Abstract
Diabetic cardiomyopathy (DbCM) is a common complication in individuals with type 2 diabetes mellitus (T2DM), and its exact pathogenesis is still debated. It was hypothesized that chronic hyperglycemia and insulin resistance activate critical cellular pathways that are responsible for numerous functional and anatomical perturbations in the heart. Interstitial inflammation, oxidative stress, myocardial apoptosis, mitochondria dysfunction, defective cardiac metabolism, cardiac remodeling, hypertrophy and fibrosis with consequent impaired contractility are the most common mechanisms implicated. Epigenetic changes also have an emerging role in the regulation of these crucial pathways. The aim of this review was to highlight the increasing knowledge on the molecular mechanisms of DbCM and the new therapies targeting specific pathways.
1. Introduction
Type 2 diabetes mellitus (T2DM) is a metabolic disorder characterized by hyperglycemia resulting from peripheral insulin resistance, failure of pancreatic beta cells or both [1]. The prevalence of T2DM is rising among adolescent population in many countries, although type 1 diabetes mellitus (T1DM) still has the highest prevalence [2]. This increase in T2DM in youths has been attributed to the rising prevalence of obesity worldwide. Excess adipose tissue and especially the accumulation of visceral fat predispose individuals to insulin resistance and T2DM [3]. Moreover, T2DM is strictly associated with increased morbidity and mortality, mainly due to long-term vascular complications.
Cardiovascular (CV) risk factors appear early in the clinical course of T2DM [4]. Children and adolescents may already show subclinical ventricular abnormalities, and insulin resistance can negatively impact on endothelial function since childhood, particularly in children born small for gestational age [5,6]. Numerous evidence suggests that early-onset T2DM causes an increased risk of CV mortality [7,8,9,10]. Furthermore, the coexistence of obesity worsens the CV risk of T2DM individuals [11].
Diabetic cardiomyopathy (DbCM) is defined by “the existence of abnormal myocardial structure and performance in the absence of other cardiac risk factors such as coronary artery disease, hypertension, and significant valvular disease, in individuals with diabetes mellitus” [12]. The prevalence of DbCM is approximately 16% in adult individuals with T2DM [13]. Heart failure is the most common T2DM complication, with a prevalence of 3.4% in adults [14].
The course of DbCM has two phases: an early stage, in which the main features are left ventricular (LV) hypertrophy and impaired diastolic function, and a later one, in which myocardial fibrosis and systolic dysfunction are observed. The exact mechanisms of DbCM are still not fully understood, especially in youth, although chronic hyperglycemia and insulin resistance with compensatory hyperinsulinemia exert toxic effects on the myocardium through direct and indirect pathways [15]. Several factors, such as inflammation, deposition of advanced glycation end products (AGEs), oxidative stress, mitochondrial dysfunction, lipotoxicity, and alterations of subcellular components, contribute to numerous functional and anatomical perturbations [16,17,18]. The aim of this narrative review was to highlight the most recent evidence on the molecular basis of pathogenetic mechanisms of cardiomyopathies in T2DM patients and the future therapeutic prospectives.
2. Inflammation
Chronic low-grade inflammation is a key component in the course of T2DM and its related complications, which contributes to peripheral disease progression and leads to structural and functional changes in cardiomyocytes [17]. This condition is mediated by cytokines that are primarily released via the activation of nuclear factor NF-κB through different mechanisms: the most relevant are represented by hyperglycemia, increased levels of fatty acids (FAs), mitochondrial dysfunction, increased reactive oxygen species (ROS), derangement in the renin–angiotensin–aldosterone system (RAAS) and the accumulation of AGEs [19]. AGEs are proteins or lipids that become glycated after exposure to glucose and accumulate in many different cell types. They act via the AGE receptor (RAGE) triggering the activation of NF-κB signaling, as shown in murine models in which diabetes increased cytosolic levels of ROS, RAGEs, Tumor Necrosis Factor alpha (TNF-α) and nuclear levels of NFkB-p65 in cardiomyocytes [20]. Indeed, NF-κB activation promotes an upregulation of pro-inflammatory cytokines, such as TNF-α, Interleukin 1 β (IL-1β) and IL-6 (Figure 1) [21].
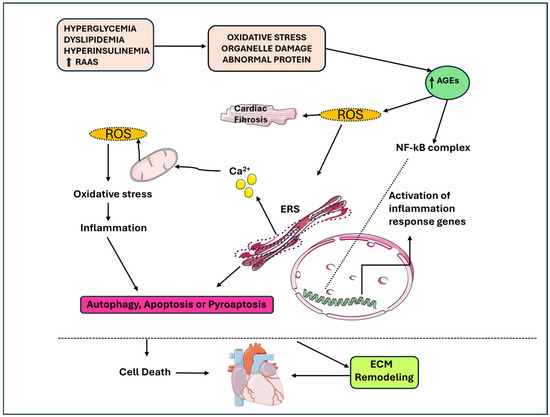
Figure 1.
The link between apoptosis, autophagy, pyro-apoptosis, and cardiomyocyte loss in DbCM, inflammation and metabolic stress contribute to the senescence of cardiomyocytes. AGEs: advanced glycation end products; DbCM: diabetic cardiomyopathy; ERS: stress endoplasmic reticulum; ROS: reactive oxygen species.
Proinflammatory cytokines causes damage to mitochondria and the endoplasmic reticulum (ER) and generate ROS and reactive nitrogen species (RNS), and thus, contribute to apoptosis, autophagy and pyroptosis, which is an inflammation-induced cell death [22,23]. The increased ROS production causes the activation of the NLR family pyrin domain containing 3 (NLRP3) inflammasome, which through caspase-1, promotes the release of IL-1β-mediated interactions [19]. Luo B. et al. explored the central role of NLRP23 in DbCM. In their study, the activation of NLRP3 inflammasome was associated with worse cardiac function in mice models of streptozotocin (STZ)-induced diabetes due to IL-1β release and caspase-1 activation. In high-glucose-treated H9c2 cardiomyocytes, increased ROS activation and phosphorylation of NF-kB p65 were found, suggesting their role in activating NLRP3 inflammasome. Furthermore, the downregulation of NLRP3 suppressed the release of IL-1β, IL-18, TNF-α and IL-6; reduced the rate of apoptosis; and inhibited caspase-1 activation, with the consequence of reduced pyroptosis. Furthermore, the inhibition of NF-kB caused reduced ROS production and NLRP3 expression, suggesting that NF-kB can be a link between ROS and NLRP3 [24].
An increased expression of TNF-α can induce the aggregation and infiltration of immune cells (e.g., macrophages, T cells, B cells, neutrophils, dendritic cells and mast cells), and damage associated molecular pattern molecules (DAMPs), which are contributing factors in determining myocardial injury in other cardiac diseases [25]. Increased IL-1β production inhibits the beta-adrenergic receptor, consequently causing decreased myocardial contractility [26]. Overall, cytokines released via the NF-κB pathway contribute to cardiomyocytes stress and damage, causing consequent remodeling.
Lower levels of adiponectin have been reported in individuals with diabetes and heart failure. Adiponectin has protective properties through the modulation of endothelial adhesion molecules, such as vascular cell adhesion protein-1 (VCAM-1), intracellular adhesion molecule-1 (ICAM-1) and E-selectin, and through the inhibition of TNF-α and NF-κB [27]. Adiponectin also enhances glucose uptake into skeletal muscle and restrains hepatic gluconeogenesis [28]. Adiponectin deficiency causes mitochondrial dysfunction in various organs, including the heart, and in obese children, a worse cardiovascular risk profile has been related to lower levels of adiponectin [29,30]. Recently, high glucose levels have been linked to an increase in the expression of miR-223-3p in cardiomyocytes, causing an increased release of pro-inflammatory cytokines and subsequent pyroptosis [31].
Transcriptomics studies demonstrated an upregulation in genes involved in the inflammatory pathways in cardiac tissues isolated from T2DM mice, and particularly inflammasome-related genes (e.g., Aim2, Nlrc4) associated with antigen presentation and langerin, which is an antigen uptake receptor most often expressed by Langerhans cells and stimulates T cell responses [32,33,34]. Inflammation and metabolic stress contribute to the senescence of both cardiomyocytes and immune cells, which causes impaired energetic and metabolic perturbations and ultimately contributing to cardiac dysfunction [35]. Overall, ROS production is increased and protection mechanisms are decreased in a diabetic heart: ROS induce oxidative damage to DNA, proteins and lipids, and may inhibit autophagy, which is a regulated process that removes dysfunctional cellular components, and mitochondria dysfunction causes energy imbalance in the heart [36].
3. Mitochondrial Dysfunction
In individuals with T2DM, defects in the mitochondrial structure and function contribute to the development of insulin resistance in skeletal muscle, adipose tissue and pancreatic β-cells [37,38]. Mitochondria dysfunction appears in the early stages of DbCM development, which induces damage through increased ROS generation and reduced ATP production, which can lead to cardiovascular diseases [39,40,41]. Damaged mitochondria cause mtDNA release with activation of the cGAS-STING pathway, which, in turn, triggers myocardial cell apoptosis and an inflammatory response, contributing to the development of myocardial cell hypertrophy and cardiac remodeling [42].
