
A Barth Syndrome Patient-Derived D75H Point Mutation in TAFAZZIN Drives Progressive Cardiomyopathy in Mice

 , , , , , ,
, , , , , ,  ,
, 
Abstract
1. Introduction
2. Results
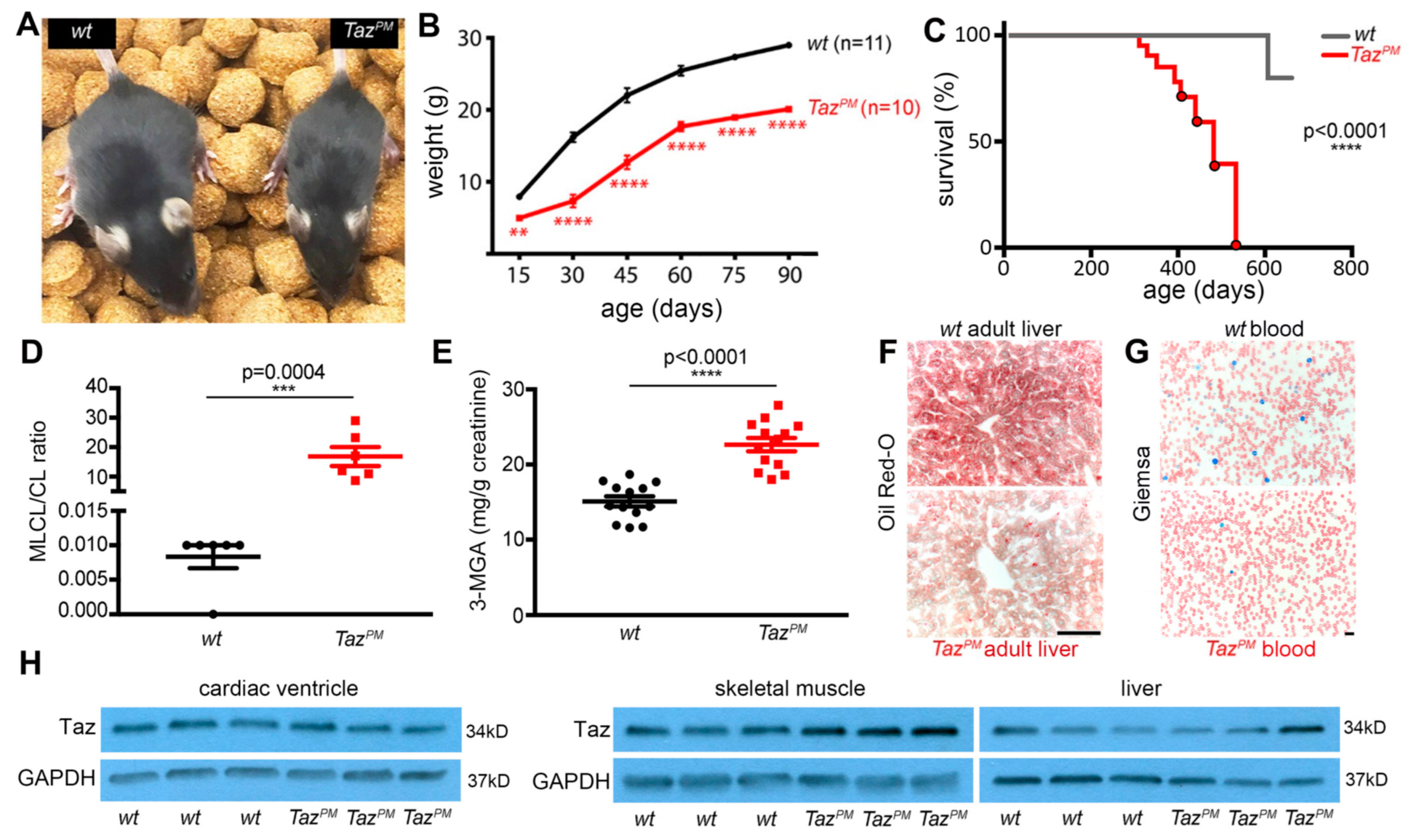
2.1. Point Mutant TazPM Mice Recapitulate Barth Syndrome Clinical Characteristics
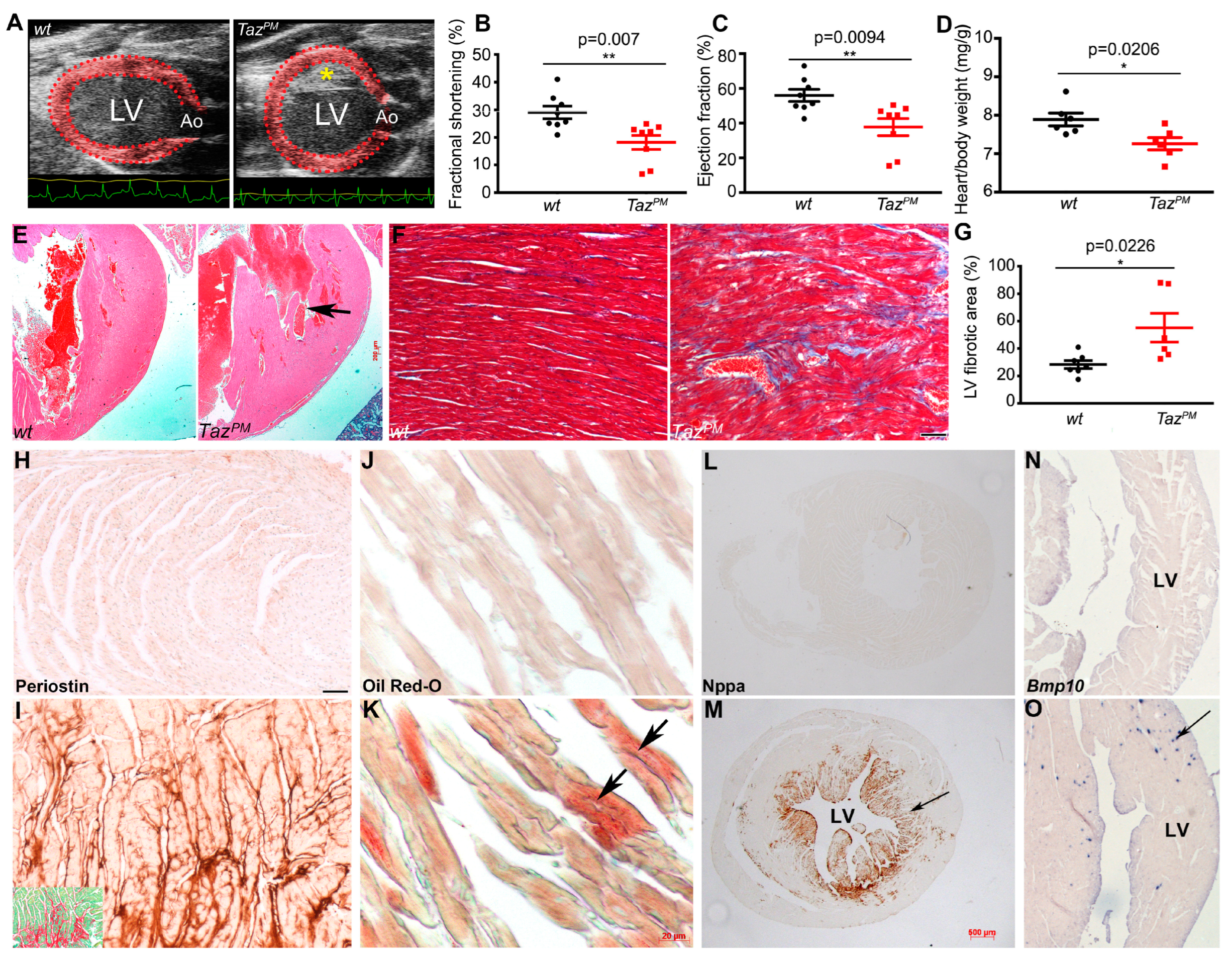
2.2. Adult Cardiac Phenotype in Surviving TazPM Male Mice
2.3. Progressive Cardiomyopathy in TazPM Males
2.4. Progressive Metabolic Shift in Surviving TazPM Male Hearts
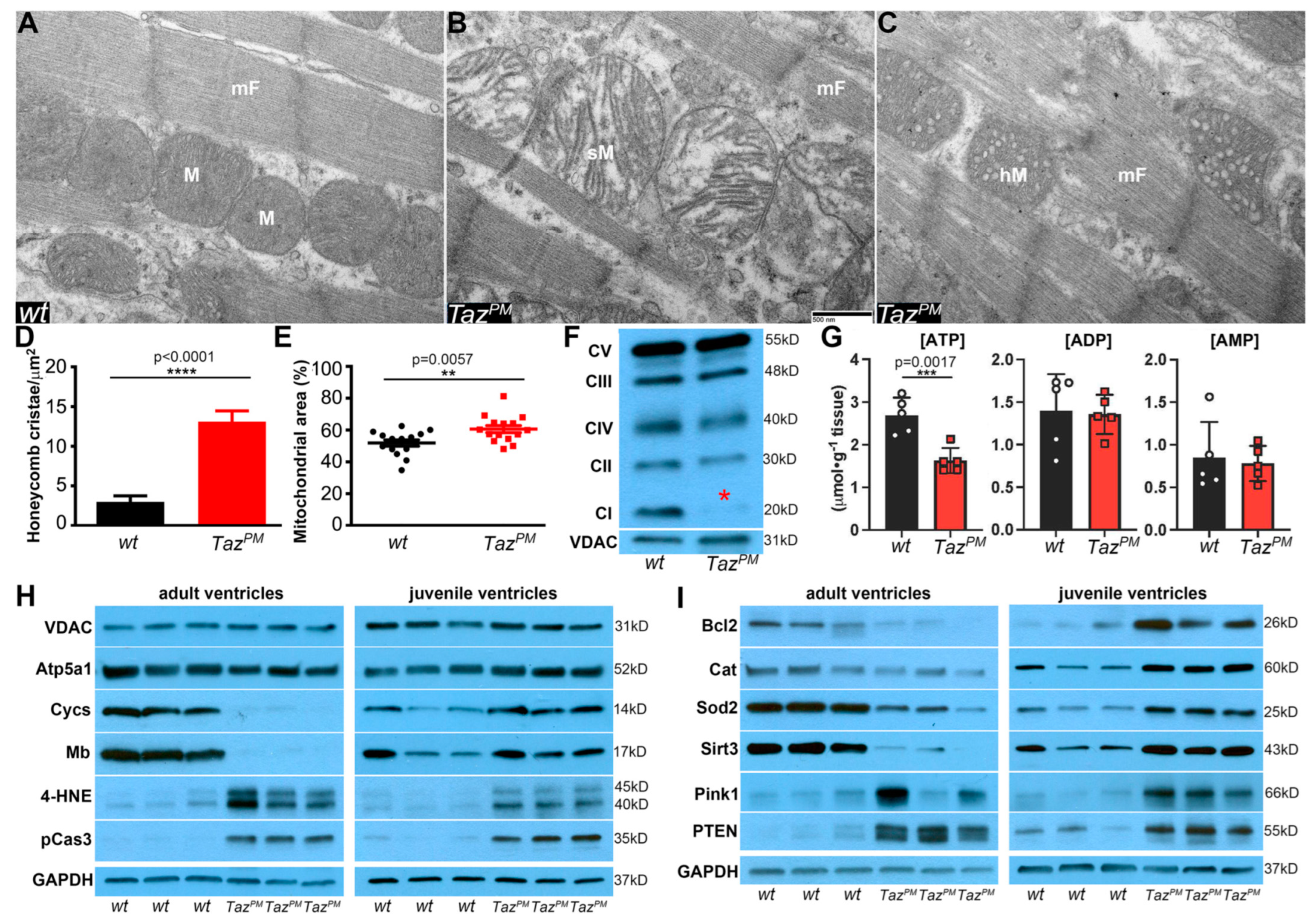
2.5. Surviving TazPM♂ Hearts Exhibit Structurally and Functionally Abnormal Mitochondria
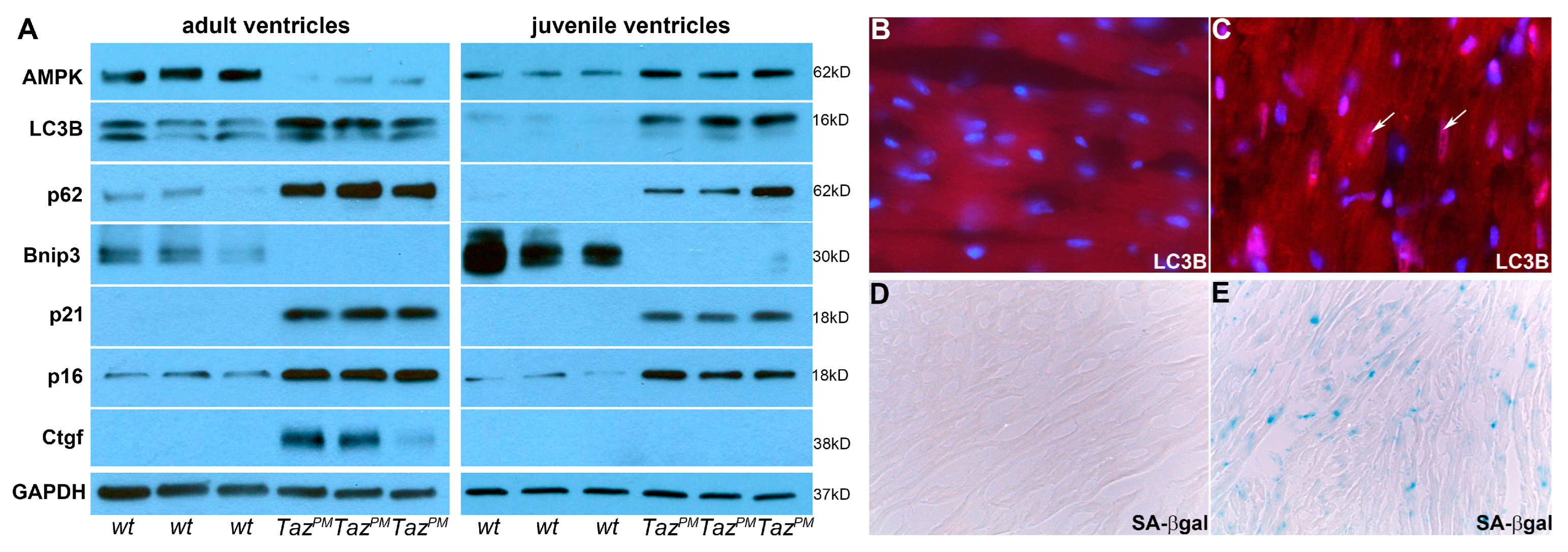
2.6. Autophagy-Mediated Senescence
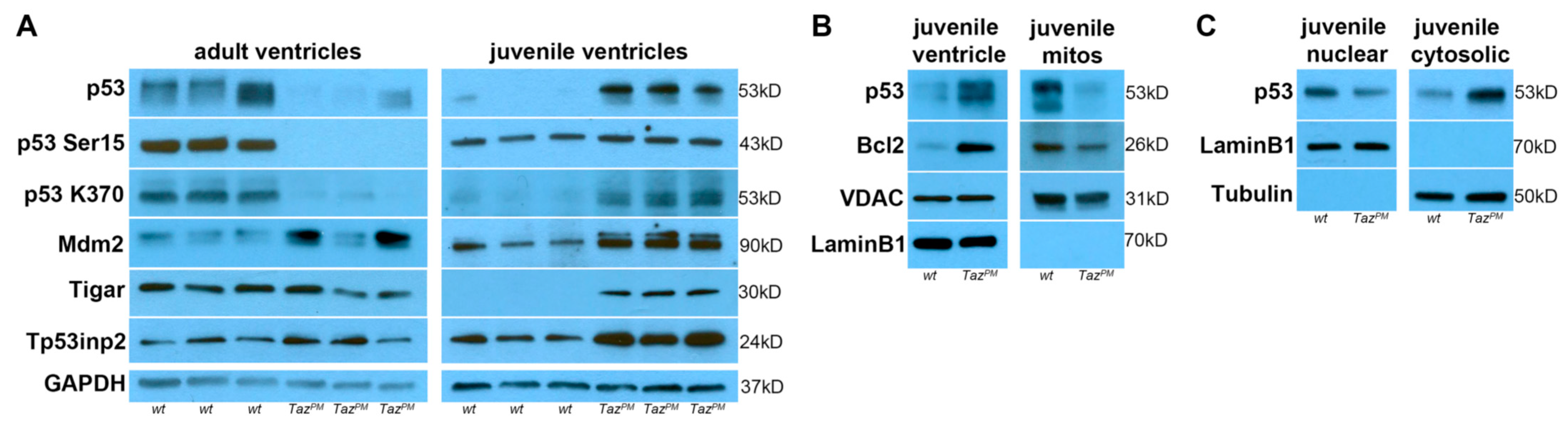
2.7. Biphasic p53 Pathway Hyperactivation
2.8. Genotype-Phenotype-Specific p53-Related Reprogramming
3. Discussion
4. Materials and Methods
4.1. Sex as a Biological Variable
4.2. Animal Models
4.3. Survival and Bodyweight
4.4. Blood Tests
4.5. Mouse Embryonic Fibroblasts and Lentiviral Transfection
4.6. Liquid Chromatography Mass Spectrometry
4.7. Transthoracic Echocardiography
4.8. Western Analysis
4.9. qPCR
4.10. Histologic Analysis, Lineage Mapping, and Molecular Marker Analysis
4.11. Transmission Electron Microscopy
4.12. NAD+/NADH and ATP/ADP/AMP Measurement
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brown, D.A.; Perry, J.B.; Allen, M.E.; Sabbah, H.N.; Stauffer, B.L.; Shaikh, S.R.; Cleland, J.G.; Colucci, W.S.; Butler, J.; Voors, A.A.; et al. Expert consensus document: Mitochondrial function as a therapeutic target in heart failure. Nat. Rev. Cardiol. 2017, 14, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Ren, M.; Phoon, C.K.; Schlame, M. Metabolism and function of mitochondrial cardiolipin. Prog. Lipid. Res. 2014, 55, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.; Ardehali, H.; Balaban, R.S.; DiLisa, F.; Dorn, G.W.; Kitsis, R.N.; Otsu, K.; Ping, P.; Rizzuto, R.; Sack, M.N.; et al. American Heart Association Council on Basic Cardiovascular Sciences, Council on Clinical Cardiology, and Council on Functional Genomics and Translational Biology. Mitochondrial Function, Biology, and Role in Disease: A Scientific Statement From the American Heart Association. Circ. Res. 2016, 118, 1960–1991. [Google Scholar] [CrossRef] [PubMed]
- Towbin, J.A.; Jefferies, J.L. Cardiomyopathies Due to Left Ventricular Noncompaction, Mitochondrial and Storage Diseases, and Inborn Errors of Metabolism. Circ. Res. 2017, 121, 838–854. [Google Scholar] [CrossRef] [PubMed]
- Barth, P.G.; Scholte, H.R.; Berden, J.A.; Van der Klei-Van Moorsel, J.M.; Luyt-Houwen, I.E.M.; Van’t Veer-Korthof, E.T.; Van der Harten, J.J.; Sobotka-Plojhar, M.A. An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J. Neurol. Sci. 1983, 62, 327–355. [Google Scholar] [CrossRef]
