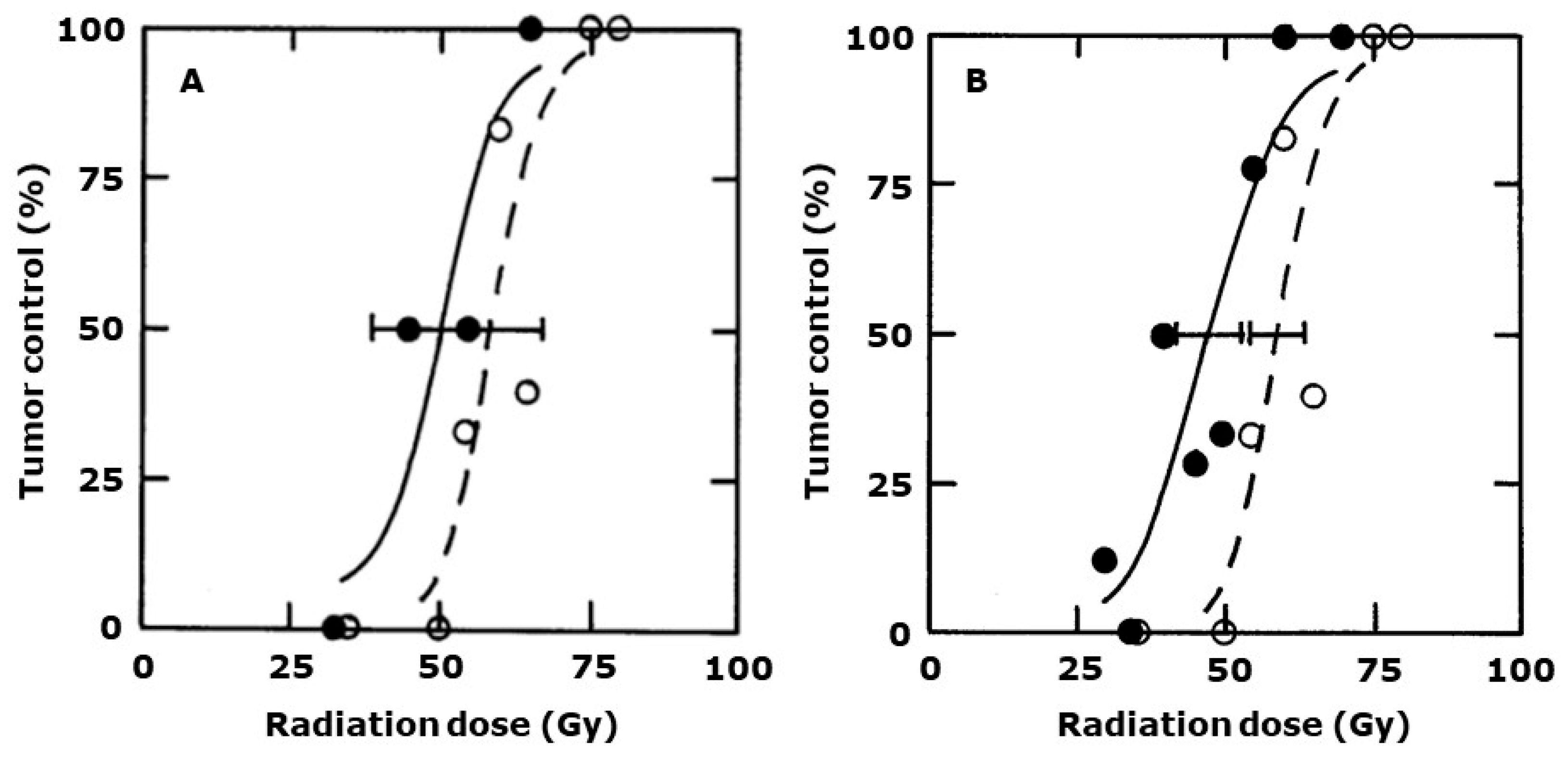
2. Results
3. Discussion
4. Materials and Methods
4.1. Animal and Tumor Model
4.2. Drug Preparation
4.3. Radiation Treatments
4.4. Tumor Response Endpoints
4.5. Statistical Analysis
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [PubMed]
- Brem, S.; Brem, H.; Folkman, J.; Finkelstein, D.; Patz, A. Prolonged tumor dormancy by prevention of neo-vascularization in the vitreous. Cancer Res. 1976, 36, 2807–2812. [Google Scholar] [PubMed]
- Hahnfeldt, P.; Panigrahy, D.; Folkman, J.; Hlatky, L. Tumor development under angiogenic signalling: A dynamic theory of tumor growth, treatment response, and postvascular dormancy. Cancer Res. 1999, 59, 4770–4775. [Google Scholar] [PubMed]
- Bergers, G.; Benjamin, L.E. Tumorigenesis and the angiogenic switch. Nat. Rev. Cancer 2003, 3, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Denekamp, J. Review article: Angiogenesis, neovascular proliferation and vascular pathophysiology as targets for cancer therapy. Br. J. Radiol. 1993, 66, 181–196. [Google Scholar] [CrossRef] [PubMed]
- Siemann, D.W.; Bibby, M.C.; Dark, G.G.; Dicker, A.; Eskens, F.A.L.M.; Horsman, M.R.; Marmé, D.; LoRusso, P.M. Differentiation and definition of vascular-targeted therapies. Clin. Cancer Res. 2005, 11, 416–420. [Google Scholar] [CrossRef] [PubMed]
- Marmé, D. The impact of anti-angiogenic agents on cancer therapy. J. Cancer Res. Clin. Oncol. 2003, 129, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Eskens, F.A. Angiogenesis inhibitors in clinical development; where are we now and where are we going? Br. J. Cancer 2004, 90, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Kerbel, R.S. Angiogenesis as a therapeutic target. Nature 2005, 438, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Denekamp, J.; Hill, S. Angiogenic attack as a therapeutic strategy for cancer. Radiother. Oncol. 1991, 20, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, P.E. Vascular targeting agents as cancer therapeutics. Clin. Cancer Res. 2004, 10, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Woglem, W.H. A critique of tumour resistance. J. Cancer Res. 1923, 7, 283–311. [Google Scholar]
- Boyland, E.; Boyland, M.E. Studies in tissue metabolism: The action of colchicine and B. typhosus extract. J. Cancer Res. 1937, 31, 454–460. [Google Scholar]
- Seed, L.; Slaughter, D.P.; Limarzi, L.R. Effect of colchicine on human carcinoma. Surgery 1940, 7, 696–709. [Google Scholar]
- Horsman, M.R.; Siemann, D.W. Pathophysiological effects of vascular targeting agents and the implications for combination with conventional therapies. Cancer Res. 2006, 66, 11520–11539. [Google Scholar] [CrossRef] [PubMed]
- Siemann, D.W.; Horsman, M.R. Vascular targeted therapies in oncology. Cell Tissue Res. 2009, 335, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Baguley, B.C.; Ching, L.M. DMXAA: An antivascular agent with multiple host responses. Int. J. Radiat. Oncol. Biol. Phys. 2002, 54, 1503–1511. [Google Scholar] [CrossRef] [PubMed]