The heart has a high energy request, consuming approximately 8% of the total ATP expenditure of the body. In the heart, a higher mitochondrial volume density is necessary to produce ATP via oxidative phosphorylation, mainly from the oxidation of FAs in physiologic conditions [43,44]. In T2DM, the rise in serum FAs and triglyceride levels promotes higher rates of FAs uptake and oxidation, with lower glucose utilization [45,46]. This is driven by cardiac-specific overexpression and the increased activity of peroxisome proliferator-activated receptor α (PPARα) [47,48]. Increased rates of fatty acid oxidation (FAO) and the shift to non-oxidative metabolism have different effects: First, if the delivery of FA to the mitochondria exceeds their capacity, acyl-CoAs accumulate and they are shifted to alternative pathways of lipid metabolism, with the production of other products. Among them, ceramides and diacylglycerols accumulate and exert cardiac toxicity, which activates protein kinase C signaling, apoptosis, ER stress and increased ROS production (Figure 1) [49,50,51,52].
Increased rates of FAO also decrease the mitochondrial efficiency of ATP regeneration and increase myocardial O2 consumption, with a total effect of decreased cardiac efficiency.
Elevated FA levels in diabetes can also impair cardiac Hypoxia-inducible factor 1-alpha (HIF-1α) accumulation, with a subsequent reduced cardiac adaptation to hypoxic conditions based on a model of insulin resistance in HL-1 cardiomyocytes, which affects cardioprotection [53].
It was also proposed that increased levels of mitochondrial calpains or monoamine oxidase (MAO) can have a role in inducing mitochondria dysfunction [54]. Furthermore, in a diabetic heart, the reduction in levels of the mitochondrial deacetylase SIRT3 promotes acetylation and, consequently, inactivation of Manganese superoxide dismutase (MnSOD), which is a metalloprotein that protects the mitochondria from ROS damage [55].
Uncoupling proteins (UCPs), which comprise a set of five similar proteins, are situated in the inner mitochondrial membranes of various tissues. They play a role in a variety of functions and cellular activities, ranging from regulating the body temperature to influencing insulin release and providing neuroprotection [56]. Amongst all tissues, skeletal muscle possesses the widest array of UCPs, with all five types present in its mitochondria. As a result, skeletal muscle has been extensively researched to enhance our understanding of UCP functions and related disorders. Over the past thirty years, UCPs have been extensively examined due to their impact on glucose and lipid metabolism. Furthermore, numerous studies that involved mice, rats and humans demonstrated the significant protective effects of mitochondrial UCPs against oxidative stress and issues with mitochondrial function. Nevertheless, their precise role remains incompletely understood. Recent research underscores the critical roles of mitochondrial proteins, such as UCP1/3 and mitochondrial protein adenine nucleotide translocase 1 (ANT1), in the pathogenesis of DbCM [57].
UCP1–UCP3 are involved in the regulation of mitochondrial proton leakage and the dissipation of the proton gradient, which can modulate reactive ROS production and protect against oxidative stress in cardiac cells [58]. On the other hand, ANT1, which is a key component of the mitochondrial ADP/ATP translocase system, plays a pivotal role in maintaining energy homeostasis by facilitating the exchange of ADP and ATP across the mitochondrial inner membrane [57].
The dysregulation of ANT1 can lead to impaired ATP production and energy deficits, which contributes to the contractile dysfunction characteristic of a diabetic heart [59]. Exploring the interplay between UCP1-3 and ANT1 could reveal novel insights into their potential as therapeutic targets to ameliorate mitochondrial dysfunction and improve cardiac outcomes in diabetic patients [60].
4. Molecular Mechanisms Determining Cardiac Remodeling
Cardiac hypertrophy and fibrosis are the main features of DbCM determined by different mechanisms [61,62]. Increased insulin levels with the consequent higher levels of insulin-like growth factor (IGF-1) activate the mitogen activated protein kinase 1 (Erk1/2) and phosphoinositide 3-kinases (PI3K) signaling pathways, which, in turn, induce cardiomyocyte hypertrophy [63]. The mammalian target of rapamycin complex 2 (mTORC2) signaling pathway functions as an effector of insulin/IGF-1; it activates and phosphorylates protein kinase B (PKB), which modulates key substrates, including the transcription factors of FoxO1/3a, the metabolic regulator (GSK3b) and the mTORC1 inhibitor (TSC2) [64], resulting in cell production and proliferation. Hyperglycemia exerts its deleterious effects mainly through chronic hexosamine biosynthetic pathway (HBP) activation; this leads to excessive O-GlcNAcylation of target proteins, which, in turn, can contribute to increased oxidative stress, impairment of DNA repair machinery, enhanced apoptotic and autophagic stimuli and mitochondrial dysfunction [65,66,67,68]. This all contributes to cardiac fibrosis, hypertrophy and increased susceptibility to ischemic injury [69,70,71,72,73]. Oxidative stress contributes to cardiac hypertrophy via the increased expression of nitrogen oxide (NOX) proteins due to the dysregulation of PPARα and angiotensin II [74,75]. In animal models, NOX 2 and NOX4 are associated with cardiac hypertrophy and fibrosis [76]. In diabetic cardiac tissue, collagen deposition in the extracellular matrix (ECM) is enhanced, mainly due to the increased deposition of collagen types I and III. The activation of Transforming Growth Factor-b1 (TGF-b1) and wingless-related integration site (WNT) pathways, together with an accelerated extracellular matrix degradation due to the remodeling of matrix metalloproteinases (MMPs) and decreased availability of nitric oxide (NO), lead to a dysregulated process, with the consequence of high cardiac collagen deposition and consequent interstitial fibrosis [77,78]. MMP release is enhanced by inflammation [79]. The activation of these processes is the consequence of the stimulation of increased renin–angiotensin–aldosterone (RAAS) activity, increased AGEs, insulin resistance and hyperglycemia [80,81,82]. AGEs accumulate in different cell types, which affects the extracellular and intracellular structure, causing crosslinks in collagen molecules, thereby making it more difficult to be degraded and stimulating cardiac fibroblasts. This, in turn, leads to increased fibrosis with consequent increased myocardial stiffness [83].
Hyperglycemia is also a direct contributor to cardiac fibrosis. In experimental models, cardiac fibroblasts exposed to high glucose synthesize excessive amounts of collagen and other ECM proteins [83]. As discussed before, other factors such as adipokines release, oxidation of FAs as a source of energy and neurohumoral pathways can activate fibroblasts and, consequently, enhance myocardial fibrosis [84]. Fibroblast activation is also triggered by the accumulation of harmful lipids in cardiomyocytes and successive activation of inflammatory and fibrogenic programs [85]. In DbCM, higher rates of apoptosis and autophagy of cardiomyocytes, fibroblasts and endothelial cells were observed, which were caused by hyperglycemia, insulin resistance, lipid peroxidation, increased RAAS activation, oxidative stress and endoplasmic stress [86]. Particularly, hyperglycemia induces cell death through the local increase of angiotensin II and by inducing lysosomal membrane damage and enhanced cathepsin D expression and lysosomal release [87,88]. Furthermore, in T2DM, an alteration of Treg/Th17 and Treg/th1 is documented and leads to a pro-inflammatory subset that contributes to cardiac cell death [89,90]. Cardiomyocyte apoptosis stimulates the deposition of collagen through cardiac fibroblast activation and proliferation and cardiac interstitial fibrosis [91]. Heart stiffness and hypertrophy causes decreased complacency and the subsequent development of systolic and diastolic dysfunction [92].
5. Myocardial Calcium Handling
Myocyte contraction needs an appropriate calcium balance, as the binding of calcium to troponin C in the sarcomere is responsible for the conformational changes that lead to heart contraction [93]. In DbCM, perturbations in calcium handling have been described [94]. L-type calcium channels (LTCCs) are voltage-gated channels located on the sarcolemma activated by the influx of sodium ions caused by the arrival of action potential at the cardiac myocytes.
The activation of LTCCs causes an influx of calcium ions. In animal models of diabetic mice with heart impairment, a decrease in the expression of LTCC was observed, with a consequent reduction in the calcium current [95].
Also, the exposure of cardiomyocyte to AGEs causes increased ROS and consequent calcium transients reduction [96]. In cardiac myocytes produced from induced pluripotent stem cells (iPSC-CMs) and exposed to a diabetic environment, calcium transient frequency and amplitude reduction was observed [97].
However, in animal models, an increase in the amplitude of calcium transients in myocytes was documented, and this effect has been considered to be caused by a dysfunction in the sodium/calcium exchanger, independently of LTCCs [98]. Furthermore, patients with T2DM have an increased expression of SLC8A1, which is a gene that encodes the sodium/calcium exchanger [99].
Calcium binds to ryanodine receptor-2 (RyR2) on the sarcoplasmic reticulum (SR), which leads to an extensive release of calcium from the SR into the cytosol; this, in turn, allows calcium to bind to troponin C with subsequent destabilization of the troponin–tropomyosin complex from the actin–myosin binding site, causing cross-bridges formation and producing tension; AGEs can contribute to impaired Ryanodine receptor (RyR2) [100,101].
SR calcium ATPase (SERCA-2) is a channel responsible for the reuptake of calcium back into the SR; in diabetic mice, a reduced expression of SERCA-2 [94,102] and an AGE decrease has led to SERCA-2 activity being observed [103].
Overall, cytosolic calcium overload can be observed in cardiomyocytes of T2DM individuals, particularly in end-stage heart failure (Table 1; Figure 2).
