

Identification and Characterization of miRNAs and lncRNAs Associated with Salinity Stress in Rice Panicles
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
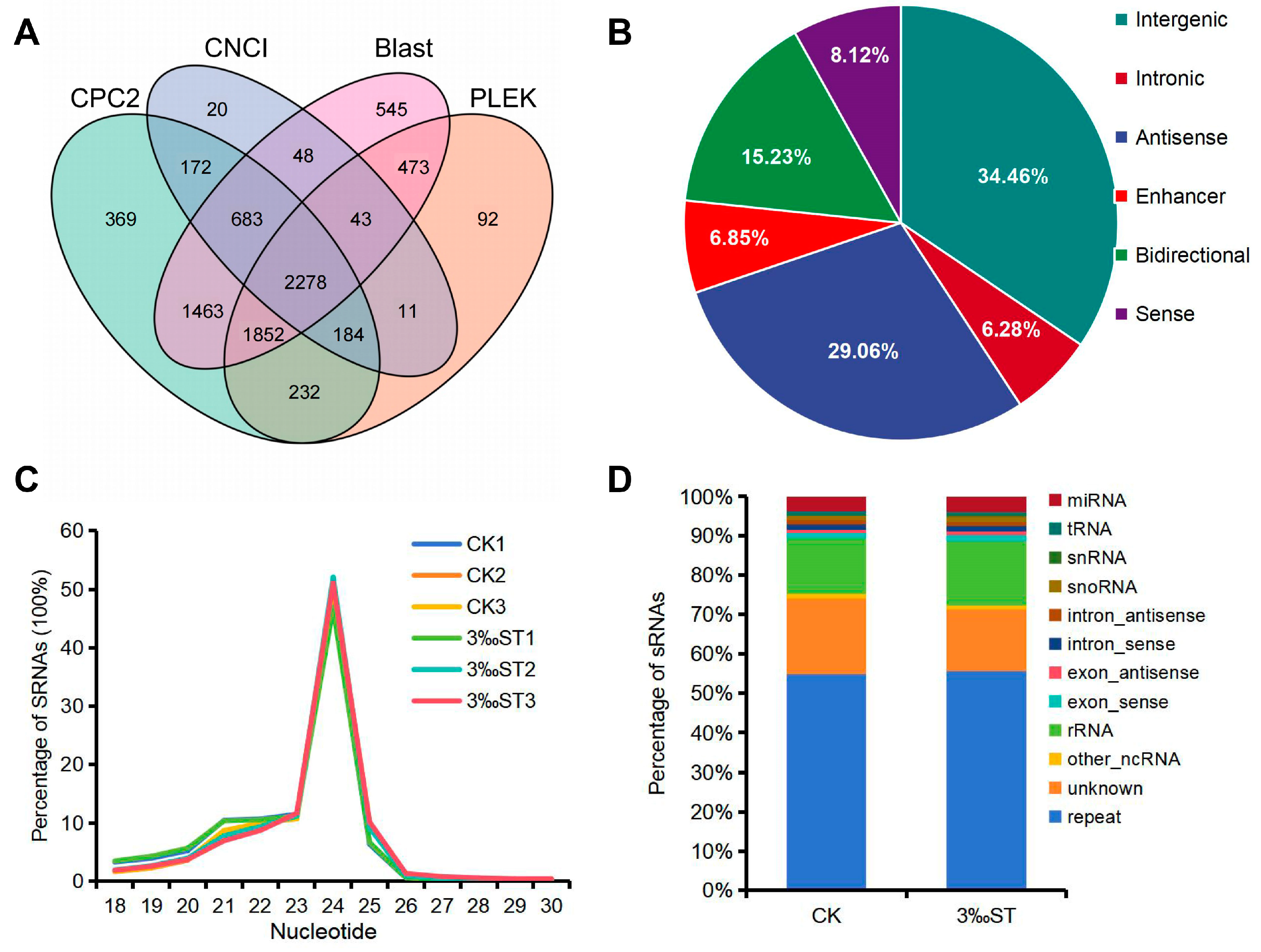
2.1. Genome-Wide Identification of lncRNAs, miRNAs, and mRNAs
2.2. Analysis of DE-lncRNAs, DE-mRNAs and DE-miRNAs
2.3. Analysis of Potential Targets of lncRNAs and miRNAs
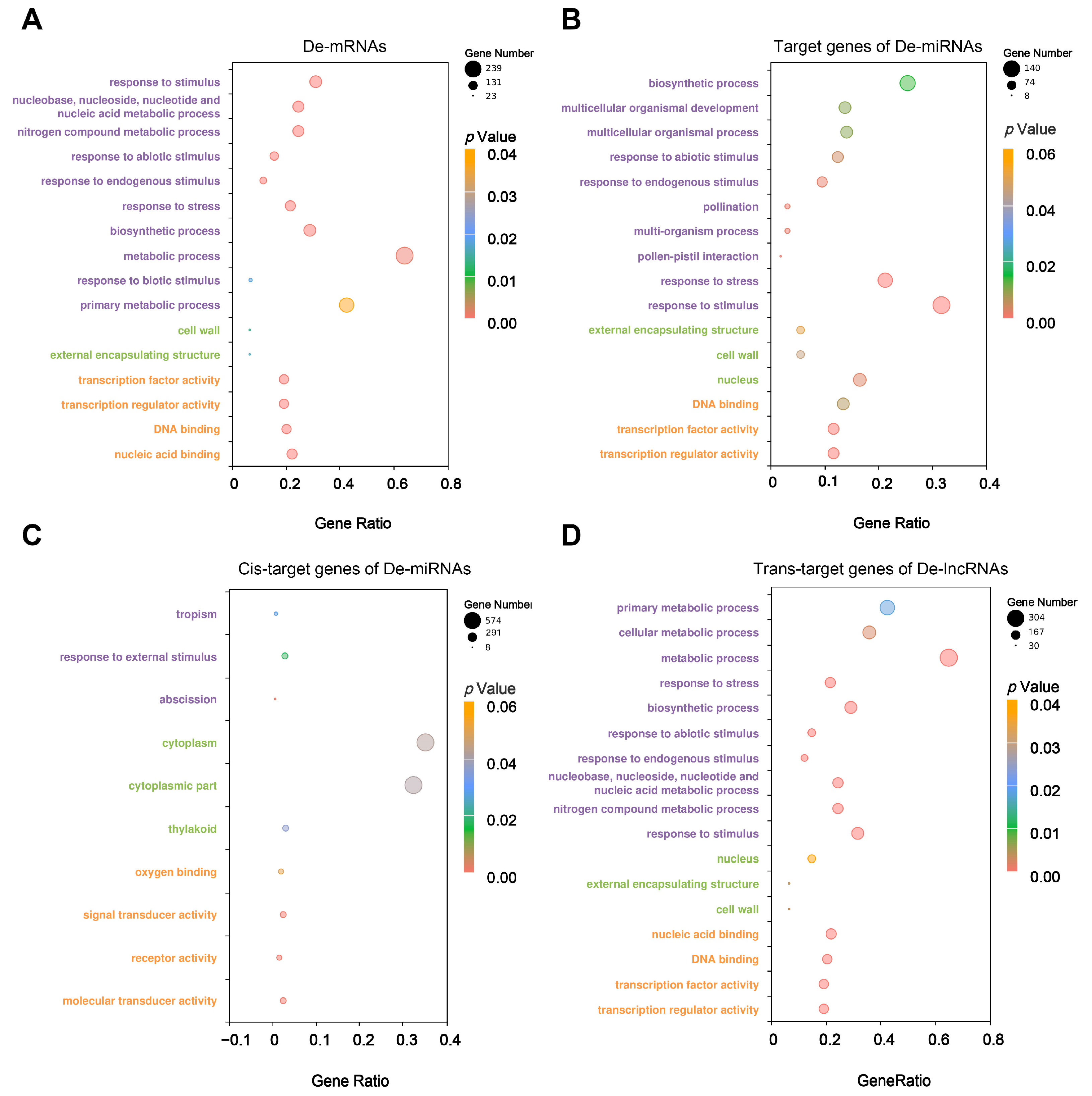
2.4. Functional Enrichment Analysis of DE-mRNAs, DE-miRNA Target Genes and DE-lncRNA Target Genes
2.5. Construction of ceRNA Network Associated with Salt Tolerance
3. Discussion
4. Materials and Methods
4.1. Plant Material and Salinity Stress Treatment
4.2. RNA Library Preparation and Sequencing
4.3. Small RNA Analysis and miRNA Identification
4.4. lncRNA Analysis
4.5. Differentially Expressed lncRNA, miRNA and mRNA under Salt Stress
4.6. Target Gene Prediction and Functional Annotation
4.7. Co-Expression Analysis and CeRNA Network Construction
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zeng, L.; Shannon, M.C.; Grieve, C.M. Evaluation of salt tolerance in rice genotypes by multiple agronomic parameters. Euphytica 2002, 127, 235–245. [Google Scholar] [CrossRef]
- Munns, R.; Tester, M. Mechanisms of salinity tolerance. Annu. Rev. Plant Biol. 2008, 59, 651–681. [Google Scholar] [CrossRef]
- Zheng, C.; Liu, C.; Liu, L.; Tan, Y.; Sheng, X.; Yu, D.; Sun, Z.; Sun, X.; Chen, J.; Yuan, D.; et al. Effect of salinity stress on rice yield and grain quality: A meta-analysis. Eur. J. Agron. 2023, 144, 126765. [Google Scholar] [CrossRef]
- Chen, T.; Shabala, S.; Niu, Y.; Chen, Z.; Shabala, L.; Meinke, H.; Venkataraman, G.; Pareek, A.; Xu, J.; Zhou, M. Molecular mechanisms of salinity tolerance in rice. Crop J. 2021, 9, 506–520. [Google Scholar] [CrossRef]
- Ponce, K.S.; Meng, L.; Guo, L.; Leng, Y.; Ye, G. Advances in sensing, response and regulation mechanism of salt tolerance in rice. Int. J. Mol. Sci. 2021, 22, 2254. [Google Scholar] [CrossRef]
- Islam, W.; Waheed, A.; Naveed, H.; Zeng, F. MicroRNAs mediated plant responses to salt stress. Cells 2022, 11, 2806. [Google Scholar] [CrossRef]