- Bione, S.; D’Adamo, P.; Maestrini, E.; Gedeon, A.K.; Bolhuis, P.A.; Toniolo, D. A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat. Genet. 1996, 12, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.L.; Bowron, A.; Gonzalez, I.L.; Groves, S.J.; Newbury-Ecob, R.; Clayton, N.; Martin, R.P.; Tsai-Goodman, B.; Garratt, V.; Ashworth, M.; et al. Barth syndrome. Orphanet J. Rare Dis. 2013, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, Y.; Xu, Y.; Ma, Q.; Lin, Z.; Schlame, M.; Bezzerides, V.J.; Strathdee, D.; Pu, W.T. AAV Gene Therapy Prevents and Reverses Heart Failure in a Murine Knockout Model of Barth Syndrome. Circ. Res. 2020, 126, 1024–1039. [Google Scholar] [CrossRef]
- Garlid, A.O.; Schaffer, C.T.; Kim, J.; Bhatt, H.; Guevara-Gonzalez, V.; Ping, P. TAZ encodes tafazzin, a transacylase essential for cardiolipin formation and central to the etiology of Barth syndrome. Gene 2020, 726, 144148. [Google Scholar] [CrossRef]
- Vernon, H.J.; Sandlers, Y.; McClellan, R.; Kelley, R.I. Clinical laboratory studies in Barth Syndrome. Mol. Genet. Metab. 2014, 112, 143–147. [Google Scholar] [CrossRef]
- Schlame, M.; Greenberg, M.L. Biosynthesis, remodeling and turnover of mitochondrial cardiolipin. Biochim. Biophys Acta Mol. Cell Biol. Lipids. 2017, 1862, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Duncan, A.L. Monolysocardiolipin (MLCL) interactions with mitochondrial membrane proteins. Biochem. Soc. Trans. 2020, 48, 993–1004. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Ye, C.; McCain, K.; Greenberg, M.L. The role of cardiolipin in cardiovascular health. Biomed. Res. Int. 2015, 2015, 891707. [Google Scholar] [CrossRef] [PubMed]
- Thiels, C.; Fleger, M.; Huemer, M.; Rodenburg, R.J.; Vaz, F.M.; Houtkooper, R.H.; Haack, T.B.; Prokisch, H.; Feichtinger, R.G.; Lucke, T.; et al. Atypical Clinical Presentations of TAZ Mutations: An Underdiagnosed Cause of Growth Retardation? JIMD Rep. 2016, 29, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, H.N.; Taylor, C.; Vernon, H.J. Temporal evolution of the heart failure phenotype in Barth syndrome and treatment with elamipretide. Future Cardiol. 2023, 19, 211–225. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.W.; Galbraith, L.; Herndon, J.D.; Lu, Y.L.; Pras-Raves, M.; Vervaart, M.; Van Kampen, A.; Luyf, A.; Koehler, C.M.; McCaffery, J.M.; et al. Defining functional classes of Barth syndrome mutation in humans. Hum. Mol. Genet. 2016, 25, 1754–1770. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhang, S.; Malhotra, A.; Edelman-Novemsky, I.; Ma, J.; Kruppa, A.; Cernicica, C.; Blais, S.; Neubert, T.A.; Ren, M.; et al. Characterization of tafazzin splice variants from humans and fruit flies. J. Biol. Chem. 2009, 284, 29230–29239. [Google Scholar] [CrossRef]