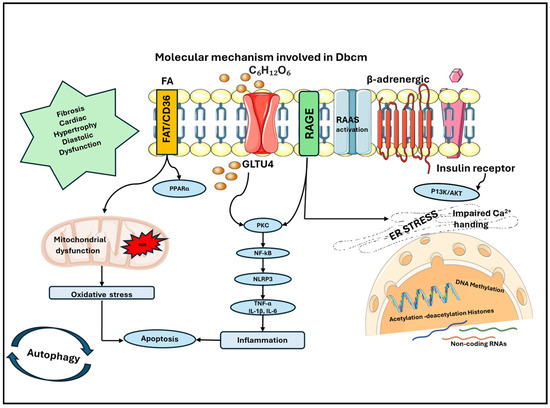
Figure 2.
Molecular mechanisms involved in the development of DbCM: accumulation of FAs contributes to mitochondrial dysfunction with increased production of ROS and consequent oxidative stress in cardiomyocytes. This causes a higher rate of apoptosis, which is also enhanced by inflammation that is sustained by the production of cytokines. Cytokines are released through the activation of NF-kB, which is mainly sustained by hyperglycemia, the accumulation of AGEs and the activation of their receptor AGE. Other contributing mechanisms are the disarrangement of RAAS, the stress on ER and epigenetics, which is induced by hyperglycemia and hyperinsulinism, which contribute to modification of the gene expression and cellular phenotype. The principal perturbations in the heart are fibrosis, cardiac hypertrophy and diastolic dysfunction, which are all main features of DbCM. AGE: advanced glycation end product; DbCM: diabetic cardiomyopathy; ER: endoplasmic reticulum; FAs: fatty acids; RAAS: renin angiotensin aldosterone system; ROS: reactive oxygen species.
6. Epigenetic Changes
Several studies indicate that elevated glucose levels likely play a significant role in the epigenetic regulation of numerous genes, which influences their expression. Consequently, epigenetic alterations could offer a biological rationale for the complexities of DbCM [104,105]. In addition, some gene variants can increase the risk of hypertension in obese children and adolescents, who are the subjects that are mainly predisposed to develop T2DM [104,105].
The main epigenetic changes represented by DNA methylation; alteration of the expression of microRNAs (miRNAs), circular RNA (circRNAs) and long non-coding RNAs (lncRNAs); and alterations to histones (acetylation and methylation) are significantly involved in the development of DbCM [106,107].
6.1. DNA Methylation/Demethylation
Hyperglycemia and the body’s reduced response to insulin (insulin resistance) are closely linked to DNA methylation. DNA methylation controls the activity of genes connected to DbCM, leading to a decline in the heart’s ability to contract, an increase in oxidative stress, cardiac restructuring, cell death in heart muscle cells (cardiomyocytes), and the initiation and advancement of DbCM. Hence, regulating DNA methylation to counteract impaired insulin sensitivity could offer an encouraging avenue for the quick management and restoration of sugar levels, potentially halting the progression of DbCM [108].
DNA methylation involves the adding of 5-methylcytosine through DNA methyltransferases (DNMTs) [109]. Irregular DNA methylation is engaged in conditions like diabetes and cardiovascular diseases, hypertension, coronary artery atherosclerosis and cardiac failure [110]. There is a positive correlation between oxidative stress and cardiac insulin resistance. A study indicated that demethylation of the Nuclear factor erythroid erythtroid 2-related factor 2 (Nrf2) promoter reduces Nrf2 activity, boosts Keap1 protein expression, hampers the transcription of various antioxidant defense genes, causes oxidative damage and worsens insulin sensitivity [111]. Furthermore, in hyperglycemic conditions, irreversible changes in DNMT activity can occur. The activation of the pro-inflammatory cytokine TNF-ɑ might raise the levels of DNA methyltransferase, which leads to the methylation of the promoter region of the SERCA2a. This results in the decrease in SERCA2a levels, which causes an overload of calcium ions and contributes to dysfunction in the relaxation phase of the heart (diastolic heart failure) and the progression of cardiac failure in the most serious cases. In conditions characterized by high blood sugar levels and insulin resistance, the RAAS is triggered. This activation triggers the methylation of the promoter of the angiotensin type II-1b (AT1b) gene, which leads to the increased expression of AT1b and the promotion of enlargement of the heart muscle (myocardial hypertrophy) [112,113].
6.2. Histone Modifications
Histone methylation, mainly on lysine and arginine residues, is regulated by enzymes that ensure orderly gene expression.
The modification of histones through processes such as methylation, acetylation and phosphorylation affects the transcriptional activity of genes, which results in changes in gene expression [114]. Histone acetylation, which is facilitated by histone acetyltransferases (HATs) and histone deacetylases (HDACs), are crucial for gene regulation and maintaining genomic wholeness. HDACs are known to play a significant role in DbCM, such as cardiac fibrosis and hypertrophy [115]. Different HDACs have different effects on DbCM; the SIRT family, especially SIRT1, is involved in controlling factors related to metabolism, adipogenesis and insulin secretion. The interplay between HATs and HDACs impacts the NF-κB transcriptional activity, thereby molding the expression of inflammatory genes downstream. When exposed to heightened glucose levels, cultured monocytes show an augmented recruitment of HATs CPB and p/CAF, leading to heightened histone lysine acetylation at promoters of inflammatory genes. The elevation in histone acetylation levels in monocytes from T2D patients compared with healthy underscores its role in inflammation. Beyond glucose, oxidized lipids are implicated in the enhancement of inflammatory gene promoter histone acetylation in a promoter histone acetylation in a CREB/p300 (HAT)-dependent manner, consequently amplifying gene expression [116]. The protein p300 proves influential in pivotal signaling pathways responding to oxidative stress induced by elevated glucose, which influences extracellular matrix (ECM) components within diabetic-affected organs.
Elevations in p300 levels due to high glucose concentrations lead to heightened histone acetylation at promoters of crucial ECM genes and vasoactive factors in endothelial cells [117]. Curcumin acting on the p300 inhibitor aided in curtailing aberrant gene expression changes triggered by hyperglycemia, especially those linked to diabetic complications and cardiomyocyte hypertrophy. These findings collectively underscore the pivotal role played by chromatin histone acetylation in dictating gene expression patterns associated with complications arising from diabetes.
6.3. Non-Coding RNAs
MicroRNAs, or miRNAs, are short, non-coding RNA molecules that play a crucial role in regulating gene expression within cells. As seen in previous studies, specific miRNA profiles can represent specific biomarkers for detecting an increased risk of developing metabolic dysfunction in obese children [118]. In diabetic heart tissues, approximately 30% of miRNA showed altered expression patterns [119]. These miRNAs are implicated in the modulation of cardiac fibrosis and hypertrophy in DbCM, and the processes oxidative stress, apoptosis and cardiac remodeling [120].
MicroRNAs are involved in either promoting or inhibiting apoptosis in cardiomyocytes. miR-203 can alleviate oxidative damage and the fibrosis of cardiac cells in DbCM by targeting PIK3CA to inhibit the PI3K/Akt signaling pathway [121]. An increased expression of miR-22 in DbCM was shown to help counteract oxidative stress [122]. The inhibition of miR-144 reduces oxidative stress, decreases STZ-induced (streptozotocin) cardiomyocyte apoptosis and enhances heart function in DbCM mice through the Nrf2 pathway [123]. The knockdown of miR-451 impaired the transition from endothelial to mesenchymal states, reduced heart fibrosis and enhanced cardiac function in diabetic mice [124]. Inversely, the amplification of miR-200b also produces protective actions. Transforming growth factor beta (TGF-β) plays a key role in the process of endothelial-to-mesenchymal transition (EMT). miR-142-3p can hinder EMT induced by high blood sugar levels and prevent the progression of diabetic cardiomyopathy to heart failure by acting on the TGF-β/Smad pathway. A reduced miR-133a expression in diabetic mouse cardiomyocytes under high glucose conditions inhibits myocardial hypertrophy when miR-133a is overexpressed [125]. Furthermore, blocking miR-199a activates PGC-1α, which enhances mitochondrial fatty acid oxidation and reverses myocardial hypertrophy [126]. A decreased miR-1 expression in cardiomyocytes under high glucose conditions can be mitigated by boosting miR-1 levels to prevent diabetes-induced oxidative stress in the cardiomyocytes. For instance, miR-1 is able to suppress IGF-1 expression, trigger cytochrome c release and enhance apoptosis in cardiomyocytes [127]. Hsp60 plays a crucial role in the protection against DbCM, and studies showed that miR-1 and miR-206 can increase cardiomyocyte apoptosis under high-glucose conditions by inhibiting Hsp60 expression [128]. An increased expression of miR-532 in the heart tissue of T2DM individuals reduces the expression of the anti-apoptotic protein ARC [129]. miR-150-5p is involved in regulating lymphocyte development and inflammatory cytokine production. The inhibition of miR-150-5p was shown to improve cardiac inflammation and fibrosis by targeting Smad7 in heart fibroblasts exposed to high glucose [130]. miRNA associated with an adverse profile includes the elevated expression of the miR-212/132 family in diabetic rat cardiomyocytes, which activates the transcription factor FoxO3 and results in myocardial hypertrophy and autophagy. miR-451 may worsen cardiac muscle cell lipotoxicity and high-fat diet-induced lipotoxicity, which leads to cardiac hypertrophy in DbCM mice by blocking the hepatic kinase B1/AMP-activated protein kinase (LKB1/AMPK) pathway [131]. miRNAs and ROS have a complex interaction in DbCM; for instance, miR-210 may influence mitochondrial damage that is closely related to oxidative stress by targeting ROS-related molecules [132]. miR-30c and miR181a can collaboratively enhance p53 and p21 gene expression, which leads to cardiac cell hypertrophy and apoptosis in rats with diabetic cardiomyopathy (DbCM) and cardiomyocytes treated with high blood sugar. In the hearts of type 2 diabetic mice lacking the leptin receptor and treated with STZ, miR-195 is significantly upregulated, which results in the downregulation of its targets SIRT1 and Bcl2, ultimately causing cardiomyocyte apoptosis [133]. Additionally, miRNAs, like miR-700, miR-223 and miR-146, also play a role in the onset of DbCM through modulating inflammatory responses, although further research is necessary in this area.

