
Exploration of Alicyclobacillus spp. Genome in Search of Antibiotic Resistance

 , , , and
, , , and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Assessment of Antibiotic Resistance
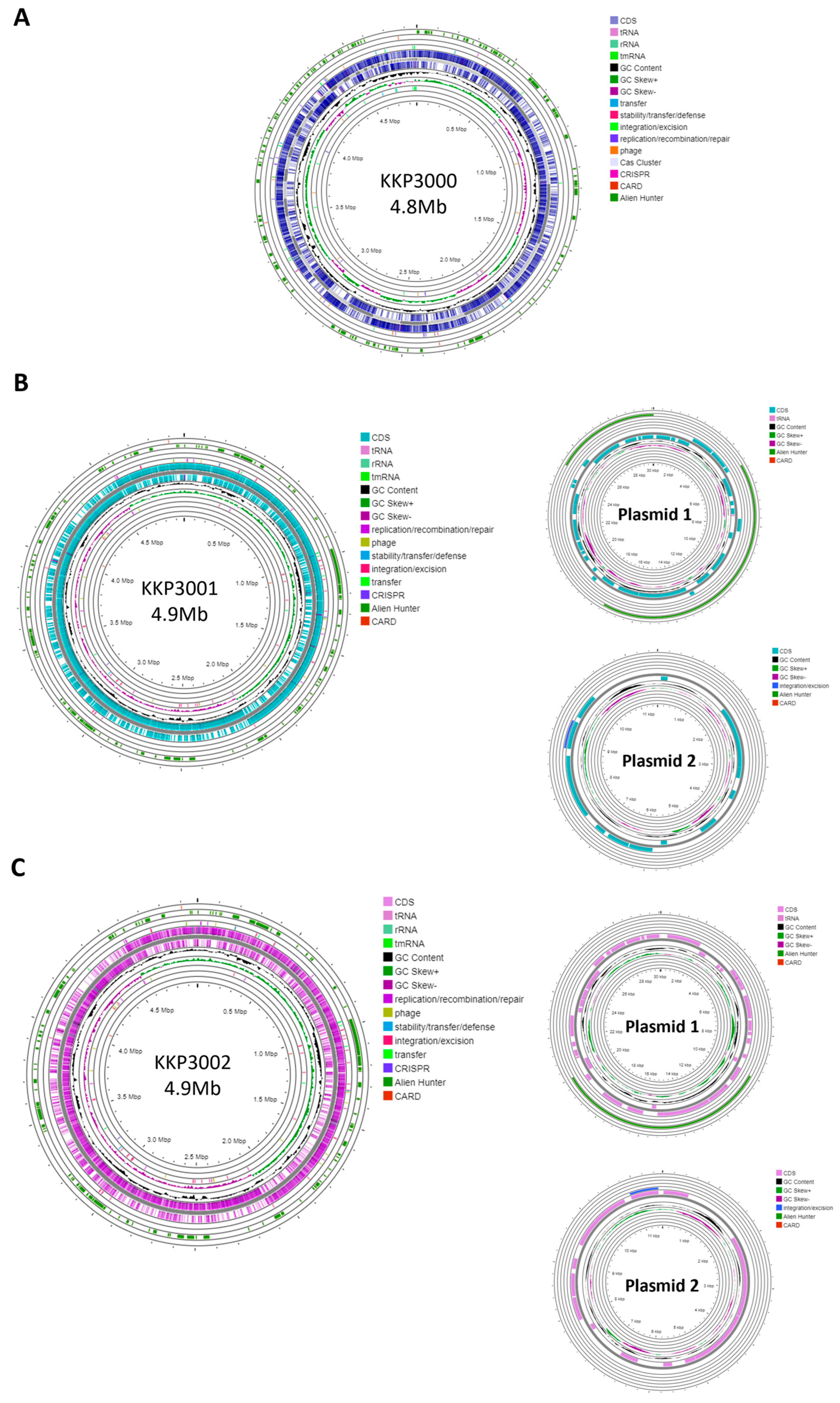
2.2. Genome Characteristics
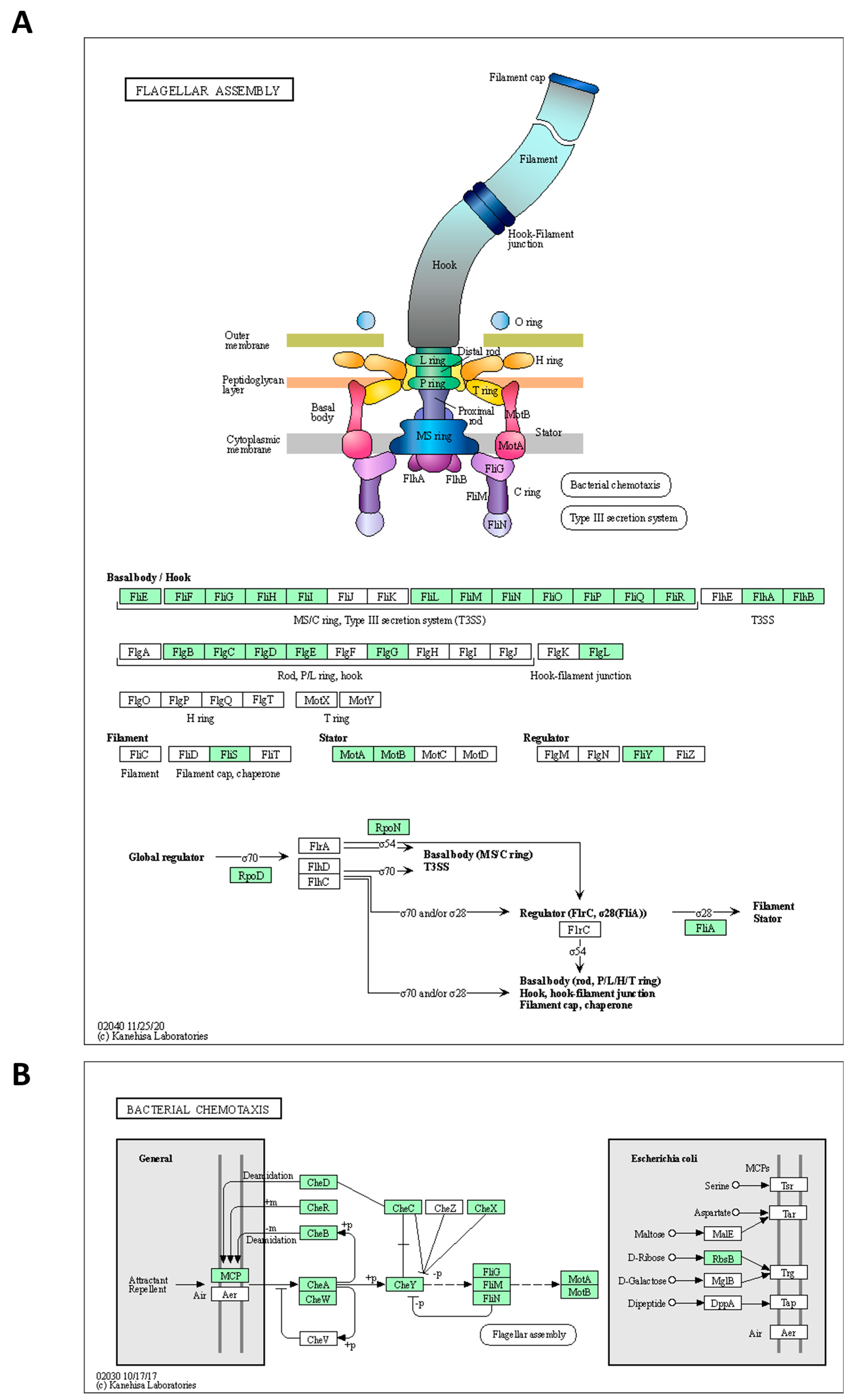
2.3. Functional Annotation
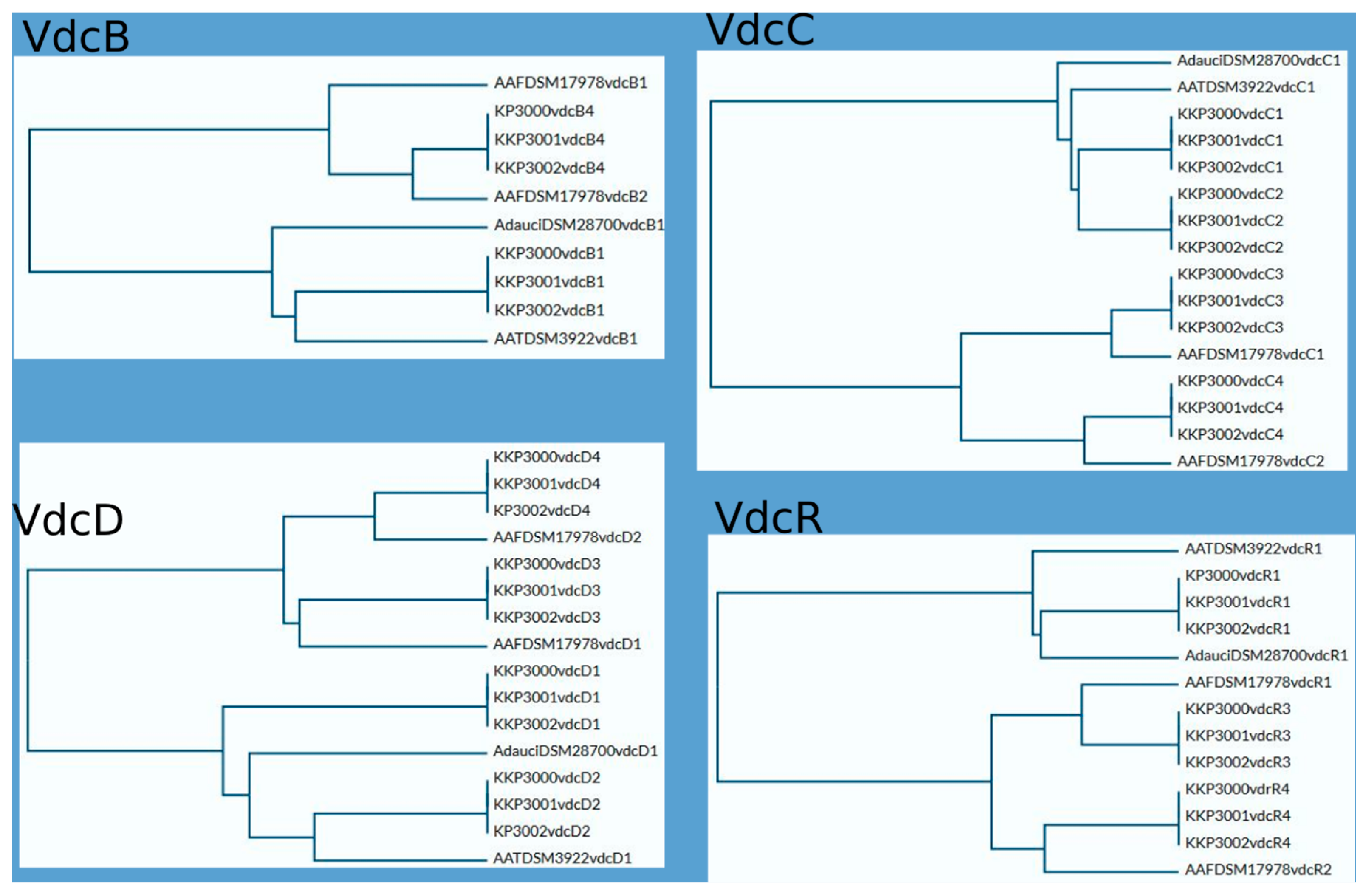
2.4. Vdc Operons
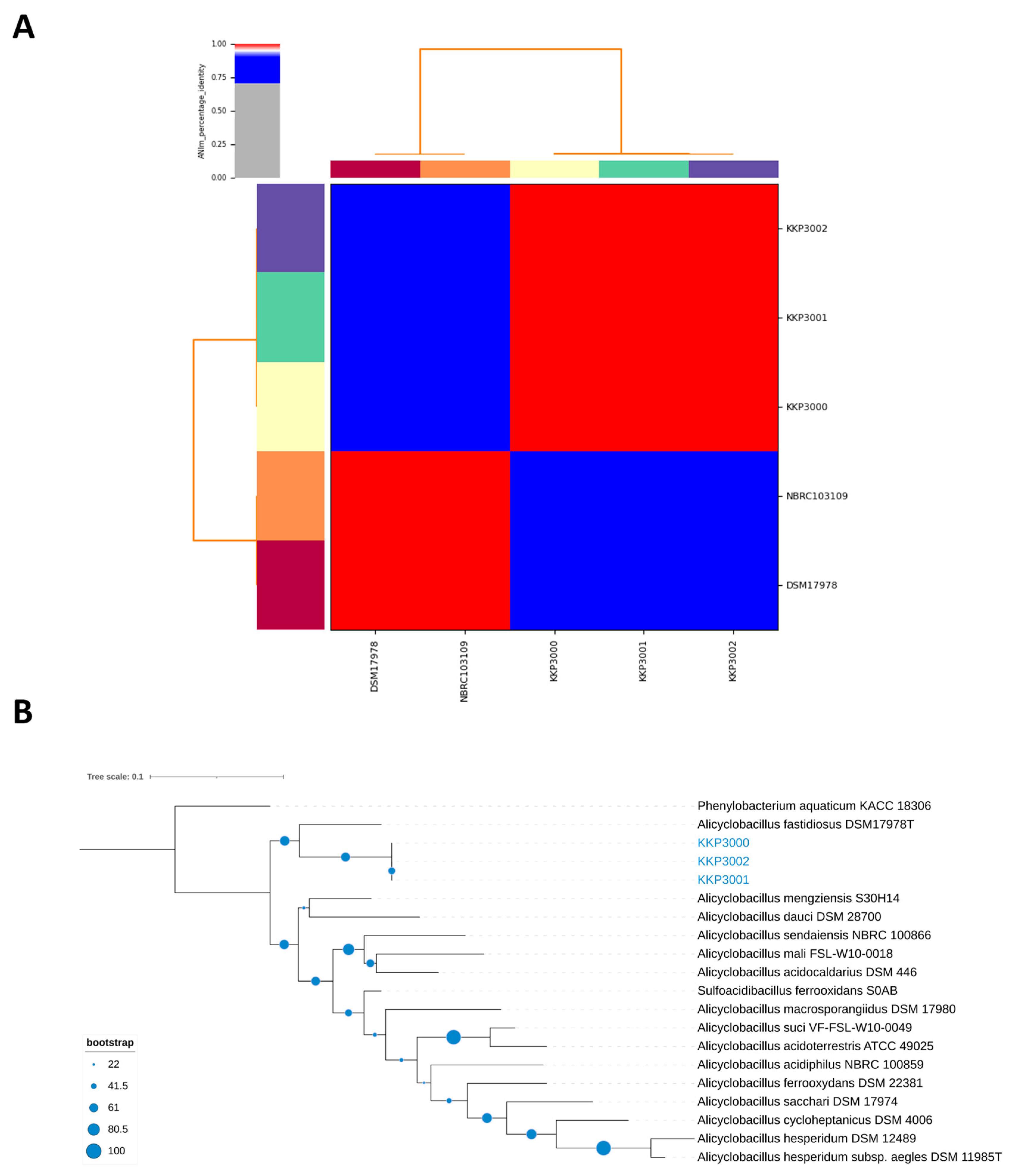
2.5. ANI, and Phylogenomic Analysis
3. Materials and Methods
3.1. Bacterial Strains and Growth Conditions
3.2. Genomic DNA Isolation and Sequencing
3.3. Antibiotic Resistance
3.4. Genome Annotation and Analysis
3.5. Phylogenomic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dekowska, A.; Niezgoda, J.; Sokołowska, B.S. Genetic Heterogeneity of Alicyclobacillus Strains Revealed by RFLP Analysis of VDC Region and RpoB Gene. Biomed. Res. Int. 2018, 2018, 9608756. [Google Scholar] [CrossRef]
- Aulitto, M.; Gallo, G.; Puopolo, R.; Mormone, A.; Limauro, D.; Contursi, P.; Piochi, M.; Bartolucci, S.; Fiorentino, G. Genomic Insight of Alicyclobacillus Mali FL18 Isolated From an Arsenic-Rich Hot Spring. Front. Microbiol. 2021, 12, 639697. [Google Scholar] [CrossRef]