- Siemann, D.W.; Chaplin, D.J.; Horsman, M.R. Realizing the potential of vascular targeted therapy: The rationale for combining vascular disrupting agents and anti-angiogenic agents to treat cancer. Cancer Investig. 2017, 35, 519–534. [Google Scholar] [CrossRef] [PubMed]
- Tozer, G.M.; Kanthou, C.; Baguley, B.C. Disrupting tumour blood vessels. Nat. Rev. Cancer 2005, 5, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Salmon, H.W.; Siemann, D.W. Effect of the second-generation vascular disrupting agent OXi4503 on tumor vascularity. Clin. Cancer Res. 2006, 12, 4090–4094. [Google Scholar] [CrossRef] [PubMed]
- Iversen, A.B.; Busk, M.; Bertelsen, L.B.; Lausten, C.; Munk, O.L.; Nielsen, T.; Wittenborn, T.R.; Bussink, J.; Lok, J.; Støkilde-Jørgensen, H.; et al. The potential of hyperpolarized 13C magnetic resonance spectroscopy to monitor the effect of combretastatin based vascular disrupting agents. Acta Oncol. 2017, 56, 1626–1633. [Google Scholar] [CrossRef] [PubMed]
- Folkes, L.K.; Christlieb, M.; Madej, E.; Stratford, M.R.L.; Wardman, P. Oxidative metabolism of combretastatin A-1 produces quinine intermediates with the potential to bind to nucleophiles and to enhance oxidative stress via free radicals. Chem. Res. Toxicol. 2007, 20, 1885–1894. [Google Scholar] [CrossRef] [PubMed]
- Gray, L.H.; Conger, A.D.; Ebert, M.; Hornsey, S.; Scott, O.C.A. The concentration of oxygen dissolved in tissues at the time of irradiation as a factor in radiotherapy. Br. J. Radiol. 1953, 26, 638–648. [Google Scholar] [CrossRef] [PubMed]
- Iversen, A.B.; Busk, M.; Horsman, M.R. Induction of hypoxia by vascular disrupting agents and the significance for their combination with radiation therapy. Acta Oncol. 2013, 52, 1320–1326. [Google Scholar] [CrossRef] [PubMed]
- Wittenborn, T.R.; Horsman, M.R. Targeting tumour hypoxia to improve outcome of stereotactic radiotherapy. Acta Oncol. 2015, 54, 1385–1392. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, T.; Murata, R.; Maxwell, R.J.; Stødkilde-Jørgensen, H.; Østergaard, L.; Horsman, M.R. Preclinical studies to predict the efficacy of the vascular changes induced by combretastatin A-4 disodium phosphate in patients. Int. J. Radiat. Oncol. Biol. Phys. 2008, 70, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Overgaard, J. Simultaneous and sequential hyperthermia and radiation treatment of an experimental tumor and its surrounding normal tissue in vivo. Int. J. Radiat. Oncol. Biol. Phys. 1980, 6, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Overgaard, J.; Overgaard, M.; Nielsen, O.S.; Pedersen, A.K.; Timothy, A.R. A comparative investigation of nimorazole and misonidazole as hypoxic radiosensitizers in a C3H mammary carcinoma in vivo. Br. J. Cancer 1982, 46, 904–911. [Google Scholar] [CrossRef]