Table 1.
Principal studies that analyzed molecular mechanisms in diabetic cardiomyopathy.
Table 1.
Principal studies that analyzed molecular mechanisms in diabetic cardiomyopathy.
| Main Mechanism | Ref. | In Vitro/In Vivo/Ex Vivo | Molecular Evidence |
|---|---|---|---|
| Oxidative stress | [20] | In vivo, STZ-induced diabetic rats | ↑ ROS, TNF-α, RAGE and NFkB-p65 |
| Inflammation, oxidative stress, cardiac hypertrophy and fibrosis | [23] | In vivo, STZ-induced diabetic mice In vitro, AC16 human myocardial cells treated with high glucose | In STZ-induced diabetic mice: ↑ cardiomyocyte area ↑ collagen deposition ↑ IL-1β, IL-6, IL-18 and TNFα ↑ activation of NLRP3 In AC16 cells: ↑ phosphorylation levels of NF-κB p65 and IκB ↑ ROS |
| Inflammation, oxidative stress, apoptosis, pyroptosis, cardiac remodeling and ventricular dysfunction | [24] | In vivo, STZ-induced diabetic mice In vitro, high-glucose-treated H9c2 cardiomyocytes | ↑ NLRP3, ASC, caspase-1 and IL-1β NLRP3 gene silencing reduces left ventricular dysfunction in DbCM and reversed myocardial remodeling In H9c2 cells: ↑ ROS and phosphorylation of NF-kB p65 In cells pretreated with NAC: ↓ ROS activity, NF-kB, NLRP3, ASC, pro-caspaspe-1, activated caspase-1, pro- IL-1β and mature IL-1β |
| Inflammation and pyroptosis | [31] | In vitro, H9c2 rat cardiomyoblast cell line treated with high glucose | ↑ LDH, IL-1β, IL-18, NLRP3, GSDMD-N, caspase-1 and IL-1β ↑ miR-223-3p |
| Inflammation and mitochondrial dysfunction | [32] | In vivo, mice overexpressing IGFII in pancreatic beta cells in hyperlipidemic background with T2DM fed with high-fat diet for 12 weeks with induced MI | Impaired mitophagy in peri-infarct regions of LV, ↑ mtDNA, activation of Aim2, NLRC4 inflammasome, caspase-I, IL-18 and cardiomyocyte death |
| Interstitial fibrosis | [33] | In vivo, STZ-induced diabetic mice | LV systolic functional impairment and ↑ interstitial fibrosis |
| Inflammation | [42] | In vivo, STZ-induced diabetic mice | Release of mtDNA and activation of cGAS-STING signaling pathway leading to the activation of NLRP3 and ↑ TNF-α, IFN-β, IL-1β and IL-18 |
| Mitochondrial dysfunction | [45] | In vivo, T2DM human patients | ↓ myocardial glucose utilization rate |
| Mitochondrial dysfunction | [48] | In vivo, STZ-induced diabetic mice | ↑ PPAR alpha, ACO and M-CPTI |
| Mitochondrial dysfunction | [53] | In vivo, STZ-induced diabetic mice In vitro, HL-1 murine cardiomyocytes | In STZ-induced diabetic mice: ↓ HIF-1α after ischemia In HL-1 cardiomyocytes: failure of hypoxia-mediated metabolic adaptation with increased lactate efflux and glucose consumption |
| Mitochondrial dysfunction and oxidative stress | [55] | In vivo, STZ-induced diabetic mice | ↑ ROS, catalase activity, SOD, TFAM, PGC-1α and CO1 ↓ electron transport chain complexes and SIRT3 activity ↑ acetylation of MnSOD |
| Mitochondrial dysfunction | [67] | In vivo, T1D mice | ↑ 8-OHdG, O-GlcNAcylation of Ogg1 activity and mtDNA damage |
| Mitochondrial dysfunction | [68] | In vivo, STZ-induced diabetic mice | ↑ mitochondrial OGT, ↓ mito-specific O-GlcNAcase (OGA), OGT is mislocalized and ↓ interaction of OGT and complex IV |
| Cardiac hypertrophy | [69] | Ex vivo, cardiomyocytes isolated from type 2 diabetic db/db mice | ↑ HBP and O-GlcNAc levels |
| Cardiac hypertrophy | [70] | In vitro, endothelial vein cells of T2DM patients | ↑ O-GlcNAc levels |
| Cardiac dysfunction | [73] | In vivo, polygenic T2DM mice In vitro, isolated coronary endothelial cells from mice | ↑ coronary endothelial cells apoptosis, ↓ coronary flow velocity reserve, ↓ cardiac contractility and ↑ p53 protein levels |
| Cardiac fibrosis and inflammation | [77] | In vivo, STZ-induced diabetic mice | LV dysfunction, diastolic stiffness; ↑ TGFβ, IL1β, and fibrosis; ↓ MMP-2 activity |
| RAS, apoptosis and cardiac fibrosis | [80] | In vivo, STZ-induced diabetic mice | ↑ intracellular ATII, angiotensinogen and renin in cardiac myocytes Superoxide production, myocyte apoptosis and cardiac fibrosis were inhibited by RAS inhibitors |
| Cardiac fibrosis | [82] | In vitro, differentiated murine mesangial cells exposed to D-glucose | ↑ incorporation of 3[H]proline ↑ stimulation of collagen types I and IV by high D-glucose |
| RAS and cardiac fibrosis | [83] | In vitro, neonatal rat ventricular fibroblasts exposed to high glucose | ↑ intracellular ATII and TGFβ, ↑ collagen-1 synthesis by cardiac fibroblasts that is inhibited by RAS inhibitors |
| Myocyte death and cardiac hypertrophy | [87] | In vivo, STZ-induced diabetic mice | ↑ myocyte apoptosis, AT, AT II, renin and AT1 |
| Myocyte death | [88] | In vitro, neonatal rat ventricular cardiomyocytes cultured with glucose | ↓ number of lysosomes with acidic pH ↑ Galectin3-RFP puncta and leakage of CTSD |
| Inflammation and myocyte death | [89] | In vivo, T2DM patients | ↓ ratios of CD4(+)CD25(hi) Treg/Th17 cells and CD4(+)CD25(hi) Treg/Th1 cells |
| Alterations of myocardial calcium handling | [95] | In vivo, sedentary db/db mice | ↑ diastolic SR-Ca(2+) leak ↓ synchrony of Ca(2+) release, transverse-tubule density, peak systolic and diastolic Ca(2+), caffeine-induced Ca(2+) release and SR Ca(2+) ATPase-mediated Ca(2+) uptake during diastole rate |
| Alterations of myocardial calcium handling and oxidative stress | [96] | In vitro, neonatal rat cardiomyocytes treated with AGEs | ↓ calcium transient amplitude and sarcoplasmic reticulum calcium content ↑ ROS, NADPH oxidase activity, activation of p38 kinase and nuclear translocation of NF-κB with the subsequent induction of inducible NO synthase expression |
| Hypertrophy, apoptosis and alterations of myocardial calcium handling | [97] | In vitro, pluripotent stem cells generated from urine epithelial cells of T2DM patients | Irregular Ca(2+) transient waveforms, decreased transient amplitude, shorter transient duration, shorter decay, slower maximal rising rate and slower maximal decay rate ↑ apoptosis, cellular hypertrophy and lipid accumulation |
| Alterations of myocardial calcium handling | [98] | In vivo, STZ-induced diabetic mice | ↑ Ca(2+) transient ↓ Na(+)/Ca(2+) exchanger current |
| Alterations of myocardial calcium handling | [99] | In vivo, T2DM patients | ↓ left ventricular ERG, KCNH2 and KCNJ3 gene expression ↑ NCX1, KCNJ2, KCNJ5 and SLC8A1 gene expression ↑ QT interval |
| AGEs and alterations of myocardial calcium handling | [101] | In vivo, STZ-induced diabetic mice | AGEs formation on intracellular RyR2 |
| Alterations of myocardial calcium handling | [102] | In vitro, HEK-293 cells | ↑ Ca(2+) uptake velocity for expressed SERCA2 by exposure to CaM kinase I1 |
| AGEs and alterations of myocardial calcium handling | [103] | In vivo, STZ-induced diabetic mice | ↓ heart relaxation ↓ SERCA2a expression ↑ PLB |
| Epigenetic changes | [107] | In vivo, STZ-induced diabetic mice In vivo, formalin-fixed and paraffin-embedded human myocardial archived tissues In vitro, neonatal rat cardiomyocytes exposed to glucose | Absence of methylation in the promoter regions of the DUSP-1 |
| Epigenetic changes | [111] | In vivo, nondiabetic women | Positive association between the methylation levels of the CpG site 783 with insulin sensitivity |
| Epigenetic changes | [112] | In vitro, HL-1 cardiomyocytes | ↓ SERCA2a RNA and protein expression ↑ methylation in the SERCA2a promoter region ↑ DNA methyltransferase 1 expression |
| Epigenetic changes | [116] | In vitro, vascular smooth muscle cells | 12(S)-HETE activated Src, focal adhesion kinase, Akt, p38MAPK, CREB, expression of monocyte chemoattractant protein-1, IL-6 genes and histone H3-Lys-9/14 acetylation on their promoters |
| Epigenetic changes | [117] | In vitro, neonatal rat cardiomyocytes exposed to glucose In vivo, STZ-induced diabetic rats | Cellular hypertrophy and ↑ mRNA expression of ANP, BNP, ANG, MEF2A, MEF2C and transcriptional coactivator p300 In STZ-induced diabetic rats: ↑ ANP, BNP, ANG, mRNA, p300, MEF2A, and MEF2C expression |
| Epigenetic changes | [119] | In vivo, STZ-induced diabetic mice | Dysregulation of 316 out of 1008 total miRNAs implicated in myocardial signalling networks that trigger apoptosis, fibrosis, hypertrophic growth, autophagy oxidative stress and heart failure |
| Epigenetic changes | [121] | In vivo, STZ-induced diabetic mice | Upregulation of miR-203 inhibited the activation of PI3K/Akt signaling pathway and ↓ PIK3CA, PI3K, Akt, CoI I, CoI III, ANP, MDA and ROS in the myocardial tissues |
| Epigenetic changes | [123] | In vivo and ex vivo, STZ-induced diabetic mice In vitro, cultured cardiomyocytes | In STZ-induced diabetic mice: ↓ miR-144 in heart tissues In cultured cardiomyocytes: high glucose exposure induced ↓ miR-144 |
| Epigenetic changes | [124] | In vivo, STZ-induced diabetic mice | miR-451 knockdown attenuated cardiac fibrosis, improved cardiac function and suppressed endothelial-to-mesenchymal transition |
| Epigenetic changes | [126] | In vivo, mice treated with anti-miR-199a | Upregulation of genes related to cytoplasmic translation and mitochondrial respiratory chain complex assembly |
| Epigenetic changes | [127] | In vitro, H9C2 cells exposed to high glucose | ↑ miR-1 expression level ↓ mitochondrial membrane potential ↑ cytochrome-c release and apoptosis |
