
A Novel De Novo Missense Mutation in KIF1A Associated with Young-Onset Upper-Limb Amyotrophic Lateral Sclerosis
 , , , ,
, , , ,  and
and {kind=link}
{kind=link}
Abstract
:1. Introduction
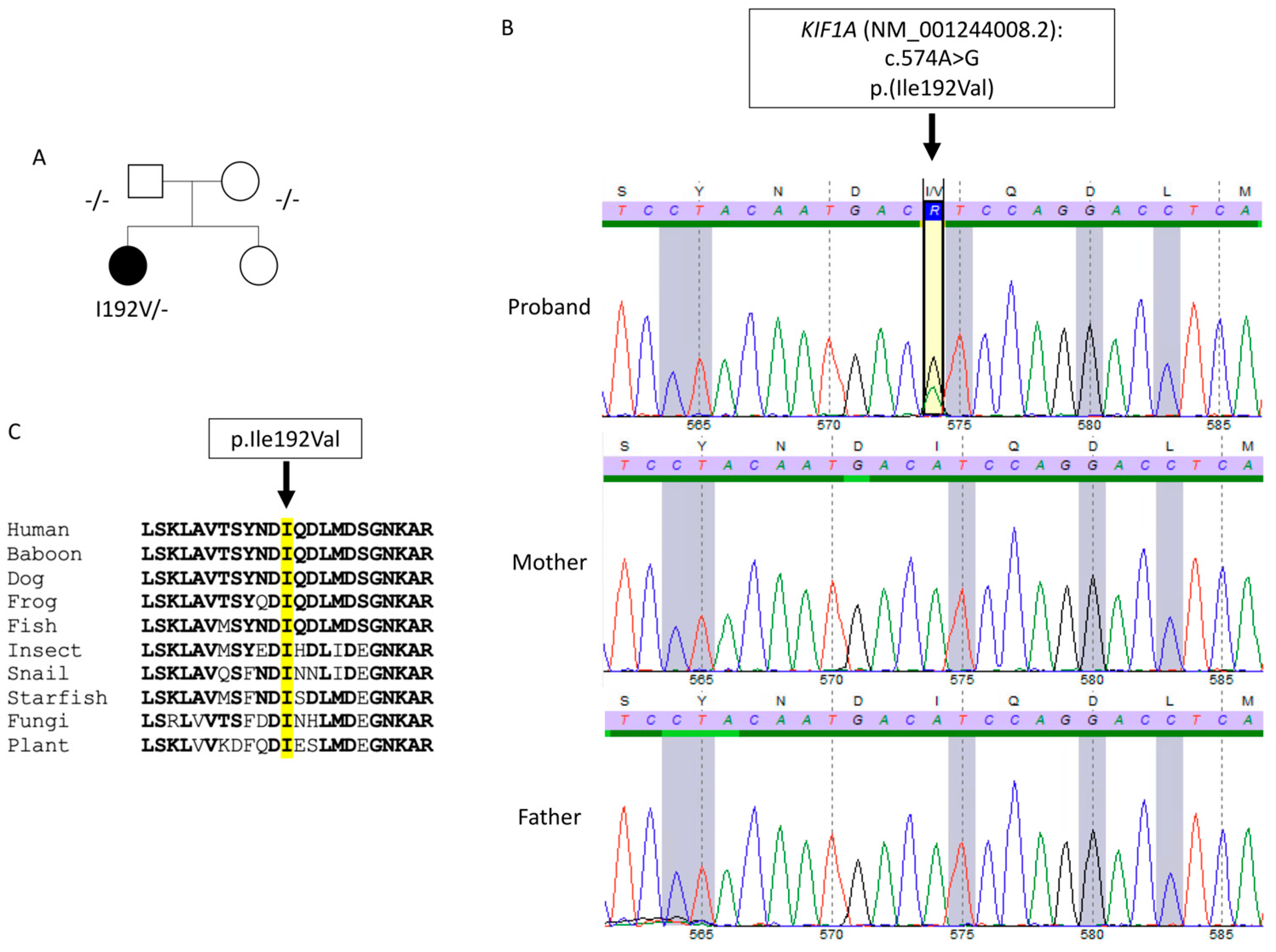
2. Case Description
3. Discussion
4. Methods
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Westeneng, H.J.; Debray, T.P.; Visser, A.E.; van Eijk, R.P.; Rooney, J.P.; Calvo, A.; Martin, S.; McDermott, C.J.; Thompson, A.G.; Pinto, S.; et al. Prognosis for patients with amyotrophic lateral sclerosis: Development and validation of a personalised prediction model. Lancet Neurol. 2018, 17, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Feldman, E.L.; Goutman, S.A.; Petri, S.; Mazzini, L.; Savelieff, M.G.; Shaw, P.J.; Sobue, G. Amyotrophic lateral sclerosis. Lancet 2022, 400, 1363–1380. [Google Scholar] [CrossRef] [PubMed]
- Ruf, W.P.; Boros, M.; Freischmidt, A.; Brenner, D.; Grozdanov, V.; de Meirelles, J.; Meyer, T.; Grehl, T.; Petri, S.; Grosskreutz, J.; et al. Spectrum and frequency of genetic variants in sporadic amyotrophic lateral sclerosis. Brain Commun. 2023, 5, fcad152. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Yamazaki, H.; Sekine-Aizawa, Y.; Hirokawa, N. The neuron-specific kinesin superfamily protein KIF1A is a unique monomeric motor for anterograde axonal transport of synaptic vesicle precursors. Cell 1995, 81, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Vecchia, S.D.; Tessa, A.; Dosi, C.; Baldacci, J.; Pasquariello, R.; Antenora, A.; Astrea, G.; Bassi, M.T.; Battini, R.; Casali, C.; et al. Monoallelic KIF1A-related disorders: A multicenter cross sectional study and systematic literature review. J. Neurol. 2022, 269, 437–450, Erratum in J. Neurol. 2023, 270, 2345–2346. [Google Scholar] [CrossRef] [PubMed]
- Olsen, C.G.; Busk, Ø.L.; Holla, Ø.L.; Tveten, K.; Holmøy, T.; Tysnes, O.B.; Høyer, H. Genetic overlap between ALS and other neurodegenerative or neuromuscular disorders. Amyotroph. Lateral Scler. Front. Degener. 2024, 25, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Liao, P.; Yuan, Y.; Liu, Z.; Hou, X.; Li, W.; Wen, J.; Zhang, K.; Jiao, B.; Shen, L.; Jiang, H.; et al. Association of variants in the KIF1A gene with amyotrophic lateral sclerosis. Transl. Neurodegener. 2022, 11, 46. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Thouvenot, E.; Demattei, C.; Lehmann, S.; Maceski-Maleska, A.; Hirtz, C.; Juntas-Morales, R.; Pageot, N.; Esselin, F.; Alphandery, S.; Vincent, T.; et al. Serum neurofilament light chain at time of diagnosis is an independent prognostic factor of survival in amyotrophic lateral sclerosis. Eur. J. Neurol. 2020, 27, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Steinacker, P.; Feneberg, E.; Weishaupt, J.; Brettschneider, J.; Tumani, H.; Andersen, P.M.; von Arnim, C.A.; Böhm, S.; Kassubek, J.; Kubisch, C.; et al. Neurofilaments in the diagnosis of motoneuron diseases: A prospective study on 455 patients. J. Neurol. Neurosurg. Psychiatry 2016, 87, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, A.; Kenna, K.P.; Renton, A.E.; Ticozzi, N.; Faghri, F.; Chia, R.; Dominov, J.A.; Kenna, B.J.; Nalls, M.A.; Keagle, P.; et al. Genome-wide Analyses Identify KIF5A as a Novel ALS Gene. Neuron 2018, 97, 1268–1283. [Google Scholar] [CrossRef] [PubMed]