- Zhu, S.; Chen, Z.; Zhu, M.; Shen, Y.; Leon, L.J.; Chi, L.; Spinozzi, S.; Tan, C.; Gu, Y.; Nguyen, A.; et al. Cardiolipin Remodeling Defects Impair Mitochondrial Architecture and Function in a Murine Model of Barth Syndrome Cardiomyopathy. Circ. Heart Fail. 2021, 14, e008289. [Google Scholar] [CrossRef]
- Chowdhury, A.; Boshnakovska, A.; Aich, A.; Methi, A.; Vergel Leon, A.M.; Silbern, I.; Lüchtenborg, C.; Cyganek, L.; Prochazka, J.; Sedlacek, R.; et al. Metabolic switch from fatty acid oxidation to glycolysis in knock-in mouse model of Barth syndrome. EMBO Mol. Med. 2023, 15, e17399. [Google Scholar] [CrossRef]
- Phoon, C.K.; Acehan, D.; Schlame, M.; Stokes, D.L.; Edelman-Novemsky, I.; Yu, D.; Xu, Y.; Viswanathan, N.; Ren, M. Tafazzin knockdown in mice leads to a developmental cardiomyopathy with early diastolic dysfunction preceding myocardial noncompaction. J. Am. Heart Assoc. 2012, 1, e000455. [Google Scholar] [CrossRef]
- Thompson, W.R.; DeCroes, B.; McClellan, R.; Rubens, J.; Vaz, F.M.; Kristaponis, K.; Avramopoulos, D.; Vernon, H.J. New targets for monitoring and therapy in Barth syndrome. Genet. Med. 2016, 18, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Donati, M.A.; Malvagia, S.; Pasquini, E.; Morrone, A.; La Marca, G.; Garavaglia, B.; Toniolo, D.; Zammarchi, E. Barth syndrome presenting with acute metabolic decompensation in the neonatal period. J. Inherit. Metab. Dis. 2006, 29, 684. [Google Scholar] [CrossRef] [PubMed]
- Snider, P.; Standley, K.N.; Wang, J.; Azhar, M.; Doetschman, T.; Conway, S.J. Origin of cardiac fibroblasts and the role of periostin. Circ. Res. 2009, 105, 934–947. [Google Scholar] [CrossRef] [PubMed]
- Piek, A.; de Boer, R.A.; Silljé, H.H. The fibrosis-cell death axis in heart failure. Heart Fail. Rev. 2016, 21, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Listenberger, L.L.; Schaffer, J.E. Mechanisms of lipoapoptosis: Implications for human heart disease. Trends Cardiovasc. Med. 2002, 12, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Kranias, E.G.; Hajjar, R.J. Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ. Res. 2012, 110, 1646–1660. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; Wende, A.R.; Abel, E.D. Cardiac Energy Metabolism in Heart Failure. Circ. Res. 2021, 128, 1487–1513. [Google Scholar] [CrossRef]
- Zachman, D.K.; Chicco, A.J.; McCune, S.A.; Murphy, R.C.; Moore, R.L.; Sparagna, G.C. The role of calcium-independent phospholipase A2 in cardiolipin remodeling in the spontaneously hypertensive heart failure rat heart. J. Lipid. Res. 2010, 51, 525–534. [Google Scholar] [CrossRef]
- Cade, W.T.; Bohnert, K.L.; Bittel, A.J.; Chacko, S.J.; Patterson, B.W.; Pacak, C.A.; Byrne, B.J.; Vernon, H.J.; Reeds, D.N. Arginine kinetics are altered in a pilot sample of adolescents and young adults with Barth syndrome. Mol. Genet. Metab. Rep. 2020, 25, 100675. [Google Scholar] [CrossRef]