| Epigenetic changes | [128] | In vivo, STZ-induced diabetic mice In vitro, cardiac muscle cell line HL-1 | In STZ-induced diabetic mice: ↓ GAS5 ↑ NLRP3, caspase-1, Pro-caspase-1, IL-1β and IL-18 GAS5 overexpression improved cardiac function and myocardial hypertrophy In cardiac muscle cell line HL-1: ↑ NLRP3, caspase-1, Pro-caspase-1, IL-1β and IL-18, |
| Epigenetic changes | [129] | In vivo, STZ-induced diabetic mice In vitro, H9C2 cells exposed to high glucose | STZ-induced diabetic mice: ↑ p53 and p21 ↓ miR-30c and miR-181a H9C2 cells exposed to high glucose: ↑ p53 and p21 ↓ miR-181a |
| Epigenetic changes | [130] | In vivo, STZ-induced diabetic mice | ↑ miR-195 expression |
| Epigenetic changes | [131] | In vivo, mice with obesity and diabetes induced by high-fat diet In vitro, neonatal rat cardiac myocytes stimulated with palmitic acid | In neonatal rat cardiac myocytes: ↑ miR-451 expression in a dose- and time-dependent manner In cardiomyocyte-specific miR-451 knockout mice: cardiac hypertrophy and contractile reserves were ameliorated, ↑ Cab39 and phosphorylated AMPK, and ↓ mTOR |
| Epigenetic changes | [133] | In vivo, T2DM human patients undergoing coronary artery bypass graft surgery In vivo, type-2 diabetic mice In vitro, human ventricular cardiomyocytes treated with high glucose | ↑ miR-532 expression in the right atrial appendage tissue, which was associated with downregulation of ARC In human ventricular cardiomyocytes (AC16) treated with high glucose: inhibition of miR-532 activity in high-glucose-cultured human cardiomyocytes prevented the downregulation of ARC and attenuated apoptotic cell death |
↑: increased; ↓: decreased. ACO, 1-Aminocyclopropane-1-Carboxylic Acid Oxidase; AIM2, absent in melanoma 2; ANG, Angiogenin; ANP, Atrial Natriuretic Peptide; ARC, apoptosis repressor with caspase recruitment domain; ASC, apoptosis associated speck like protein; AT, angiotensin; ATP, Adenosine triphosphate; BNP, Natriuretic Peptide B; Ca, calcium; CaMKII, Ca(2+)/calmodulin-dependent protein kinase II; CO1, cytochrome oxydase I; CPT1B, carnitine palmitoyltransferase 1B; CREB, Cyclic adenosine monophosphate response element-binding protein; CTSD, cathepsin D; DbCM, diabetic cardiomyopathy; DUSP1, Dual Specificity Phosphatase 1; FDG, fluorine-18 fluorodeoxyglucose; GSDMD, Gasdermin D; HBP, hexosamine biosynthesis pathway; HIF, hypoxia inducible factor; IFN, interferon; IGF, insulin-like growth factor; Il, interleukin; KCNJ, potassium inwardly rectifying channel subfamily J; LDH, lactate dehydrogenase; LV, left ventricle; LVFS, left-ventricular fractional shortening; MAPK, mitogen-activated protein kinases; membrane carnitine palmitoyltransferase, M-CPT I; MEF, Myocyte Enhancer Factor; MI, myocardial infarction; miR, microRNA; MnSOD, Manganese Superoxide Dismutase; mtDNA, mitochondrial DNA; mTOR, phosphorylated mammalian target of rapamycin; Na, sodium; NAC, N-acetylcysteine; NADPH, Nicotinamide adenine dinucleotide phosphate; NCX, Na(+)-Ca(2+) exchanger; NFkB, nuclear factor-kB; NLRC4, NLR Family CARD Domain Containing 4; NLRP3, NOD-like receptor family pyrin domain containing 3; NO, nitric oxide; Nrf2, nuclear factor-erythroid 2-related factor 2; OGT, O-GlcNAc transferase; PET, Positron Emission Tomography; PGC1alpha, Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha; PIK3, Phosphoinositide 3-kinase; PLB, Phospholamban; PPAR, peroxisome proliferator-activated receptor; PPARGC1A, PPAR coactivator 1-alpha; PP2, Protein phosphatase 2; RAGE, Advanced glycated end products receptor; RAS, renin angiotensin system; ROS, reactive oxygen species; SERCA, Sarco-Endoplasmic Reticulum Calcium ATPase; SLC8A1, Solute Carrier Family 8 Member A 1; SIRT3, NAD-dependent deacetylase sirtuin-3; SOD, Superoxide Dismutase; SR, sarcoplasmic reticulum; STING, stimulator of interferon genes; STZ, streptozotocin; TFAM, Mitochondrial transcription factor A; TGF, Transforming growth factor; TNF-α, Tumor Necrosis factor alpha; T1D, type 1 diabetes; T2DM, type 2 diabetes mellitus; 8-OHdG, 8-hydroxy-2′-deoxyguanosine.
7. Prospective Treatments
To date, there is not a specific therapeutic approach for DbCM in clinical practice; the increasing knowledge on molecular mechanisms of DbCM may open new possibilities for developing therapies that target specific pathways (Table 2). Some drugs already used for the treatment of T2DM were studied to explore their potential role on DbCM, such as Glucagon-like Peptide-1 (GLP-1). Receptor agonists ameliorate cardiomyocytes’ calcium handling and have anti-inflammatory effects [134,135]. Metformin, in addition to its classical actions, can improve mitochondrial biogenesis and enhance antioxidant activity, decrease mitochondrial ROS production, upregulate mitophagy and normalize calcium handling [136,137,138,139]. Clinical studies are also exploring the potential role on DbCM of drugs used for other diseases. Cyclic guanosine monophosphate-phosphodiesterase type 5 (PDE5) inhibitors, fenofibrate and alpha-lipoic acid can inhibit NLRP3 inflammasome-mediated pyroptosis [140]. Levosimendan and istaroxime may have a role in improving calcium handling in cardiomyocytes by stabilizing the calcium-troponin C complex and increasing the expression of SERCA-2 and sodium-calcium exchanger-1, respectively [141,142]. The inhibition of HDACs can attenuate cardiac hypertrophy and fibrosis and can improve cardiac function and reverse cardiac remodeling [143,144]. The most promising medications in treating of DbCM and cardiovascular disease in T2DM patients are sodium-glucose cotransporter-2 inhibitors (SGLT-2is), also known as gliflozins. SGLT2is drastically reduce the risk of cardiovascular death patients with heart failure according to a recent series of large clinical trials, including the Dapagliflozin and Prevention of Adverse Outcomes in Heart Failure (DAPA-HF), the Empagliflozin Outcome Trial in Patients with Chronic Heart Failure and a Reduced Ejection Fraction (EMPEROR-Reduced), and the Empagliflozin Outcome Trial in Patients with Chronic Heart Failure with Preserved Ejection Fraction (EMPEROR-Preserved) [145,146,147,148,149]. SGLT-2is have antioxidant and anti-inflammatory properties, can mitigate cardiac fibrosis and have a positive impact on calcium handling [140,150,151,152,153]. SGLT-2is prevent glucose reabsorption in the proximal tubule kidneys, increasing urinary glucose excretion and lowering plasma glucose. Through a variety of tissue- and organ-specific anti-inflammatory effects, SGLT2is may be able to provide cardiovascular protection. For instance, hyperuricemia, which is frequently observed in people with heart disease [154], seems to support the pathophysiology of cardiovascular disease by promoting the proliferation of vascular smooth muscle cells and raising the synthesis of pro-oxidative [155]. Given its demonstrated ability to lower uric acid levels, SGLT2is may have the potential to improve this diseased route. By increasing the uric acid excretion in urine, SGLT2is lower uric acid levels. Another proposed mechanism involves epicardial adipose tissue. Epicardial adipose tissue secretes high levels of activin A, which is a cytokine that enhances inflammation, in people with DbCM [156]. It was demonstrated that dapagliflozin and canagliflozin reduce epicardial adipose tissue in people with type 2 diabetes (T2D), which lowers vascular inflammation and fibrosis. SGLT2is significantly decreased the left ventricular mass in patients with DbCM according to a recent meta-analysis of a double-blinded, placebo-controlled, randomized trial that evaluated left-ventricular remodeling by cardiac magnetic resonance imaging [157]. Moreover, studies that employed murine models of myocytes demonstrated that dapagliflozin and empagliflozin can prevent cardiac fibrosis [158,159]. Similarly, according to preliminary data on human myofibroblasts, empagliflozin may directly influence extracellular matrix reorganization through cell-mediated collagen remodeling and the reduction of myofibroblast activity [160]. In T2DM, there is a slight and sustained increase in blood ketones. In this condition, the heart uses more beta-hydroxybutyrate—which is a ketone—than usual to produce energy instead of fatty acids [161]. It enhances the mitochondrial level conversion of oxygen consumption into work efficiency [162]. It was proposed that SGLT2is cause the metabolism to shift toward the oxidation of FAs and promote ketogenesis based on research with dapagliflozin and empagliflozin. Because of this change, beta-hydroxybutyrate plasma levels are higher. This may provide substantial cardioprotection for T2DM patients [152,163]. Moreover, hemoconcentration induced by SGLT2is increases oxygen release. Thus, such a metabolic substrate change can provide cardioprotection [164]. In animal models, the suppression of overexpressed miRNA in heart failure was studied with promising results [165]. Specific miRNA and lncRNA profiles could also be potentially used as biomarkers for early detection of DbCM, or to identify diabetic individuals with an increased risk of developing cardiac dysfunction. It was hypothesized that miR-9, miR-21, miR-29, miR-30d, miR-34a, miR144, miR150, miR-320 and miR378 in particular could be associated with a higher risk of cardiomyopathy [120]. Lastly, a complete knowledge of molecular mechanisms of DbCM could help to better understand the different phenotypes of early-onset DMT2 and the pathological basis of the worst cardiac complications, with the aim of developing tailored treatments.