- Soustelle, L.; Aimond, F.; López-Andrés, C.; Brugioti, V.; Raoul, C.; Layalle, S. ALS-Associated KIF5A Mutation Causes Locomotor Deficits Associated with Cytoplasmic Inclusions, Alterations of Neuromuscular Junctions, and Motor Neuron Loss. J. Neurosci. 2023, 43, 8058–8072. [Google Scholar] [CrossRef] [PubMed]
- Pant, D.C.; Parameswaran, J.; Rao, L.; Loss, I.; Chilukuri, G.; Parlato, R.; Shi, L.; Glass, J.D.; Bassell, G.J.; Koch, P.; et al. ALS-linked KIF5A ΔExon27 mutant causes neuronal toxicity through gain-of-function. EMBO Rep. 2022, 23, e54234. [Google Scholar] [CrossRef] [PubMed]
- Nakano, J.; Chiba, K.; Niwa, S. An ALS-associated KIF5A mutant forms oligomers and aggregates and induces neuronal toxicity. Genes Cells. 2022, 27, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Ueda, T.; Takeuchi, T.; Fujikake, N.; Suzuki, M.; Minakawa, E.N.; Ueyama, M.; Fujino, Y.; Kimura, N.; Nagano, S.; Yokoseki, A.; et al. Dysregulation of stress granule dynamics by DCTN1 deficiency exacerbates TDP-43 pathology in Drosophila models of ALS/FTD. Acta Neuropathol. Commun. 2024, 12, 20. [Google Scholar] [CrossRef] [PubMed]
- Kikkawa, M.; Okada, Y.; Hirokawa, N. 15 A resolution model of the monomeric kinesin motor, KIF1A. Cell 2000, 100, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Hirokawa, N. A processive single-headed motor: Kinesin superfamily protein KIF1A. Science 1999, 283, 1152–1157. [Google Scholar] [CrossRef] [PubMed]
- Chiba, K.; Kita, T.; Anazawa, Y.; Niwa, S. Insight into the regulation of axonal transport from the study of KIF1A-associated neurological disorder. J. Cell Sci. 2023, 136, jcs260742. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.T. A call to arms against ultra-rare diseases. Nat. Biotechnol. 2021, 39, 671–677. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernard, E.; Cluse, F.; Bohic, A.; Hermier, M.; Raoul, C.; Leblanc, P.; Guissart, C. A Novel De Novo Missense Mutation in KIF1A Associated with Young-Onset Upper-Limb Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2024, 25, 8170. https://doi.org/10.3390/ijms25158170
Bernard E, Cluse F, Bohic A, Hermier M, Raoul C, Leblanc P, Guissart C. A Novel De Novo Missense Mutation in KIF1A Associated with Young-Onset Upper-Limb Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences. 2024; 25(15):8170. https://doi.org/10.3390/ijms25158170
Chicago/Turabian StyleBernard, Emilien, Florent Cluse, Adrien Bohic, Marc Hermier, Cédric Raoul, Pascal Leblanc, and Claire Guissart. 2024. "A Novel De Novo Missense Mutation in KIF1A Associated with Young-Onset Upper-Limb Amyotrophic Lateral Sclerosis" International Journal of Molecular Sciences 25, no. 15: 8170. https://doi.org/10.3390/ijms25158170