- Ikon, N.; Ryan, R.O. Cardiolipin and mitochondrial cristae organization. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1156–1163. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Schenk, R.; Collins, N.L.; Schenk, N.A.; Beard, D.A. Integrated Functions of Cardiac Energetics, Mechanics, and Purine Nucleotide Metabolism. Compr. Physiol. 2023, 14, 5345–5369. [Google Scholar] [CrossRef] [PubMed]
- Breitzig, M.; Bhimineni, C.; Lockey, R.; Kolliputi, N. 4-Hydroxy-2-nonenal: A critical target in oxidative stress? Am. J. Physiol. Cell Physiol. 2016, 311, C537–C543. [Google Scholar] [CrossRef] [PubMed]
- Kaludercic, N.; Maiuri, M.C.; Kaushik, S.; Fernández, Á.F.; de Bruijn, J.; Castoldi, F.; Chen, Y.; Ito, J.; Mukai, R.; Murakawa, T.; et al. Comprehensive autophagy evaluation in cardiac disease models. Cardiovasc. Res. 2020, 116, 483–504. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Adboli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021, 17, 382. [Google Scholar] [CrossRef]
- Tong, M.; Saito, T.; Zhai, P.; Oka, S.I.; Mizushima, W.; Nakamura, M.; Ikeda, S.; Shirakabe, A.; Sadoshima, J. Mitophagy Is Essential for Maintaining Cardiac Function during High Fat Diet-Induced Diabetic Cardiomyopathy. Circ. Res. 2019, 124, 1360–1371. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.S.; Lee, R.T.; Garbern, J.C. Senescence mechanisms and targets in the heart. Cardiovasc. Res. 2022, 118, 1173–1187. [Google Scholar] [CrossRef] [PubMed]
- Dulić, V.; Beney, G.E.; Frebourg, G.; Drullinger, L.F.; Stein, G.H. Uncoupling between phenotypic senescence and cell cycle arrest in aging p21-deficient fibroblasts. Mol. Cell Biol. 2000, 20, 6741–6754. [Google Scholar] [CrossRef] [PubMed]
- Matoba, S.; Kang, J.G.; Patino, W.D.; Wragg, A.; Boehm, M.; Gavrilova, O.; Hurley, P.J.; Bunz, F.; Hwang, P.M. p53 regulates mitochondrial respiration. Science 2006, 312, 1650–1653. [Google Scholar] [CrossRef]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef]
- Hu, W.; Feng, Z.; Levine, A.J. The Regulation of Multiple p53 Stress Responses is Mediated through MDM2. Genes Cancer 2012, 3, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.H.; Clarke, M.F. Regulation of p53 localization. Eur. J. Biochem. 2001, 268, 2779–2783. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Bonora, M.; Sorrentino, G.; Missiroli, S.; Poletti, F.; Suski, J.M.; Galindo Ramirez, F.; Rizzuto, R.; Di Virgilio, F.; Zito, E.; et al. p53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner. Proc. Natl. Acad. Sci. USA 2015, 112, 1779–1784. [Google Scholar] [CrossRef] [PubMed]