- Leonardo, I.C.; Crespo, M.T.B.; Gaspar, F.B. Unveiling the Complete Genome Sequence of Alicyclobacillus Acidoterrestris DSM 3922T, a Taint-Producing Strain. G3 Genes Genomes Genet. 2022, 12, jkac225. [Google Scholar] [CrossRef]
- Sourri, P.; Tassou, C.C.; Nychas, G.J.E.; Panagou, E.Z. Fruit Juice Spoilage by Alicyclobacillus: Detection and Control Methods—A Comprehensive Review. Foods 2022, 11, 747. [Google Scholar] [CrossRef]
- Zhang, B.; Wu, Y.F.; Song, J.L.; Huang, Z.S.; Wang, B.J.; Liu, S.J.; Jiang, C.Y. Alicyclobacillus Fodiniaquatilis Sp. Nov, Isolated from Acid Mine Water. Int. J. Syst. Evol. Microbiol. 2015, 65, 4915–4920. [Google Scholar] [CrossRef]
- Parte, A.C.; Carbasse, J.S.; Meier-Kolthoff, J.P.; Reimer, L.C.; Göker, M. List of Prokaryotic Names with Standing in Nomenclature (LPSN) Moves to the DSMZ. Int. J. Syst. Evol. Microbiol. 2020, 70, 5607–5612. [Google Scholar] [CrossRef]
- Shymialevich, D.; Wójcicki, M.; Świder, O.; Średnicka, P.; Sokołowska, B. Characterization and Genome Study of a Newly Isolated Temperate Phage Belonging to a New Genus Targeting Alicyclobacillus Acidoterrestris. Genes 2023, 14, 1303. [Google Scholar] [CrossRef]
- Smit, Y.; Cameron, M.; Venter, P.; Witthuhn, R.C. Alicyclobacillus Spoilage and Isolation–A Review. Food Microbiol. 2011, 28, 331–349. [Google Scholar]