- Yang, H.; Cui, Y.; Feng, Y.; Hu, Y.; Liu, L.; Duan, L. Long non-Coding RNAs of plants in response to abiotic stresses and their regulating roles in promoting environmental adaption. Cells 2023, 12, 729. [Google Scholar] [CrossRef]
- Axtell, M.J.; Snyder, J.A.; Bartel, D.P. Common functions for diverse small RNAs of land plants. Plant Cell 2007, 19, 1750–1769. [Google Scholar] [CrossRef]
- Gao, P.; Bai, X.; Yang, L.; Lv, D.; Li, Y.; Cai, H.; Ji, W.; Guo, D.; Zhu, Y. Over-expression of osa-MIR396c decreases salt and alkali stress tolerance. Planta 2010, 231, 991–1001. [Google Scholar] [CrossRef]
- Gao, P.; Bai, X.; Yang, L.; Lv, D.; Pan, X.; Li, Y.; Cai, H.; Ji, W.; Chen, Q.; Zhu, Y. osa-MIR393: A salinity- and alkaline stress-related microRNA gene. Mol. Biol. Rep. 2011, 38, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Zhou, G.; Zhang, Z.; Li, W.; Peng, Y.; Xie, X. Moderate salinity stress reduces rice grain yield by influencing expression of grain number- and grain filling-associated genes. J. Plant Growth Regul. 2021, 40, 1111–1120. [Google Scholar] [CrossRef]
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef] [PubMed]
- Chekanova, J.A. Long non-coding RNAs and their functions in plants. Curr. Opin. Plant Biol. 2015, 27, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Dong, J.; Deng, F.; Wang, W.; Cheng, Y.; Song, L.; Hu, M.; Shen, J.; Xu, Q.; Shen, F. The long non-coding RNA lncRNA973 is involved in cotton response to salt stress. BMC Plant Biol. 2019, 19, 458. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Jiang, X.; Niu, F.; Sun, X.; Hu, Z.; Gao, F.; Zhang, H.; Jiang, Q. Overexpression of lncRNA77580 regulates drought and salinity stress responses in soybean. Plants 2023, 12, 181. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Wan, H.; Li, Z.; Ai, N.; Zhou, B. Long noncoding RNA TRABA suppresses β-glucosidase-encoding BGLU24 to promote salt tolerance in cotton. Plant Physiol. 2024, 194, 1120–1138. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Shen, J.; Xu, Q.; Dong, J.; Song, L.; Wang, W.; Shen, F. Long noncoding RNA lncRNA354 functions as a competing endogenous RNA of miR160b to regulate ARF genes in response to salt stress in upland cotton. Plant Cell Environ. 