- Stambolic, V.; MacPherson, D.; Sas, D.; Lin, Y.; Snow, B.; Jang, Y.; Benchimol, S.; Mak, T.W. Regulation of PTEN transcription by p53. Mol. Cell. 2001, 8, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Perera, G.; Power, L.; Larson, A.; Codden, C.J.; Awata, J.; Batorsky, R.; Strathdee, D.; Chin, M.T. Single Cell Transcriptomic Analysis in a Mouse Model of Barth Syndrome Reveals Cell-Specific Alterations in Gene Expression and Intercellular Communication. Int. J. Mol. Sci. 2023, 24, 11594. [Google Scholar] [CrossRef] [PubMed]
- Wüst, R.C.I.; Coolen, B.F.; Held, N.M.; Daal, M.R.R.; Alizadeh Tazehkandi, V.; Baks-Te Bulte, L.; Wiersma, M.; Kuster, D.W.D.; Brundel, B.J.J.M.; van Weeghel, M.; et al. The Antibiotic Doxycycline Impairs Cardiac Mitochondrial and Contractile Function. Int. J. Mol. Sci. 2021, 22, 4100. [Google Scholar] [CrossRef] [PubMed]
- Kagan, V.E.; Tyurin, V.A.; Jiang, J.; Tyurina, Y.Y.; Ritov, V.B.; Amoscato, A.A.; Osipov, A.N.; Belikova, N.A.; Kapralov, A.A.; Kini, V.; et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat. Chem. Biol. 2005, 1, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.E.; Nixon, C.; Steward, C.G.; Gauvreau, K.; Maisenbacher, M.; Fletcher, M.; Geva, J.; Byrne, B.J.; Spencer, C.T. The Barth Syndrome Registry: Distinguishing disease characteristics and growth data from a longitudinal study. Am. J. Med. Genet. A 2012, 158A, 2726–2732. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Wang, L.; Chi, B. Upregulation of periostin prevents P53-mediated apoptosis in SGC-7901 gastric cancer cells. Mol. Biol. Rep. 2013, 40, 1677–1683. [Google Scholar] [CrossRef]
- Liu, X.; Burke, R.M.; Lighthouse, J.K.; Baker, C.D.; Dirkx RAJr Kang, B.; Chakraborty, Y.; Mickelsen, D.M.; Twardowski, J.J.; Mello, S.S.; Ashton, J.M. p53 Regulates the Extent of Fibroblast Proliferation and Fibrosis in Left Ventricle Pressure Overload. Circ. Res. 2023, 133, 271–287. [Google Scholar] [CrossRef]
- Chen, X.; Ashraf, S.; Ashraf, N.; Harmancey, R. UCP3 (Uncoupling Protein 3) Insufficiency Exacerbates Left Ventricular Diastolic Dysfunction During Angiotensin II-Induced Hypertension. J. Am. Heart Assoc. 2021, 10, e022556. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Xu, S.; Xu, J.; Xin, Y.; Lu, Y.; Zhang, H.; Zhou, B.; Xu, H.; Sheu, S.S.; Tian, R.; et al. Elevated MCU Expression by CaMKIIδB Limits Pathological Cardiac Remodeling. Circulation 2022, 145, 1067–1083. [Google Scholar] [CrossRef] [PubMed]
- Schiller, J.; Laube, E.; Wittig, I.; Kühlbrandt, W.; Vonck, J.; Zickermann, V. Insights into complex I assembly: Function of NDUFAF1 and a link with cardiolipin remodeling. Sci. Adv. 2022, 8, eadd3855. [Google Scholar] [CrossRef] [PubMed]
- Piquereau, J.; Ventura-Clapier, R. Maturation of Cardiac Energy Metabolism During Perinatal Development. Front. Physiol. 2018, 9, 959. [Google Scholar] [CrossRef] [PubMed]