- Porębska, I.; Sokołowska, B.; Skąpska, S.; Rzoska, S.J. Treatment with High Hydrostatic Pressure and Supercritical Carbon Dioxide to Control Alicyclobacillus Acidoterrestris Spores in Apple Juice. Food Control 2017, 73, 24–30. [Google Scholar] [CrossRef]
- Sourri, P.; Dekowska, A.; Joanna Bucka-Kolendo, J.; Nychas, G.-J.; Tassou, C.; Doulgeraki, A.I. Identification of Guaiacol Producing Alicyclobacillus Recovered from Commercial Orange Juices Distributed in Greek Markets. Ital. J. Food Sci. 2024, 36, 205–215. [Google Scholar] [CrossRef]
- Połaska, M.; Dekowska, A.; Sokołowska, B. Isolation and Identification of Guaiacol-Producing Alicyclobacillus Fastidiosus Strains from Orchards in Poland. Acta Biochim. Pol. 2021, 68, 301–307. [Google Scholar] [CrossRef]
- Van Luong, T.S.; Moir, C.; Bowman, J.P.; Chandry, P.S. Heat Resistance and Genomics of Spoilage Alicyclobacillus spp. Isolated from Fruit Juice and Fruit-Based Beverages. Food Microbiol. 2021, 94, 103662. [Google Scholar] [CrossRef]
- Dekowska, A. Zastosowanie Metod Biologii Molekularnej W Diagnostyce Bakterii Z Rodzaju Alicyclobacillus. 2011, Volume 66. Available online: https://www.ibprs.pl/wp-content/uploads/2018/08/PNiTPRS-2011-2-rozdzia-iii.pdf (accessed on 21 March 2024).