- Hill, S.A.; Tozer, G.M.; Pettit, G.R.; Chaplin, D.J. Preclinical evaluation of the antitumour activity of the novel vascular targeting agent Oxi4503. Anticancer Res. 2002, 22, 1453–1458. [Google Scholar] [PubMed]
- Holwell, S.E.; Cooper, P.A.; Thompson, M.J.; Pettit, G.R.; Lippert, J.W., III; Martin, S.W.; Bibby, M.C. Anti-tumor and anti-vascular effects of the novel tubulin-binding agent combretastatin A-1 phosphate. Anticancer Res. 2002, 22, 3933–3940. [Google Scholar] [PubMed]
- Salmon, H.W.; Mladinich, C.; Siemann, D.W. Evaluations of vascular disrupting agents CA4P and Oxi4503 in renal cell carcinoma (Caki-1) using a silicon based microvascular casting technique. Eur. J. Cancer 2006, 42, 3073–3078. [Google Scholar] [CrossRef] [PubMed]
- Siemann, D.W.; Shi, W. Dual targeting of tumor vasculature: Combining avastin and vascular disrupting agents (CA4P or Oxi4503). Anticancer Res. 2008, 28, 2027–2032. [Google Scholar] [PubMed]
- Holwell, S.E.; Cooper, P.A.; Grosios, K.; Lippert, J.W., III; Pettit, G.R.; Shnyder, S.D.; Bibby, M.C. Combretastatin A-1 phosphate a novel tubulin-binding agent with in vivo anti vascular effects in experimental tumours. Anticancer Res. 2002, 22, 707–712. [Google Scholar] [PubMed]
- Sheng, Y.; Hua, J.; Pinney, K.G.; Garner, C.M.; Kane, R.R.; Prezioso, J.A.; Chaplin, D.J.; Edvardsen, K. Combretastatin family member Oxi4503 induces tumor vascular collapse through the induction of endothelial apoptosis. Int. J. Cancer 2004, 111, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Rice, L.; Pampo, C.; Lepler, S.; Rojiani, A.M.; Siemann, D.W. Support of a free radical mechanism for enhanced antitumor efficacy of the microtubule disruptor OXi4503. Microvasc. Res. 2011, 81, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Laufer, J.; Johnson, P.; Zhang, E.; Treeby, B.; Cox, B.; Pedley, B.; Beard, P. In vivo preclinical photoacoustic imaging of tumor vasculature development and therapy. J. Biomed. Opt. 2012, 17, 056016. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.P.; Ogunlade, O.; Lythgoe, M.F.; Beard, P.; Pedley, R.B. Longitudinal photoacoustic imaging of the pharmacodynamic effect of vascular targeted therapy on tumors. Clin. Cancer Res. 2019, 25, 7436–7447. [Google Scholar] [CrossRef] [PubMed]
- Chan, L.S.; Malcontenti-Wilson, C.; Muralidharan, V.; Christophi, C. Effet of vascular targeting agent Oxi4503 on tumor cell kinetics in a mouse model of colorectal liver metastasis. Anticancer Res. 2007, 27, 2317–2323. [Google Scholar] [PubMed]
- Hua, J.; Sheng, Y.; Pinney, K.G.; Garner, C.M.; Kane, R.R.; Prezioso, J.A.; Pettit, G.R.; Chaplin, D.J.; Edvardsen, K. Oxi4503, a novel vascular targeting agent: Effects on blood flow and antitumor activity in comparison to combretastatin A-4 phosphate. Antocancer Res. 2003, 23, 1433–1440. [Google Scholar]
- Malcontenti-Wilson, C.; Chan, L.; Nikfarjam, M.; Muralidharan, V.; Christophi, C. Vascular targeting agent Oxi4503 inhibits tumor growth in a colorectal liver metastases model. J. Gastroenterol. Hepatol. 2007, 23, e96–e104. [Google Scholar] [CrossRef] [PubMed]