2021, 44, 3302–3321. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Hussian, S.; Nishad, J.; Dubey, H.; Bisht, D.S.; Sharma, T.R.; Mondal, T.K. Identification and functional prediction of long non-coding RNAs of rice (Oryza sativa L.) at reproductive stage under salinity stress. Mol. Biol. Rep. 2021, 48, 2261–2271. [Google Scholar] [CrossRef]
- Rehman, O.U.; Uzair, M.; Farooq, M.S.; Saleem, B.; Attacha, S.; Attia, K.A.; Farooq, U.; Fiaz, S.; El-Kallawy, W.H.; Kimiko, I.; et al. Comprehensive insights into the regulatory mechanisms of lncRNA in alkaline-salt stress tolerance in rice. Mol. Biol. Rep. 2023, 50, 7381–7392. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.J.; Wuriyanghan, H.; Zhang, Y.Q.; Duan, K.X.; Chen, H.W.; Li, Q.T.; Lu, X.; He, S.J.; Ma, B.; Zhang, W.K.; et al. An S-domain receptor-like kinase, OsSIK2, confers abiotic stress tolerance and delays dark-induced leaf senescence in rice. Plant Physiol. 2013, 163, 1752–1765. [Google Scholar] [CrossRef]
- Liu, D.; Chen, X.; Liu, J.; Ye, J.; Guo, Z. The rice ERF transcription factor OsERF922 negatively regulates resistance to Magnaporthe oryzae and salt tolerance. J. Exp. Bot. 2012, 63, 3899–3911. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Guan, Y.; Wu, Y.; Chen, H.; Chen, F.; Chu, C. Overexpression of a rice OsDREB1F gene increases salt, drought, and low temperature tolerance in both Arabidopsis and rice. Plant Mol. Biol. 2008, 67, 589–602. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Dai, X.; Zhang, W.H. A R2R3-type MYB gene, OsMYB2, is involved in salt, cold, and dehydration tolerance in rice. J. Exp. Bot. 2012, 63, 2541–2556. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cui, S.; Wu, F.; Yan, S.; Lin, X.; Du, X.; Chong, K.; Schilling, S.; Theissen, G.; Meng, Z. Functional conservation of MIKC*-Type MADS box genes in Arabidopsis and rice pollen maturation. Plant Cell 2013, 25, 1288–1303. [Google Scholar] [CrossRef] [PubMed]