Table 2.
Drugs with potential effects on DbCM.
8. Conclusions
Patients with T2DM require multidisciplinary surveillance, including cardiac follow-up, to exclude the development of DbCM. The pathogenetic basis of DbCM involves multiple molecular pathways and metabolic changes. Knowing these mechanisms helps to establish effective therapies that may prevent the development of DbCM or treat overt disease conditions. Providing that lifestyle changes and adequate diet should always be pursued to aim for optimal glycemic control, several therapeutic options may be employed. Among them, SGLT-2is already represent a proven effective therapeutic option in improving the outcome of T2DM patients with heart disease. By acting on multiple pathogenic pathways, we can assist with reducing the mortality and morbidity due to DbCM.
Author Contributions
Conceptualization, S.G. and M.F.F.; methodology, all authors of the manuscript; writing—original draft preparation, S.G., G.M., D.M. and G.D.; writing— review and editing, all authors of the manuscript; supervision, M.F.F. and G.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Goyal, R.; Singhal, M.; Jialal, I. Type 2 Diabetes. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Armocida, B.; Monasta, L.; Sawyer, S.M.; Bustreo, F.; Onder, G.; Castelpietra, G.; Pricci, F.; Minardi, V.; Giacomozzi, C.; Abbafati, C.; et al. The Burden of Type 1 and Type 2 Diabetes among Adolescents and Young Adults in 24 Western European Countries, 1990–2019: Results from the Global Burden of Disease Study 2019. Int. J. Public Health 2024, 68, 1606491. [Google Scholar] [CrossRef] [PubMed]
- Valaiyapathi, B.; Gower, B.; Ashraf, P.A. Pathophysiology of Type 2 Diabetes in Children and Adolescents. Curr. Diabetes Rev. 2020, 16, 220–229. [Google Scholar] [PubMed]
- Pastore, I.; Bolla, A.M.; Montefusco, L.; Lunati, M.E.; Rossi, A.; Assi, E.; Zuccotti, G.V.; Fiorina, P. The impact of diabetes mellitus on cardiovascular risk onset in children and adolescents. Int. J. Mol. Sci. 2020, 21, 4928. [Google Scholar] [CrossRef] [PubMed]
- Miniello, V.L.; Faienza, M.F.; Scicchitano, P.; Cortese, F.; Gesualdo, M.; Zito, A.; Basile, M.; Recchia, P.; Leogrande, D.; Viola, D.; et al. Insulin resistance and endothelial function in children and adolescents. Int. J. Cardiol. 2014, 174, 343–347. [Google Scholar] [CrossRef]
- Faienza, M.F.; Brunetti, G.; Delvecchio, M.; Zito, A.; De Palma, F.; Cortese, F.; Nitti, A.; Massari, E.; Gesualdo, M.; Ricci, G.; et al. Vascular Function and Myocardial Performance Indices in Children Born Small for Gestational Age. Circ. J. 2016, 80, 958–963. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, Y.; Zhang, J.; Zhang, R.; Zhao, L.; Ren, H.; Zou, Y.; Wang, T.; Wang, J.; Zhao, Y.; et al. Early-onset of type 2 diabetes mellitus is a risk factor for diabetic nephropathy progression: A biopsy-based study. Aging 2021, 13, 8146–8154. [Google Scholar] [CrossRef] [PubMed]
- Nanayakkara, N.; Curtis, A.J.; Heritier, S.; Gadowski, A.M.; Pavkov, M.E.; Kenealy, T.; Owens, D.R.; Thomas, R.L.; Song, S.; Wong, J. Impact of age at type 2 diabetes mellitus diagnosis on mortality and vascular complications: Systematic review and meta-analyses. Diabetologia 2021, 64, 275–287. [Google Scholar] [CrossRef]
- Zou, X.; Zhou, X.; Ji, L.; Yang, W.; Lu, J.; Weng, J.; Jia, W.; Shan, Z.; Liu, J.; Tian, H. The characteristics of newly diagnosed adult early-onset diabetes: A population-based cross-sectional study. Sci. Rep. 2017, 7, 46534. [Google Scholar] [CrossRef]
- Ke, C.; Shah, B.R.; Thiruchelvam, D.; Echouffo-Tcheugui, J.B. Association between Age at Diagnosis of Type 2 Diabetes and Hospitalization for Heart Failure: A Population-Based Study. J. Am. Heart Assoc. 2024, 13, e030683. [Google Scholar] [CrossRef]
- Lindberg, L.; Danielsson, P.; Persson, M.; Marcus, C.; Hagman, E. Association of childhood obesity with risk of early all-cause and cause-specific mortality: A Swedish prospective cohort study. PLoS Med. 2020, 17, e1003078. [Google Scholar] [CrossRef]
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic cardiomyopathy: An update of mechanisms contributing to this clinical entity. Circ. Res. 2018, 122, 624–638. [Google Scholar] [CrossRef]
- Swiatkiewicz, I.; Patel, N.T.; Villarreal-Gonzalez, M.; Taub, P.R. Prevalence of diabetic cardiomyopathy in patients with type 2 diabetes in a large academic medical center. BMC Med. 2024, 22, 195. [Google Scholar] [CrossRef]
- NHS Digital. National Diabetes Audit, 2017–2018. Available online: https://files.digital.nhs.uk/91/084B1D/National%20Diabetes%20Audit%2C%202017-18%2C%20Report%202a.pdf (accessed on 13 December 2019).