- Staflin, K.; Järnum, S.; Hua, J.; Honeth, G.; Kannisto, P.; Lindvall, M. Combretastatin A-1 phosphate potentiates the antitumor activity of carboplatin and paclitaxel in a severe combined immunodeficiency disease (SCID) mouse model of human ovarian carcinoma. Int. J. Gynecol. Cancer 2006, 16, 1557–1564. [Google Scholar] [CrossRef]
- Dalal, S.; Burchill, S.A. Preclinical evaluation of vascular-disrupting agents in Ewig’s sarcoma family of tumours. Eur. J. Cancer 2009, 45, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Shnyder, S.D.; Cooper, P.A.; Pettit, G.R.; Lippert, J.W., III; Bibby, M.C. Combretastatin A-1 phosphate potentiates the antitumour activity of cisplatin in a murine adenocarcinoma model. Anticancer Res. 2003, 23, 1619–1624. [Google Scholar] [PubMed]
- Nguyen, L.; Fifis, T.; Christophi, C. Vascular disruptive agent OXi4503 and anti-angiogenic agent sunitinib combination treatment prolong survival of mice with CRC liver metastasis. BMC Cancer 2016, 16, 533. [Google Scholar] [CrossRef] [PubMed]
- Horsman, M.R. The therapeutic potential of using the vascular disrupting agent OXi4503 to enhance mild temperature thermoradiation. Int. J. Hyperth. 2015, 31, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Joiner, M.C.; van der Kogel, A.J.; Steel, G.G. Introduction: The significance of radiobiology and radiotherapy in cancer treatment. In Basic Clinical Radiobiology, 5th ed.; Joiner, M.C., van der Kogel, A.J., Eds.; CRC Press: Boca Raton, FL, USA, 2019; pp. 1–8. [Google Scholar]
- Overgaard, K.; Overgaard, J. Investigations on the possibility of a thermic tumour therapy—1. Short-wave treatment of a transplanted isologous mouse mammary carcinoma. Eur. J. Cancer 1972, 8, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Clémenson, C.; Chargari, C.; Deutsch, E. Combination of vascular disrupting agents and ionizing radiation. Crit. Rev. Oncol. Haematol. 2013, 86, 143–169. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. Available online: http://www.cancer.gov/clinicaltrials (accessed on 5 June 2023).

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Horsman, M.R. Targeting the Tumor Vascular Supply to Enhance Radiation Therapy Administered in Single or Clinically Relevant Fractionated Schedules. Int. J. Mol. Sci. 2024, 25, 8078. https://doi.org/10.3390/ijms25158078
Horsman MR. Targeting the Tumor Vascular Supply to Enhance Radiation Therapy Administered in Single or Clinically Relevant Fractionated Schedules. International Journal of Molecular Sciences. 2024; 25(15):8078. https://doi.org/10.3390/ijms25158078
Chicago/Turabian StyleHorsman, Michael R. 2024. "Targeting the Tumor Vascular Supply to Enhance Radiation Therapy Administered in Single or Clinically Relevant Fractionated Schedules" International Journal of Molecular Sciences 25, no. 15: 8078. https://doi.org/10.3390/ijms25158078