- Tariq, M.; Paszkowski, J. DNA and histone methylation in plants. Trends Genet. 2004, 20, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Niu, S.; Yan, Y.; Zhou, G.; Peng, Y.; He, Y.; Zhou, J.; Li, Y.; Xie, X. Moderate salinity stress affects rice quality by influencing expression of amylose- and protein-content-associated genes. Int. J. Mol. Sci. 2024, 25, 4042. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Maekawa, M.; Shimamoto, K.; Kyozuka, J. The LAX1 and FRIZZY PANICLE 2 genes determine the inflorescence architecture of rice by controlling rachis-branch and spikelet development. Dev. Biol. 2001, 231, 364–373. [Google Scholar] [CrossRef]
- Ko, S.S.; Li, M.J.; Sun-Ben, K.M.; Ho, Y.C.; Lin, Y.J.; Chuang, M.H.; Hsing, H.X.; Lien, Y.C.; Yang, H.T.; Chang, H.C.; et al. The bHLH142 transcription factor coordinates with TDR1 to modulate the expression of EAT1 and regulate pollen development in rice. Plant Cell 2014, 26, 2486–2504. [Google Scholar] [CrossRef]
- Gao, Z.; Zeng, D.; Cheng, F.; Tian, Z.; Guo, L.; Su, Y.; Yan, M.; Jiang, H.; Dong, G.; Huang, Y.; et al. ALK, the key gene for gelatinization temperature, is a modifier gene for gel consistency in rice. J. Integr. Plant Biol. 2011, 53, 756–765. [Google Scholar] [CrossRef]
- Ren, D.; Yu, H.; Rao, Y.; Xu, Q.; Zhou, T.; Hu, J.; Zhang, Y.; Zhang, G.; Zhu, L.; Gao, Z.; et al. ‘Two-floret spikelet’ as a novel resource has the potential to increase rice yield. Plant Biotechnol. J. 2018, 16, 351–353. [Google Scholar] [CrossRef]
- Song, S.; Wang, G.; Hu, Y.; Liu, H.; Bai, X.; Qin, R.; Xing, Y. OsMFT1 increases spikelets per panicle and delays heading date in rice by suppressing Ehd1, FZP and SEPALLATA-like genes. J. Exp. Bot. 2018, 69, 4283–4293. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zheng, Y.P.; Zhou, X.H.; Yang, X.M.; He, X.R.; Feng, Q.; Zhu, Y.; Li, G.B.; Wang, H.; Zhao, J.H.; et al. Rice miR1432 fine-tunes the balance of yield and blast disease resistance via different modules. Rice 2021, 14, 87. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Li, L.; Yang, X.; Yu, Y.; Gao, L.; Mo, B.; Chen, X.; Liu, L. MicroRNA1432 regulates rice drought stress tolerance by targeting the CALMODULIN-LIKE2 gene. Plant Physiol. 2024, 195, 1954–1968. [Google Scholar] [CrossRef] [PubMed]