- Calmettes, G.; John, S.A.; Weiss, J.N.; Ribalet, B. Hexokinase-mitochondrial interactions regulate glucose metabolism differentially in adult and neonatal cardiac myocytes. J. Gen. Physiol. 2013, 142, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Swift, L.M.; Burke, M.; Guerrelli, D.; Reilly, M.; Ramadan, M.; McCullough, D.; Prudencio, T.; Mulvany, C.; Chaluvadi, A.; Jaimes, R.; et al. Age-dependent changes in electrophysiology and calcium handling: Implications for pediatric cardiac research. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H354–H365. [Google Scholar] [CrossRef]
- Ryan, J.J.; Archer, S.L. Emerging concepts in the molecular basis of pulmonary arterial hypertension: Part I: Metabolic plasticity and mitochondrial dynamics in the pulmonary circulation and right ventricle in pulmonary arterial hypertension. Circulation 2015, 131, 1691–1702. [Google Scholar] [CrossRef]
- Chin, M.T.; Conway, S.J. Role of Tafazzin in Mitochondrial Function, Development and Disease. J. Dev. Biol. 2020, 8, 10. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.J.; Thompson, C.B. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar] [CrossRef]
- Tang, Y.; Zhao, W.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation is indispensable for p53 activation. Cell 2008, 133, 612–626. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.; Bono, E.; Ghigo, A. The Interplay between Autophagy and Senescence in Anthracycline Cardiotoxicity. Curr. Heart Fail. Rep. 2021, 18, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Coryell, P.R.; Goraya, S.K.; Griffin, K.A.; Redick, M.A.; Sisk, S.R.; Purvis, J.E. Autophagy regulates the localization and degradation of p16INK4a. Aging Cell 2020, 19, e13171. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yazawa, E.; Keating, E.M.; Mazumdar, N.; Hauschild, A.; Ma, Q.; Wu, H.; Xu, Y.; Shi, X.; Strathdee, D.; et al. Genetic modifiers modulate phenotypic expression of tafazzin deficiency in a mouse model of Barth syndrome. Hum. Mol. Genet. 2023, 32, 2055–2067. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Hou, T.; Gao, T.; Lu, X.; Yang, Q.; Zhu, Q.; Li, Z.; Liu, C.; Mu, G.; Liu, G.; et al. p53 cooperates with SIRT6 to regulate cardiolipin de novo biosynthesis. Cell Death Dis. 2018, 9, 941. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, A.; Pillai, V.B.; Samant, S.; Gupta, M.; Gupta, M.P. The nuclear and mitochondrial sirtuins, Sirt6 and Sirt3, regulate each other’s activity and protect the heart from developing obesity-mediated diabetic cardiomyopathy. FASEB J. 2019, 33, 10872–10888. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Offenbacher, R.; Corral, N.H.; Bansal, N.; Ding, J.; Gennarini, L.; Ostrodka, L.; Tal, A. A 9-year-old male with Barth syndrome and cardiac transplant presenting with hyperviscosity syndrome caused by EBV-negative plasmacytoid posttransplant lymphoproliferative disorder. Pediatr. Blood Cancer 2021, 68, e29264. [Google Scholar] [CrossRef] [PubMed]