- Goto, K.; Mochida, K.; Kato, Y.; Asahara, M.; Fujita, R.; An, S.Y.; Kasai, H.; Yokota, A. Proposal of Six Species of Moderately Thermophilic, Acidophilic, Endospore-Forming Bacteria: Alicyclobacillus contaminans sp. Nov., Alicyclobacillus fastidiosus sp. Nov., Alicyclobacillus kakegawensis sp. Nov., Alicyclobacillus macrosporangiidus sp. Nov., Alicyclobacillus sacchari sp. Nov. and Alicyclobacillus shizuokensis sp. Nov. Int. J. Syst. Evol. Microbiol. 2007, 57, 1276–1285. [Google Scholar] [CrossRef]
- Tianli, Y.; Jiangbo, Z.; Yahong, Y. Spoilage by Alicyclobacillus Bacteria in Juice and Beverage Products: Chemical, Physical, and Combined Control Methods. Compr. Rev. Food Sci. Food Saf. 2014, 13, 771–797. [Google Scholar] [CrossRef]
- Osopale, B.A.; Witthuhn, C.R.; Albertyn, J.; Oguntoyinbo, F.A. Inhibitory Spectrum of Diverse Guaiacol-Producing Alicyclobacillus Acidoterrestris by Poly Dimethyl Ammonium Chloride Disinfectant. LWT 2017, 84, 241–247. [Google Scholar] [CrossRef]
- EFSA Panel on Additives and Products or Substances used in Animal Feed (FEEDAP). Guidance on the Assessment of Bacterial Susceptibility to Antimicrobials of Human and Veterinary Importance. EFSA J. 2012, 10, 2740. [Google Scholar] [CrossRef]
- European Food Safety Authority (EFSA). Opinion of the Scientific Panel on Additives and Products or Substances Used in Animal Feed (FEEDAP) on the Updating of the Criteria Used in the Assessment of Bacteria for Resistance to Antibiotics of Human or Veterinary Importance. EFSA J. 2005, 3, 223. [Google Scholar] [CrossRef]
- Adimpong, D.B.; Sørensen, K.I.; Thorsen, L.; Stuer-Lauridsen, B.; Abdelgadir, W.S.; Nielsen, D.S.; Derkx, P.M.F.; Jespersen, L. Antimicrobial Susceptibility of Bacillus Strains Isolated from Primary Starters for African Traditional Bread Production and Characterization of the Bacitracin Operon and Bacitracin Biosynthesis. Appl. Env. Microbiol. 2012, 78, 7903–7914. [Google Scholar] [CrossRef]
- Kashmir, S.V.S.; Hotchkiss, R.D. Evidence of Tandem Duplication of Genes in a Merodiploid Region of Pneumococcal Mutants Resistant to Sulfonamide. Genetics 1975, 81, 21–31. [Google Scholar] [CrossRef]
- Brochet, M.; Couvé, E.; Zouine, M.; Poyart, C.; Glaser, P. A Naturally Occurring Gene Amplification Leading to Sulfonamide and Trimethoprim Resistance in Streptococcus Agalactiae. J. Bacteriol. 2008, 190, 672–680. [Google Scholar] [CrossRef]
- Bertini, A.; Poirel, L.; Bernabeu, S.; Fortini, D.; Villa, L.; Nordmann, P.; Carattoli, A. Multicopy BlaOXA-58 Gene as a Source of High-Level Resistance to Carbapenems in Acinetobacter Baumannii. Antimicrob. Agents Chemother. 2007, 51, 2324–2328. [Google Scholar] [CrossRef]
- Okubo, T.; Ae, R.; Noda, J.; Iizuka, Y.; Usui, M.; Tamura, Y. Detection of the Sul2–StrA–StrB Gene Cluster in an Ice Core from Dome Fuji Station, East Antarctica. J. Glob. Antimicrob. Resist. 2019, 17, 72–78. [Google Scholar] [CrossRef]
- D’Costa, V.M.; King, C.E.; Kalan, L.; Morar, M.; Sung, W.W.L.; Schwarz, C.; Froese, D.; Zazula, G.; Calmels, F.; Debruyne, R.; et al. Antibiotic Resistance Is Ancient. Nature 2011, 477, 457–461. [Google Scholar] [CrossRef]
- Chen, B.; Yuan, K.; Chen, X.; Yang, Y.; Zhang, T.; Wang, Y.; Luan, T.; Zou, S.; Li, X. Metagenomic Analysis Revealing Antibiotic Resistance Genes (ARGs) and Their Genetic Compartments in the Tibetan Environment. Env. Sci. Technol. 2016, 50, 6670–6679. [Google Scholar] [CrossRef]
- D’Costa, V.M.; McGrann, K.M.; Hughes, D.W.; Wright, G.D. Sampling the Antibiotic Resistome. Science 2006, 311, 374–377. [Google Scholar] [CrossRef]
- Aminov, R.I.; Mackie, R.I. Evolution and Ecology of Antibiotic Resistance Genes. FEMS Microbiol. Lett. 2007, 271, 147–161. [Google Scholar] [CrossRef]
- Davies, J. Origins and Evolution of Antibiotic Resistance. Microbiologia 1996, 12, 9–16. [Google Scholar] [CrossRef]
- Hopwood, D.A. How Do Antibiotic-Producing Bacteria Ensure Their Self-Resistance before Antibiotic Biosynthesis Incapacitates Them? Mol. Microbiol. 2007, 63, 937–940. [Google Scholar] [CrossRef]