- Gévaudant, F.; Duby, G.; von Stedingk, E.; Zhao, R.; Morsomme, P.; Boutry, M. Expression of a constitutively activated plasma membrane H+-ATPase alters plant development and increases salt tolerance. Plant Physiol. 2007, 144, 1763–1776. [Google Scholar] [CrossRef]
- Cao, Y.; Zhang, M.; Liang, X.; Li, F.; Shi, Y.; Yang, X.; Jiang, C. Natural variation of an EF-hand Ca2+-binding-protein coding gene confers saline-alkaline tolerance in maize. Nat. Commun. 2020, 11, 186. [Google Scholar] [CrossRef] [PubMed]
- Vitart, V.; Baxter, I.; Doerner, P.; Harper, J.F. Evidence for a role in growth and salt resistance of a plasma membrane H+-ATPase in the root endodermis. Plant J. 2010, 27, 191–201. [Google Scholar] [CrossRef]
- Borghesi, E.; Gonzalez-Miret, M.L.; Escudero-Gilete, M.L.; Malorgio, F.; Heredia, F.J.; Melendez-Martinez, A.J. Effects of salinity stress on carotenoids, anthocyanins, and color of diverse tomato genotypes. J. Agric. Food Chem. 2011, 59, 11676–11682. [Google Scholar] [CrossRef]
- Payyavula, R.S.; Navarre, D.A.; Kuhl, J.C.; Pantoja, A.; Pillai, S.S. Differential effects of environment on potato phenylpropanoid and carotenoid expression. BMC Plant Biol. 2012, 12, 39. [Google Scholar] [CrossRef] [PubMed]
- Gay, F.; Maraval, I.; Roques, S.; Gunata, Z.; Boulanger, R.; Audebert, A.; Mestres, C. Effect of salinity on yield and 2-acetyl-1-pyrroline content in the grains of three fragrant rice cultivars (Oryza sativa L.) in Camargue (France). Field Crop. Res. 2010, 117, 154–160. [Google Scholar] [CrossRef]
- Lu, J.; Wang, C.; Zhang, F.; Zeng, D.; Zhou, Y. Comparative microRNA profiling reveals microRNAs involved in rice resistant response to bacterial blight. Crop J. 2021, 9, 834–842. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Kuang, Z.; Wang, Y.; Li, L.; Yang, X. miRDeep-P2: Accurate and fast analysis of the microRNA transcriptome in plants. Bioinformatics 2019, 35, 2521–2522. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.J.; Yang, D.C.; Kong, L.; Hou, M.; Meng, Y.Q.; Wei, L.; Gao, G. CPC2: A fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017, 45, W12–W16. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- McGinnis, S.; Madden, T.L. BLAST: At the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 2004, 32, W20–W25. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Zhang, J.; Zhou, Z. PLEK: A tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinform. 2014, 15, 311. [Google Scholar] [CrossRef]