- Oyarbide, U.; Kodger, J.; del Rio, S.; Sandlers, Y.; Corey, S.J. No Cardiomyopathy or Skeletal Muscle Weakness in an Organismal Model of Barth Syndrome—Is Metabolism the Basis for Its Neutropenia? Blood 2022, 140 (Suppl. S1), 5472. [Google Scholar] [CrossRef]
- Edwards, D.; Sierra Potchanant, E.; Huang, X.; Sun, Z.; Capitano, M.; Miller, C.; He, Y.; Broxmeyer, H.E.; Nalepa, G. Patient-Tailored Mouse Genome Editing Recapitulates Hematopoietic and Systemic Manifestations of Barth Syndrome. Blood 2017, 130 (Suppl. S1), 775. [Google Scholar] [CrossRef]
- Dinca, A.A.; Chien, W.M.; Chin, M.T. Identification of novel mitochondrial localization signals in human Tafazzin, the cause of the inherited cardiomyopathic disorder Barth syndrome. J. Mol. Cell Cardiol. 2018, 114, 83–92. [Google Scholar] [CrossRef]
- Vyas, P.M.; Tomamichel, W.J.; Pride, P.M.; Babbey, C.M.; Wang, Q.; Mercier, J.; Martin, E.M.; Payne, R.M. A TAT-frataxin fusion protein increases lifespan and cardiac function in a conditional Friedreich’s ataxia mouse model. Hum. Mol. Genet. 2012, 21, 1230–1247. [Google Scholar] [CrossRef] [PubMed]
- Lajiness, J.D.; Snider, P.; Wang, J.; Feng, G.S.; Krenz, M.; Conway, S.J. SHP-2 deletion in postmigratory neural crest cells results in impaired cardiac sympathetic innervation. Proc. Natl. Acad Sci. USA 2014, 111, E1374–E1382. [Google Scholar] [CrossRef] [PubMed]
- Debacq-Chainiaux, F.; Erusalimsky, J.D.; Campisi, J.; Toussaint, O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat. Protoc. 2009, 4, 1798–1806. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Shi, S.; Acosta, L.; Li, W.; Lu, J.; Bao, S.; Chen, Z.; Yang, Z.; Schneider, M.D.; Chien, K.R.; et al. BMP10 is essential for maintaining cardiac growth during murine cardiogenesis. Development 2004, 131, 2219–2231. [Google Scholar] [CrossRef]
- Law, A.S.; Hafen, P.S.; Brault, J.J. Liquid chromatography method for simultaneous quantification of ATP and its degradation products compatible with both UV-Vis and mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2022, 1206, 123351. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parental Cross: wt Male x TazPM Female | ||||
|---|---|---|---|---|
| Genotype | Expected | Observed | Chi2 p Value | Fertile |
| wt ♂ | 70 (25%) | 82 (29.4%) | 0.252 | 100% |
| TazPM ♂ | 70 (25%) | 38 (13.6%) | <0.0001 | 0% |
| wt ♀ | 70 (25%) | 67 (23.9%) | 0.271 | 100% |
| TazPM ♀ | 70 (25%) | 69 (24.6%) | 0.285 | 100% |
| Adult Wt♂ (n = 8) | Adult TazPM♂ (n = 8) | Juvenile Wt♂ (n = 3) | Juvenile TazPM♂ (n = 3) | |
|---|---|---|---|---|
| Body weight (g) | 39.6 ± 2.7 | 33.19 ± 3.4 * | 26.7 ± 2.2 | 20.9 ± 1.3 * |
| Heart rate @ (bpm) | 487.3 ± 17.5 | 496.11 ± 37.7 | 467.9 ± 9.1 | 424.5 ± 29.9 |
| LV mass (mg) | 228.7 ± 22.1 | 189.1 ± 11.8 * | 107.9 ± 17.6 | 424.5 ± 29.9 |