- Huo, J.-L.; Feng, Q.; Pan, S.; Fu, W.-J.; Liu, Z.; Liu, Z. Diabetic cardiomyopathy: Early diagnostic biomarkers, pathogenetic mechanisms, and therapeutic interventions. Cell Death Discov. 2023, 9, 256. [Google Scholar] [CrossRef] [PubMed]
- Sudo, S.Z.; Montagnoli, T.L.; Rocha, B.D.S.; Santos, A.D.; de Sá, M.P.; Zapata-Sudo, G. Diabetes-induced cardiac autonomic neuropathy: Impact on heart function and prognosis. Biomedicines 2022, 10, 3258. [Google Scholar] [CrossRef]
- Ramesh, P.; Yeo, J.L.; Brady, E.M.; McCann, G.P. Role of inflammation in diabetic cardiomyopathy. Ther. Adv. Endocrinol. Metab. 2022, 13, 20420188221083530. [Google Scholar] [CrossRef]
- Ritchie, R.H.; Abel, E.D. Basic mechanisms of diabetic heart disease. Circ. Res. 2020, 126, 1501–1525. [Google Scholar] [CrossRef] [PubMed]
- Frati, G.; Schirone, L.; Chimenti, I.; Yee, D.; Biondi-Zoccai, G.; Volpe, M.; Sciarretta, S. An overview of the inflammatory signalling mechanisms in the myocardium underlying the development of diabetic cardiomyopathy. Cardiovasc. Res. 2017, 113, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Aragno, M.; Mastrocola, R.; Medana, C.; Catalano, M.G.; Vercellinatto, I.; Danni, O.; Boccuzzi, G. Oxidative stress-dependent impairment of cardiac-specific transcription factors in experimental diabetes. Endocrinology 2006, 147, 5967–5974. [Google Scholar] [CrossRef]
- Wang, N.; Liang, H.; Zen, K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front. Immunol. 2014, 5, 614. [Google Scholar] [CrossRef]
- Zeng, C.; Wang, R.; Tan, H. Role of pyroptosis in cardiovascular diseases and its therapeutic implications. Int. J. Biol. Sci. 2019, 15, 1345. [Google Scholar] [CrossRef]
- Qu, X.-F.; Zhai, B.-Z.; Hu, W.-L.; Lou, M.-H.; Chen, Y.-H.; Liu, Y.-F.; Chen, J.-G.; Mei, S.; You, Z.-Q.; Liu, Z. Pyrroloquinoline quinone ameliorates diabetic cardiomyopathy by inhibiting the pyroptosis signaling pathway in C57BL/6 mice and AC16 cells. Eur. J. Nutr. 2022, 61, 1823–1836. [Google Scholar] [CrossRef] [PubMed]
- Luo, B.; Li, B.; Wang, W.; Liu, X.; Xia, Y.; Zhang, C.; Zhang, M.; Zhang, Y.; An, F. NLRP3 gene silencing ameliorates diabetic cardiomyopathy in a type 2 diabetes rat model. PLoS ONE 2014, 9, e104771. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.-M.; Liu, Y.; White, D.; Su, Y.; Drew, B.G.; Bruce, C.R.; Kiriazis, H.; Xu, Q.; Jennings, N.; Bobik, A.; et al. Deletion of macrophage migration inhibitory factor protects the heart from severe ischemia–reperfusion injury: A predominant role of anti-inflammation. J. Mol. Cell. Cardiol. 2011, 50, 991–999. [Google Scholar] [CrossRef] [PubMed]
- Van Tassell, B.W.; Seropian, I.M.; Toldo, S.; Mezzaroma, E.; Abbate, A. Interleukin-1β induces a reversible cardiomyopathy in the mouse. Inflamm. Res. 2013, 62, 637–640. [Google Scholar] [CrossRef] [PubMed]
- Simons, P.J.; van den Pangaart, P.S.; Aerts, J.M.; Boon, L. Pro-inflammatory delipidizing cytokines reduce adiponectin secretion from human adipocytes without affecting adiponectin oligomerization. J. Endocrinol. 2007, 192, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Qiu, S.; Yang, G.; Wu, Q. Adiponectin and metabolic cardiovascular diseases: Therapeutic opportunities and challenges. Genes Dis. 2023, 10, 1525–1536. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Zhang, H.; Liu, P.; Wang, H.; Liu, J.; Gao, C.; Liu, Y.; Lian, K.; Yang, L.; Sun, L. Impaired mitochondrial biogenesis due to dysfunctional adiponectin-AMPK-PGC-1α signaling contributing to increased vulnerability in diabetic heart. Basic Res. Cardiol. 2013, 108, 329. [Google Scholar] [CrossRef]
- Ciccone, M.M.; Faienza, M.F.; Altomare, M.; Nacci, C.; Montagnani, M.; Valente, F.; Cortese, F.; Gesualdo, M.; Zito, A.; Mancarella, R.; et al. Endothelial and Metabolic Function Interactions in Overweight/Obese Children. J. Atheroscler. Thromb. 2016, 23, 950–959. [Google Scholar] [CrossRef]
- Zhao, S.; Tan, Y.; Qin, J.; Xu, H.; Liu, L.; Wan, H.; Zhang, C.; Fan, W.; Qu, S. MicroRNA-223–3p promotes pyroptosis of cardiomyocyte and release of inflammasome factors via downregulating the expression level of SPI1 (PU. 1). Toxicology 2022, 476, 153252. [Google Scholar] [CrossRef]
- Devi, T.D.; Babu, M.; Mäkinen, P.; Kaikkonen, M.U.; Heinaniemi, M.; Laakso, H.; Ylä-Herttuala, E.; Rieppo, L.; Liimatainen, T.; Naumenko, N. Aggravated postinfarct heart failure in type 2 diabetes is associated with impaired mitophagy and exaggerated inflammasome activation. Am. J. Pathol. 2017, 187, 2659–2673. [Google Scholar] [CrossRef]
- Lother, A.; Bondareva, O.; Saadatmand, A.R.; Pollmeier, L.; Härdtner, C.; Hilgendorf, I.; Weichenhan, D.; Eckstein, V.; Plass, C.; Bode, C. Diabetes changes gene expression but not DNA methylation in cardiac cells. J. Mol. Cell. Cardiol. 2021, 151, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Kesherwani, V.; Shahshahan, H.R.; Mishra, P.K. Cardiac transcriptome profiling of diabetic Akita mice using microarray and next generation sequencing. PLoS ONE 2017, 12, e0182828. [Google Scholar] [CrossRef]
- Henson, S.M.; Aksentijevic, D. Senescence and type 2 diabetic cardiomyopathy: How young can you die of old age? Front. Pharmacol. 2021, 12, 716517. [Google Scholar] [CrossRef] [PubMed]
- Kenny, H.C.; Abel, E.D. Heart Failure in Type 2 Diabetes Mellitus. Circ. Res. 2019, 124, 121–141. [Google Scholar] [CrossRef] [PubMed]
- Morino, K.; Petersen, K.F.; Shulman, G.I. Molecular mechanisms of insulin resistance in humans and their potential links with mitochondrial dysfunction. Diabetes 2006, 55 (Suppl. 2), S9–S15. [Google Scholar] [CrossRef]
- Gollmer, J.; Zirlik, A.; Bugger, H. Mitochondrial mechanisms in diabetic cardiomyopathy. Diabetes Metab. J. 2020, 44, 33. [Google Scholar] [CrossRef]
- Dabravolski, S.A.; Sadykhov, N.K.; Kartuesov, A.G.; Borisov, E.E.; Sukhorukov, V.N.; Orekhov, A.N. The role of mitochondrial abnormalities in diabetic cardiomyopathy. Int. J. Mol. Sci. 2022, 23, 7863. [Google Scholar] [CrossRef] [PubMed]
- Forte, M.; Schirone, L.; Ameri, P.; Basso, C.; Catalucci, D.; Modica, J.; Chimenti, C.; Crotti, L.; Frati, G.; Rubattu, S. The role of mitochondrial dynamics in cardiovascular diseases. Br. J. Pharmacol. 2021, 178, 2060–2076. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Xu, Y.; Liu, Y.; Gao, J.; Feng, L.; Zhang, Y.; Shi, L.; Zhang, M.; Guo, D.; Qi, B. Mitochondrial quality control in the maintenance of cardiovascular homeostasis: The roles and interregulation of UPS, mitochondrial dynamics and mitophagy. Oxidative Med. Cell. Longev. 2021, 2021, 3960773. [Google Scholar] [CrossRef]
- Yan, M.; Li, Y.; Luo, Q.; Zeng, W.; Shao, X.; Li, L.; Wang, Q.; Wang, D.; Zhang, Y.; Diao, H.; et al. Mitochondrial damage and activation of the cytosolic DNA sensor cGAS-STING pathway lead to cardiac pyroptosis and hypertrophy in diabetic cardiomyopathy mice. Cell Death Discov. 2022, 8, 258. [Google Scholar] [CrossRef]
- Barth, E.; Stämmler, G.; Speiser, B.; Schaper, J. Ultrastructural quantitation of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man. J. Mol. Cell. Cardiol. 1992, 24, 669–681. [Google Scholar] [CrossRef]
- Opie, L.H. Heart Physiology: From Cell to Circulation; Lippincott Williams Wilkins: Philadelphia, PA, USA, 2004. [Google Scholar]
- Ohtake, T.; Yokoyama, I.; Watanabe, T.; Momose, T.; Serezawa, T.; Nishikawa, J.; Sasaki, Y. Myocardial glucose metabolism in noninsulin-dependent diabetes mellitus patients evaluated by FDG-PET. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 1995, 36, 456–463. [Google Scholar]
- Nakamura, M.; Sadoshima, J. Cardiomyopathy in obesity, insulin resistance and diabetes. J. Physiol. 2020, 598, 2977–2993. [Google Scholar] [CrossRef] [PubMed]
- Barger, P.M.; Kelly, D.P. PPAR signaling in the control of cardiac energy metabolism. Trends Cardiovasc. Med. 2000, 10, 238–245. [Google Scholar] [CrossRef]
- Finck, B.N.; Lehman, J.J.; Leone, T.C.; Welch, M.J.; Bennett, M.J.; Kovacs, A.; Han, X.; Gross, R.W.; Kozak, R.; Lopaschuk, G.D. The cardiac phenotype induced by PPARα overexpression mimics that caused by diabetes mellitus. J. Clin. Investig. 2002, 109, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Zlobine, I.; Gopal, K.; Ussher, J.R. Lipotoxicity in obesity and diabetes-related cardiac dysfunctioniochim. Biochim. Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2016, 1861, 1555–1568. [Google Scholar]