- Wright, G.D. The Antibiotic Resistome: The Nexus of Chemical and Genetic Diversity. Nat. Rev. Microbiol. 2007, 5, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Li, L.; Ashbolt, N.; Wang, X.; Cui, Y.; Zhu, X.; Xu, Y.; Yang, Y.; Mao, D.; Luo, Y. Arctic Antibiotic Resistance Gene Contamination, a Result of Anthropogenic Activities and Natural Origin. Sci. Total Environ. 2018, 621, 1176–1184. [Google Scholar] [CrossRef] [PubMed]
- Piskovsky, V.; Oliveira, N.M. Bacterial Motility Can Govern the Dynamics of Antibiotic Resistance Evolution. Nat. Commun. 2023, 14, 5584. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.H.F.; Tien, Y.C.; Stedtfeld, R.D.; Topp, E. Impacts of Multi-Year Field Exposure of Agricultural Soil to Macrolide Antibiotics on the Abundance of Antibiotic Resistance Genes and Selected Mobile Genetic Elements. Sci. Total Environ. 2020, 727, 138520. [Google Scholar] [CrossRef] [PubMed]
- Georgakakos, C.B.; Martínez, C.E.; Helbling, D.E.; Walter, M.T. More Movement with Manure: Increased Mobility of Erythromycin through Agricultural Soil in the Presence of Manure. J. Water Health 2023, 21, 1143–1157. [Google Scholar] [CrossRef] [PubMed]
- Lessardl, I.A.; Pratt, S.D.; Mccaffertyl, D.G.; Bussiere, D.E.; Hutchins, C.; Wanner, B.L.; Katz, L.; Walsh’, C.T. Homologs of the Vancomycin Resistance D-Ala-D-Ala Dipeptidase VanX in Streptomyces Toyocaensis, Escherichia Co/i and Synechocystis: Attributes of Catalytic Efficiency, Stereoselectivity and Regulation with Implications for Function. Chem. Biol. 1998, 5, 489–504. [Google Scholar] [CrossRef] [PubMed]
- Zurfluh, K.; Treier, A.; Schmitt, K.; Stephan, R. Mobile Fosfomycin Resistance Genes in Enterobacteriaceae—An Increasing Threat. Microbiologyopen 2020, 9, e1135. [Google Scholar] [CrossRef] [PubMed]
- Maravić, G.; Bujnicki, J.M.; Feder, M.; Pongor, S.; Flögel, M. Alanine-Scanning Mutagenesis of the Predicted RRNA-Binding Domain of ErmC’ Redefines the Substrate-Binding Site and Suggests a Model for Protein-RNA Interactions. Nucleic Acids Res. 2003, 31, 4941–4949. [Google Scholar] [CrossRef] [PubMed]
- Shemesh, M.; Pasvolsky, R.; Zakin, V. External PH Is a Cue for the Behavioral Switch That Determines Surface Motility and Biofilm Formation of Alicyclobacillus Acidoterrestris. J. Food Prot. 2014, 77, 1418–1423. [Google Scholar] [CrossRef] [PubMed]
- Cai, R.; Yuan, Y.; Wang, Z.; Guo, C.; Liu, B.; Liu, L.; Wang, Y.; Yue, T. Precursors and Metabolic Pathway for Guaiacol Production by Alicyclobacillus Acidoterrestris. Int. J. Food Microbiol. 2015, 214, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liang, Y.; Wang, Q.; Jia, H.; Yue, T.; Yuan, Y.; Gao, Z.; Cai, R. Integrated Analysis of Transcriptome and Proteome for Exploring the Mechanism of Guaiacol Production by Alicyclobacillus Acidoterrestris. Food Res. Int. 2021, 148, 110621. [Google Scholar] [CrossRef] [PubMed]
- Niwa, M.; Kuriyama, A.A. Acidoterrestris Rapid Detection Kit. Fruit Process. 2003, 13, 328–331. [Google Scholar]
- Baumgart, J. Handbook of Culture Media for Food Microbiology; Elsevier Science: Amsterdam, The Netherlands, 2003. [Google Scholar]
- Bucka-Kolendo, J.; Kiousi, D.E.; Wojtczak, A.; Doulgeraki, A.I.; Galanis, A.; Sokołowska, B. Depiction of the In Vitro and Genomic Basis of Resistance to Hop and High Hydrostatic Pressure of Lactiplantibacillus Plantarum Isolated from Spoiled Beer. Genes 2023, 14, 1710. [Google Scholar] [CrossRef] [PubMed]
- Kiousi, D.E.; Bucka-Kolendo, J.; Wojtczak, A.; Sokołowska, B.; Doulgeraki, A.I.; Galanis, A. Genomic Analysis and In Vitro Investigation of the Hop Resistance Phenotype of Two Novel Loigolactobacillus Backii Strains, Isolated from Spoiled Beer. Microorganisms 2023, 11, 280. [Google Scholar] [CrossRef] [PubMed]
- Wójcicki, M.; Świder, O.; Średnicka, P.; Shymialevich, D.; Ilczuk, T.; Koperski, Ł.; Cieślak, H.; Sokołowska, B.; Juszczuk-Kubiak, E. Newly Isolated Virulent Salmophages for Biocontrol of Multidrug-Resistant Salmonella in Ready-to-Eat Plant-Based Food. Int. J. Mol. Sci. 2023, 24, 10134. [Google Scholar] [CrossRef] [PubMed]
- Rychen, G.; Aquilina, G.; Azimonti, G.; Bampidis, V.; de Lourdes Bastos, M.; Bories, G.; Chesson, A.; Cocconcelli, P.S.; Flachowsky, G.; Gropp, J.; et al. Guidance on the Characterisation of Microorganisms Used as Feed Additives or as Production Organisms. EFSA J. 2018, 16, 5206. [Google Scholar] [CrossRef]