- Zhang, T.; Liang, Q.; Li, C.; Fu, S.; Kundu, J.K.; Zhou, X.; Wu, J. Transcriptome Analysis of Rice Reveals the lncRNA-mRNA Regulatory Network in Response to Rice Black-Streaked Dwarf Virus Infection. Viruses 2020, 12, 951. [Google Scholar] [CrossRef]
- Li, J.; Ma, W.; Zeng, P.; Wang, J.; Geng, B.; Yang, J.; Cui, Q. LncTar: A tool for predicting the RNA targets of long noncoding RNAs. Brief. Bioinform. 2015, 16, 806–812. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Liu, C.; Gu, L.; Mo, B.; Cao, X.; Chen, X. TarHunter, a tool for predicting conserved microRNA targets and target mimics in plants. Bioinformatics 2018, 34, 1574–1576. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, C.; Wang, Y.; He, Y.; Peng, Y.; Xie, L.; Li, Y.; Sun, W.; Zhou, J.; Zheng, C.; Xie, X. Identification and Characterization of miRNAs and lncRNAs Associated with Salinity Stress in Rice Panicles. Int. J. Mol. Sci. 2024, 25, 8247. https://doi.org/10.3390/ijms25158247
Jiang C, Wang Y, He Y, Peng Y, Xie L, Li Y, Sun W, Zhou J, Zheng C, Xie X. Identification and Characterization of miRNAs and lncRNAs Associated with Salinity Stress in Rice Panicles. International Journal of Molecular Sciences. 2024; 25(15):8247. https://doi.org/10.3390/ijms25158247
Chicago/Turabian StyleJiang, Conghui, Yulong Wang, Yanan He, Yongbin Peng, Lixia Xie, Yaping Li, Wei Sun, Jinjun Zhou, Chongke Zheng, and Xianzhi Xie. 2024. "Identification and Characterization of miRNAs and lncRNAs Associated with Salinity Stress in Rice Panicles" International Journal of Molecular Sciences 25, no. 15: 8247. https://doi.org/10.3390/ijms25158247
APA StyleJiang, C., Wang, Y., He, Y., Peng, Y., Xie, L., Li, Y., Sun, W., Zhou, J., Zheng, C., & Xie, X. (2024). Identification and Characterization of miRNAs and lncRNAs Associated with Salinity Stress in Rice Panicles. International Journal of Molecular Sciences, 25(15), 8247. https://doi.org/10.3390/ijms25158247