| FS (%) | 228.7 ± 22.1 | 18.9 ± 3.1 ** | 45.3 ± 2.5 | 35.6 ± 1.5 ** |
| EF (%) | 58.0 ± 1.4 | 38.7 ± 2.8 ** | 75.7 ± 2.8 | 65.0 ± 1.9 * |
| LVIDd (mm) | 4.4 ± 0.1 | 3.2 ± 0.2 * | 3.4 ± 0.1 | 3.7 ± 0.1 |
| LVIDs (mm) | 2.9 ± 0.4 | 1.7 ± 0.2 * | 2.0 ± 0.1 | 2.5 ± 0.1 |
| LV Vold (ml) | 88.7 ± 3.2 | 128.7 ± 7.3 ** | 57.7 ± 3.8 | 61.9 ± 3.8 |
| LV Vols (ml) | 38.2 ± 4.2 | 59.0 ± 9.6 ** | 14.3 ± 2.3 | 22.4 ± 1.5 ** |
| Gene | Forward PCR Primer | Reverse PCR Primer |
|---|---|---|
| Gapdh | AAGGGCATCTTGGGCTACAC | CATTGAGAGCAATGCCAGCC |
| PPia | GGGTGGTGACTTTACACGCC | CTTGCCATCCAGCCATTCAG |
| Taz | TGTGTCGAGGAGATGGTGTC | ACACTCAGCAATCAGCCGTC |
| Prkaa1 | CCAAGGGGCATCTTGGGAAA | TCCTCCCAACAACGGCTTAC |
| Prkaa2 | CACGGTGTGGTCGTTAGTGA | AGCATGTGGTACAAGCCCAG |
| Lc3b | TTGTCATCGTGGGAACTGGG | GGTGGCAAGGTATCGACCAA |
| p62 | GGCCTACAGCTCCTAGGGAA | GAAGCTACATGGGGGTCCTG |
| Bnip3 | TACCCACGAACCCCACTTTG | GCCTGCAACAAAACTGACCA |
| p21 | TACCGTGGGTGTCAAAGCAC | GAGGACTCGGGACAATGCAG |
| p16 | GACCGACGGGCATAGCTTC | AAGAAAAAGGCGGGCTGAGG |
| p53 | CTCAGACTGACTGCCTCTGC | ACTACTCAGAGAGGGGGCTG |
| Mdm2 | GGCATGAATTGCATCTGGTGG | ACCTGGGACAAACTGCAACA |
| Tigar | TGAGGAAATGCAGGGTGAGC | AACACTGCTGCTCAGATGCT |
| Tp53inp2 | ATGAAGTGGATGGCTGGCTC | CTGCCGGTGACATAAACGGA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Snider, P.L.; Sierra Potchanant, E.A.; Sun, Z.; Edwards, D.M.; Chan, K.-K.; Matias, C.; Awata, J.; Sheth, A.; Pride, P.M.; Payne, R.M.; et al. A Barth Syndrome Patient-Derived D75H Point Mutation in TAFAZZIN Drives Progressive Cardiomyopathy in Mice. Int. J. Mol. Sci. 2024, 25, 8201. https://doi.org/10.3390/ijms25158201
Snider PL, Sierra Potchanant EA, Sun Z, Edwards DM, Chan K-K, Matias C, Awata J, Sheth A, Pride PM, Payne RM, et al. A Barth Syndrome Patient-Derived D75H Point Mutation in TAFAZZIN Drives Progressive Cardiomyopathy in Mice. International Journal of Molecular Sciences. 2024; 25(15):8201. https://doi.org/10.3390/ijms25158201
Chicago/Turabian StyleSnider, Paige L., Elizabeth A. Sierra Potchanant, Zejin Sun, Donna M. Edwards, Ka-Kui Chan, Catalina Matias, Junya Awata, Aditya Sheth, P. Melanie Pride, R. Mark Payne, and et al. 2024. "A Barth Syndrome Patient-Derived D75H Point Mutation in TAFAZZIN Drives Progressive Cardiomyopathy in Mice" International Journal of Molecular Sciences 25, no. 15: 8201. https://doi.org/10.3390/ijms25158201
APA StyleSnider, P. L., Sierra Potchanant, E. A., Sun, Z., Edwards, D. M., Chan, K.-K., Matias, C., Awata, J., Sheth, A., Pride, P. M., Payne, R. M., Rubart, M., Brault, J. J., Chin, M. T., Nalepa, G., & Conway, S. J. (2024). A Barth Syndrome Patient-Derived D75H Point Mutation in TAFAZZIN Drives Progressive Cardiomyopathy in Mice. International Journal of Molecular Sciences, 25(15), 8201. https://doi.org/10.3390/ijms25158201