- Seemann, T. Genome Analysis Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Carattoli, A.; Zankari, E.; Garciá-Fernández, A.; Larsen, M.V.; Lund, O.; Villa, L.; Aarestrup, F.M.; Hasman, H. In Silico Detection and Typing of Plasmids Using PlasmidFinder and Plasmid Multilocus Sequence Typing. Antimicrob. Agents Chemother. 2014, 58, 3895. [Google Scholar] [CrossRef] [PubMed]
- Siguier, P.; Perochon, J.; Lestrade, L.; Mahillon, J.; Chandler, M. ISfinder: The Reference Centre for Bacterial Insertion Sequences. Nucleic Acids Res. 2006, 34, D32–D36. [Google Scholar] [CrossRef] [PubMed]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A Better, Faster Version of the PHAST Phage Search Tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for Predictions of Phenotypes from Genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, S.; Voldby Larsen, M.; Møller Aarestrup, F.; Lund, O. PathogenFinder--Distinguishing Friend from Foe Using Bacterial Whole Genome Sequence Data. PLoS ONE 2013, 8, e77302. [Google Scholar] [CrossRef]
- Grant, J.R.; Enns, E.; Marinier, E.; Mandal, A.; Herman, E.K.; Chen, C.Y.; Graham, M.; Van Domselaar, G.; Stothard, P. Proksee: In-Depth Characterization and Visualization of Bacterial Genomes. Nucleic Acids Res. 2023, 51, W484–W492. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the Sensitivity of Progressive Multiple Sequence Alignment through Sequence Weighting, Position-Specific Gap Penalties and Weight Matrix Choice. Nucleic Acids Res. 1994, 22, 4673. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree of Life (ITOL) v3: An Online Tool for the Display and Annotation of Phylogenetic and Other Trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Göker, M. TYGS Is an Automated High-Throughput Platform for State-of-the-Art Genome-Based Taxonomy. Nat. Commun. 2019, 10, 2182. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Schaffer, A.A.; Aravind, L.; Madden, T.L.; Shavirin, S.; Spouge, J.L.; Wolf, Y.I.; Koonin, E.V.; Altschul, S.F. Improving the accuracy of PSI-BLAST protein database searches with composition-based statistics and other refinements. Nucleic Acids Res. 2001, 29, 2994–3005. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Minimum Inhibition Concentrations Tested [μg/mL] | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Bacteria | AM | GM | KM | SM | EM | CM | TC | CL | VA |
| KKP 3000 | 0.094 | 0.047 | 0.19 | 0.5 | >256 | 0.5 | 0.016 | 0.5 | 0.19 |
| KKP 3001 | 0.064 | 0.064 | 0.38 | 0.5 | >256 | 0.5 | 0.016 | 0.75 | 0.19 |
| KKP 3002 | 0.047 | 0.047 | 0.5 | 0.5 | >256 | 0.75 | 0.016 | 0.75 | 0.25 |
| A. fastidiosus DSM 17978 | 0.016 | 0.064 | 0.5 | 0.5 | >256 | 0.047 | 0.016 | 0.38 | 0.19 |
| Genome Characteristics | Alicyclobacillus KKP 3000 | Alicyclobacillus KKP 3001 | Alicyclobacillus KKP 3002 |
|---|---|---|---|
| Length | 4,859,599 bp | 4,972,367 bp | 4,977,437 bp |
| GC content | 52.95% | 53% | 52.92% |
| CDSs | 4872 | 4924 | 4928 |
| rRNAs | 3 | 45 | 45 |
| tRNAs | 40 | 131 | 131 |
| No. of CRISPR Arrays | 1 | 1 | 1 |
| IS elements | 52 | 14 | 14 |
| Phages | |||
| Intact | 0 | 0 | 0 |
| Incomplete | 5 | 7 | 5 |
| Questionable | 0 | 1 | 1 |
| Antibiotic resistance genes | |||
| Perfect hits | 0 | 0 | 0 |
| Strict hits | 5 | 5 | 5 |
| Loose hits | 302 | 297 | 297 |
| Virulence genes | 0 | 0 | 0 |
| Plasmids | 0 | 2 | 2 |
| Clusters of Orthologous Groups | Alicyclobacillus KKP 3000 | Alicyclobacillus KKP 3001 | Alicyclobacillus KKP 3002 |
|---|---|---|---|
| C-Energy production and conversion | 212 (4.96%) | 214 (5.5%) | 214 (5.5%) |
| D-Cell cycle control and mitosis | 59 (1.38%) | 58 (1.49%) | 58 (1.49%) |
| E-Amino Acid metabolism and transport | 482 (11.29%) | 434 (11.15%) | 434 (11,15%) |
| F-Nucleotide metabolism and transport | 99 (2.32%) | 99 (2.54%) | 99 (2.54%) |
| G-Carbohydrate metabolism and transport | 425 (9.95%) | 320 (8.22%) | 320 (8.22%) |
| H-Coenzyme metabolism | 183 (4.29%) | 163 (4.19%) | 163 (4.19%) |
| I-Lipid metabolism | 158 (3.7%) | 156 (4.01%) | 156 (4.01%) |
| J-Translation | 201 (4.7%) | 187 (4.80%) | 187 (4.8%) |
| K-Transcription | 374 (8.76%) | 352 (9.04%) | 352 (9.04%) |
| L-Replication and repair | 218 (5.11%) | 265 (6.81%) | 268 (6.88%) |
| M-Cell wall/membrane/envelope biogenesis | 217 (5.08%) | 193 (4.96%) | 193 (4.96%) |
| N-Cell motility | 47 (1.1%) | 47 (1.21%) | 47 (1.21%) |
| O-Post-translational modification, protein turnover, chaperone functions | 90 (2.11%) | 80 (2.06%) | 80 (2.05%) |
| P-Inorganic ion transport and metabolism | 278 (6.51%) | 201 (5.16%) | 200 (5.14%) |
| Q-Secondary Structure | 118 (2.76%) | 67 (1.72%) | 67 (1.72%) |
| T-Signal Transduction | 151 (3.54%) | 111 (2.85%) | 111 (2.85%) |
| U-Intracellular trafficking and secretion | 49 (1.15%) | 31 (0.80%) | 31 (0.8%) |
| V-Defence mechanisms | 56 (1.31%) | 58 (1.49%) | 58 (1.49%) |
| S-Function Unknown | 696 (16.3%) | 697 (17.91%) | 696 (17.9%) |
| No annotation | 157 (3.68%) | 159 (4.09%) | 159 (4.09%) |
| Total (%) | 4270 (100%) | 3892 (100%) | 3893 (100%) |
| Alicyclobacillus KKP 3000 | Alicyclobacillus KKP 3001 | Alicyclobacillus KKP 3002 | Predicted Protein | Gene |
|---|---|---|---|---|
| Sporulation and Germination | ||||
| KFAPOJEI_02576 | PLKKBLCC_02372 | PKEAFELD_02372 | stage III sporulation protein AG | spoIIIAG |
| KFAPOJEI_02575 | PLKKBLCC_02373 | PKEAFELD_02373 | Stage III sporulation protein AF | spoIIIAF |
| KFAPOJEI_02574 | PLKKBLCC_02374 | PKEAFELD_02374 | stage III sporulation protein AE | spoIIIAE |
| KFAPOJEI_02573 | PLKKBLCC_02375 | PKEAFELD_02375 | Stage III sporulation protein AD | spoIIIAD |
| KFAPOJEI_02572 | PLKKBLCC_02376 | PKEAFELD_02376 | stage III sporulation protein AC | spoIIIAC |
| KFAPOJEI_02571 | PLKKBLCC_02377 | PKEAFELD_02377 | Stage III sporulation protein AB | spoIIIAB |
| KFAPOJEI_02570 | PLKKBLCC_02378 | PKEAFELD_02378 | stage III sporulation protein AA | spoIIIAA |
| KFAPOJEI_03030 | PLKKBLCC_01460 | PKEAFELD_01459 | Sporulation sigma-E factor-processing peptidase | spoIIGA |
| KFAPOJEI_02435 | PLKKBLCC_04809 | PKEAFELD_04813 | Stage II sporulation protein R | spoIIR |
| KFAPOJEI_01887 | PLKKBLCC_04452 | PKEAFELD_04456 | Spore cortex biosynthesis protein YabQ | yabQ |
| KFAPOJEI_01888 | PLKKBLCC_04453 | PKEAFELD_04457 | Spore coat protein | yabP |
| KFAPOJEI_02376 | PLKKBLCC_04750 | PKEAFELD_04754 | Spore Coat Protein X and V domain | cotX |
| KFAPOJEI_02378 | PLKKBLCC_04752 | PKEAFELD_04756 | Spore Coat Protein X and V domain | cotX |
| KFAPOJEI_02939 | PLKKBLCC_01782 | PKEAFELD_01782 | Outer spore coat protein E | cotE |
| KFAPOJEI_03520 | PLKKBLCC_03012 | PKEAFELD_03013 | Spore coat associated protein JA | cotJA |
| KFAPOJEI_03586 | PLKKBLCC_01820 | PKEAFELD_01821 | Bacillus/Clostridium GerA spore germination protein | spoVAF |
| KFAPOJEI_02916 | PLKKBLCC_01759 | PKEAFELD_01759 | Dipicolinate synthase subunit B | spoVFB |
| KFAPOJEI_00796 | PLKKBLCC_02257 | PKEAFELD_02257 | Stage IV sporulation protein A | spoIVA |
| KFAPOJEI_00087 | PLKKBLCC_03559 | PKEAFELD_03564 | Spore germination protein | gerBA |
| KFAPOJEI_03728 | PLKKBLCC_04348 | PKEAFELD_04352 | Spore gernimation protein | gerD |
| KFAPOJEI_01384 | PLKKBLCC_02819 | - | Germination protease | gpr |
| KFAPOJEI_00085 | PLKKBLCC_03561 | PKEAFELD_03566 | PFAM spore germination B3 GerAC family protein | yfkR |
| KFAPOJEI_02645 | PLKKBLCC_02303 | PKEAFELD_02303 | Sporulation protein YtfJ | ytfJ |
| KFAPOJEI_02649 | PLKKBLCC_02299 | PKEAFELD_02299 | Sporulation lipoprotein YhcN/YlaJ | yhcN |
| KFAPOJEI_00243 | PLKKBLCC_03402 | PKEAFELD_03408 | Small, acid-soluble spore protein D | sspD |
| KFAPOJEI_01029 | PLKKBLCC_03882 | PKEAFELD_03887 | Small, acid-soluble spore protein Tlp | tlp |
| KFAPOJEI_02647 | PLKKBLCC_02301 | PKEAFELD_02301 | Spore maturation protein B | spmB |
| Flagella formation and Chemotaxis | ||||
| KFAPOJEI_02858 | PKEAFELD_01701 | PLKKBLCC_01701 | Flagellar basal body rod protein FlgB | flgB |
| KFAPOJEI_02859 | PKEAFELD_01702 | PLKKBLCC_01702 | Flagellar basal-body rod protein FlgC | flgC |
| KFAPOJEI_02860 | PKEAFELD_01703 | PLKKBLCC_01703 | Flagellar hook-basal body complex protein FliE | fliE |
| KFAPOJEI_02861 | PKEAFELD_01704 | PLKKBLCC_01704 | Flagellar M-ring protein | fliF |
| KFAPOJEI_02862 | PKEAFELD_01705 | PLKKBLCC_01705 | Flagellar motor switch protein FliG | fliG |
| KFAPOJEI_02863 | PKEAFELD_01706 | PLKKBLCC_01706 | Flagellar assembly protein FliH | fliH |
| KFAPOJEI_02864 | PKEAFELD_01707 | PLKKBLCC_01707 | Flagellum-specific ATP synthase | fliI |
| KFAPOJEI_02866 | PKEAFELD_01709 | PLKKBLCC_01709 | Flagellar protein FlbB | flbB |
| KFAPOJEI_02868 | PKEAFELD_01711 | PLKKBLCC_01711 | Minor extracellular protease Vpr | flgD |
| KFAPOJEI_02870 | PKEAFELD_01713 | PLKKBLCC_01713 | Flagellar basal-body rod protein FlgG | flgG |
| KFAPOJEI_02871 | PKEAFELD_01714 | PLKKBLCC_01714 | Flagellar protein FlbD | flbD |
| KFAPOJEI_02872 | PKEAFELD_01715 | PLKKBLCC_01715 | Flagellar protein FliL | fliL |
| KFAPOJEI_02873 | PKEAFELD_01716 | PLKKBLCC_01716 | Flagellar motor switch protein FliM | fliM |
| KFAPOJEI_02874 | PKEAFELD_01717 | PLKKBLCC_01717 | Flagellar motor switch protein FliN | fliY |
| KFAPOJEI_02875 | PKEAFELD_01718 | PLKKBLCC_01718 | Chemotaxis protein CheY | cheY |
| KFAPOJEI_02876 | PKEAFELD_01719 | PLKKBLCC_01719 | Flagellar biosynthesis protein FliO | fliO |
| KFAPOJEI_02877 | PKEAFELD_01720 | PLKKBLCC_01720 | Flagellar biosynthetic protein FliP | fliP |
| KFAPOJEI_02878 | PKEAFELD_01721 | PLKKBLCC_01721 | Flagellar biosynthetic protein FliQ | fliQ |
| KFAPOJEI_02879 | PKEAFELD_01722 | PLKKBLCC_01722 | Flagellar biosynthetic protein FliR | fliR |
| KFAPOJEI_02880 | PKEAFELD_01723 | PLKKBLCC_01723 | Flagellar biosynthetic protein FlhB | flhB |
| KFAPOJEI_02881 | PKEAFELD_01724 | PLKKBLCC_01724 | Flagellar biosynthesis protein FlhA | flhA |
| KFAPOJEI_01101 | PKEAFELD_03959 | PLKKBLCC_03954 | Pilus assembly protein PilZ | pilZ |
| KFAPOJEI_04448 | PKEAFELD_04051 | PLKKBLCC_04046 | Transcriptional regulator of flagella formation YvyF | yvyF |
| KFAPOJEI_04452 | PKEAFELD_04047 | PLKKBLCC_04042 | Flagellar hook-associated protein 1 | flgK |
| KFAPOJEI_04453 | PKEAFELD_04046 | PLKKBLCC_04041 | Flagellar hook-associated protein 3 | flgL |
| KFAPOJEI_04454 | PKEAFELD_04045 | PLKKBLCC_04040 | Flagellin | fliC |
| KFAPOJEI_04455 | PKEAFELD_04044 | PLKKBLCC_04039 | Flagellar hook-associated protein 2 | fliD |
| KFAPOJEI_04457 | PKEAFELD_04042 | PLKKBLCC_04037 | Flagellar secretion chaperone FliS | fliS |
| KFAPOJEI_01101 | PKEAFELD_03959 | PLKKBLCC_02269 | Pilus assembly protein PilZ | pilZ |
| KFAPOJEI_03756 | PKEAFELD_00013 | PLKKBLCC_00013 | Motility protein A | motA |
| KFAPOJEI_03757 | PKEAFELD_00014 | PLKKBLCC_00014 | Motility protein B | motB |
| PKEAFELD_02776 | PKEAFELD_02776 | PLKKBLCC_02776 | Putative sensory transducer protein YfmS | yfmS |
| PKEAFELD_00239 | PKEAFELD_00239 | PLKKBLCC_00239 | Inhibitor of MCP methylation, homolog of CheC | cheX |
| PKEAFELD_01727 | PKEAFELD_01727 | PLKKBLCC_01727 | CheB methylesterase | cheB |
| PKEAFELD_01728 | PKEAFELD_01728 | PLKKBLCC_01728 | Signal transducing histidine kinase, homodimeric domain | cheA |
| PKEAFELD_01729 | PKEAFELD_01729 | PLKKBLCC_01729 | Two component signalling adaptor domain | cheW |
| PKEAFELD_01730 | PKEAFELD_01730 | PLKKBLCC_01730 | CheY-P phosphatase CheC | cheC |
| PKEAFELD_01731 | PKEAFELD_01731 | PLKKBLCC_01731 | Chemoreceptor glutamine deamidase CheD | cheD |
| PKEAFELD_02249 | PKEAFELD_02249 | PLKKBLCC_02249 | Methyltransferase, chemotaxis proteins | cheR |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bucka-Kolendo, J.; Kiousi, D.E.; Dekowska, A.; Mikołajczuk-Szczyrba, A.; Karadedos, D.M.; Michael, P.; Galanis, A.; Sokołowska, B. Exploration of Alicyclobacillus spp. Genome in Search of Antibiotic Resistance. Int. J. Mol. Sci. 2024, 25, 8144. https://doi.org/10.3390/ijms25158144
Bucka-Kolendo J, Kiousi DE, Dekowska A, Mikołajczuk-Szczyrba A, Karadedos DM, Michael P, Galanis A, Sokołowska B. Exploration of Alicyclobacillus spp. Genome in Search of Antibiotic Resistance. International Journal of Molecular Sciences. 2024; 25(15):8144. https://doi.org/10.3390/ijms25158144
Chicago/Turabian StyleBucka-Kolendo, Joanna, Despoina Eugenia Kiousi, Agnieszka Dekowska, Anna Mikołajczuk-Szczyrba, Dimitrios Marinos Karadedos, Panagiotis Michael, Alex Galanis, and Barbara Sokołowska. 2024. "Exploration of Alicyclobacillus spp. Genome in Search of Antibiotic Resistance" International Journal of Molecular Sciences 25, no. 15: 8144. https://doi.org/10.3390/ijms25158144