- Brindley, D.N.; Kok, B.P.; Kienesberger, P.C.; Lehner, R.; Dyck, J.R. Shedding light on the enigma of myocardial lipotoxicity: The involvement of known and putative regulators of fatty acid storage and mobilization. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E897–E908. [Google Scholar] [CrossRef] [PubMed]
- Sletten, A.C.; Peterson, L.R.; Schaffer, J.E. Manifestations and mechanisms of myocardial lipotoxicity in obesity. J. Intern. Med. 2018, 284, 478–491. [Google Scholar] [CrossRef] [PubMed]
- Gollmer, J.; Zirlik, A.; Bugger, H. Established and Emerging Mechanisms of Diabetic Cardiomyopathy. J. Lipid Atheroscler. 2019, 8, 26–47. [Google Scholar] [CrossRef]
- Dodd, M.S.; Sousa Fialho, M.D.L.; Montes Aparicio, C.N.; Kerr, M.; Timm, K.N.; Griffin, J.L.; Luiken, J.; Glatz, J.F.C.; Tyler, D.J.; Heather, L.C. Fatty Acids Prevent Hypoxia-Inducible Factor-1α Signaling through Decreased Succinate in Diabetes. JACC Basic Transl. Sci. 2018, 3, 485–498. [Google Scholar] [CrossRef]
- Sturza, A.; Duicu, O.M.; Vaduva, A.; Dănilă, M.D.; Noveanu, L.; Varró, A.; Muntean, D.M. Monoamine oxidases are novel sources of cardiovascular oxidative stress in experimental diabetes. Can. J. Physiol. Pharmacol. 2015, 93, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Sultana, M.R.; Bagul, P.K.; Katare, P.B.; Anwar Mohammed, S.; Padiya, R.; Banerjee, S.K. Garlic activates SIRT-3 to prevent cardiac oxidative stress and mitochondrial dysfunction in diabetes. Life Sci. 2016, 164, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Cater, M.; Krizancic Bombek, L. Protective Role of Mitochondrial Uncoupling Proteins against Age-Related Oxidative Stress in Type 2 Diabetes Mellitus. Antioxidants 2022, 11, 1473. [Google Scholar] [CrossRef]
- Spinelli, S.; Guida, L.; Passalacqua, M.; Magnone, M.; Cossu, V.; Sambuceti, G.; Marini, C.; Sturla, L.; Zocchi, E. Abscisic Acid and Its Receptors LANCL1 and LANCL2 Control Cardiomyocyte Mitochondrial Function, Expression of Contractile, Cytoskeletal and Ion Channel Proteins and Cell Proliferation via ERRalpha. Antioxidants 2023, 12, 1692. [Google Scholar] [CrossRef]
- Dietrich, M.O.; Horvath, T.L. The role of mitochondrial uncoupling proteins in lifespan. Pflugers Arch. Eur. J. Physiol. 2010, 459, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.K.; Garikipati, V.N.S.; Kishore, R. Mitochondrial dysfunction and its impact on diabetic heart. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1098–1105. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Liu, C.; Zhu, P.; Li, Y. Novel Insights Into Molecular Mechanism of Mitochondria in Diabetic Cardiomyopathy. Front. Physiol. 2020, 11, 609157. [Google Scholar] [CrossRef] [PubMed]
- Mohan, M.; Dihoum, A.; Mordi, I.R.; Choy, A.M.; Rena, G.; Lang, C.C. Left Ventricular Hypertrophy in Diabetic Cardiomyopathy: A Target for Intervention. Front. Cardiovasc. Med. 2021, 8, 746382. [Google Scholar] [CrossRef] [PubMed]
- Pan, K.L.; Hsu, Y.C.; Chang, S.T.; Chung, C.M.; Lin, C.L. The Role of Cardiac Fibrosis in Diabetic Cardiomyopathy: From Pathophysiology to Clinical Diagnostic Tools. Int. J. Mol. Sci. 2023, 24, 8604. [Google Scholar] [CrossRef]
- Sundgren, N.C.; Giraud, G.D.; Schultz, J.M.; Lasarev, M.R.; Stork, P.J.; Thornburg, K.L. Extracellular signal-regulated kinase and phosphoinositol-3 kinase mediate IGF-1 induced proliferation of fetal sheep cardiomyocytes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 285, R1481–R1489. [Google Scholar] [CrossRef]
- Hu, X.; Bai, T.; Xu, Z.; Liu, Q.; Zheng, Y.; Cai, L. Pathophysiological Fundamentals of Diabetic Cardiomyopathy. Compr. Physiol. 2017, 7, 693–711. [Google Scholar] [PubMed]
- Cairns, M.; Joseph, D.; Essop, M.F. The dual role of the hexosamine biosynthetic pathway in cardiac physiology and pathophysiology. Front. Endocrinol. 2022, 13, 984342. [Google Scholar] [CrossRef] [PubMed]
- Chatham, J.C.; Zhang, J.; Wende, A.R. Role of O-Linked N-Acetylglucosamine Protein Modification in Cellular (Patho)Physiology. Physiol. Rev. 2021, 101, 427–493. [Google Scholar] [CrossRef]
- Cividini, F.; Scott, B.T.; Dai, A.; Han, W.; Suarez, J.; Diaz-Juarez, J.; Diemer, T.; Casteel, D.E.; Dillmann, W.H. O-GlcNAcylation of 8-Oxoguanine DNA Glycosylase (Ogg1) Impairs Oxidative Mitochondrial DNA Lesion Repair in Diabetic Hearts. J. Biol. Chem. 2016, 291, 26515–26528. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.S.; Ma, J.; Hart, G.W. Diabetes-associated dysregulation of O-GlcNAcylation in rat cardiac mitochondria. Proc. Natl. Acad. Sci. USA 2015, 112, 6050–6055. [Google Scholar] [CrossRef]
- Marsh, S.A.; Dell’Italia, L.J.; Chatham, J.C. Activation of the hexosamine biosynthesis pathway and protein O-GlcNAcylation modulate hypertrophic and cell signaling pathways in cardiomyocytes from diabetic mice. Amino Acids 2011, 40, 819–828. [Google Scholar] [CrossRef] [PubMed]
- Masaki, N.; Feng, B.; Bretón-Romero, R.; Inagaki, E.; Weisbrod, R.M.; Fetterman, J.L.; Hamburg, N.M. O-GlcNAcylation Mediates Glucose-Induced Alterations in Endothelial Cell Phenotype in Human Diabetes Mellitus. J. Am. Heart Assoc. 2020, 9, e014046. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.L.; Xue, R.Q.; Xi, H.; Ming, Z.; Yu, X.J.; Liu, L.Z.; Wu, Q.; Si, Y.; Li, D.L.; Zang, W.J. Cholinergic drugs ameliorate endothelial dysfunction by decreasing O-GlcNAcylation via M3 AChR-AMPK-ER stress signaling. Life Sci. 2019, 222, 1–12. [Google Scholar] [CrossRef]
- Karimi, M.; Pavlov, V.I.; Ziegler, O.; Sriram, N.; Yoon, S.Y.; Agbortoko, V.; Alexandrova, S.; Asara, J.; Sellke, F.W.; Sturek, M.; et al. Robust effect of metabolic syndrome on major metabolic pathways in the myocardium. PLoS ONE 2019, 14, e0225857. [Google Scholar] [CrossRef]
- Si, R.; Zhang, Q.; Tsuji-Hosokawa, A.; Watanabe, M.; Willson, C.; Lai, N.; Wang, J.; Dai, A.; Scott, B.T.; Dillmann, W.H.; et al. Overexpression of p53 due to excess protein O-GlcNAcylation is associated with coronary microvascular disease in type 2 diabetes. Cardiovasc. Res. 2020, 116, 1186–1198. [Google Scholar] [CrossRef]
- Ramachandra, C.J.A.; Cong, S.; Chan, X.; Yap, E.P.; Yu, F.; Hausenloy, D.J. Oxidative stress in cardiac hypertrophy: From molecular mechanisms to novel therapeutic targets. Free Radic. Biol. Med. 2021, 166, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Okabe, K.; Matsushima, S.; Ikeda, S.; Ikeda, M.; Ishikita, A.; Tadokoro, T.; Enzan, N.; Yamamoto, T.; Sada, M.; Deguchi, H.; et al. DPP (Dipeptidyl Peptidase)-4 Inhibitor Attenuates Ang II (Angiotensin II)-Induced Cardiac Hypertrophy via GLP (Glucagon-Like Peptide)-1-Dependent Suppression of Nox (Nicotinamide Adenine Dinucleotide Phosphate Oxidase) 4-HDAC (Histone Deacetylase) 4 Pathway. Hypertension 2020, 75, 991–1001. [Google Scholar]
- Zhao, Q.D.; Viswanadhapalli, S.; Williams, P.; Shi, Q.; Tan, C.; Yi, X.; Bhandari, B.; Abboud, H.E. NADPH oxidase 4 induces cardiac fibrosis and hypertrophy through activating Akt/mTOR and NFkappaB signaling pathways. Circulation 2015, 131, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Westermann, D.; Rutschow, S.; Jäger, S.; Linderer, A.; Anker, S.; Riad, A.; Unger, T.; Schultheiss, H.P.; Pauschinger, M.; Tschöpe, C. Contributions of inflammation and cardiac matrix metalloproteinase activity to cardiac failure in diabetic cardiomyopathy: The role of angiotensin type 1 receptor antagonism. Diabetes 2007, 56, 641–646. [Google Scholar] [CrossRef]
- Jia, G.; Habibi, J.; DeMarco, V.G.; Martinez-Lemus, L.A.; Ma, L.; Whaley-Connell, A.T.; Aroor, A.R.; Domeier, T.L.; Zhu, Y.; Meininger, G.A.; et al. Endothelial Mineralocorticoid Receptor Deletion Prevents Diet-Induced Cardiac Diastolic Dysfunction in Females. Hypertension 2015, 66, 1159–1167. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol. Asp. Med. 2019, 65, 70–99. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.P.; Le, B.; Khode, R.; Baker, K.M.; Kumar, R. Intracellular angiotensin II production in diabetic rats is correlated with cardiomyocyte apoptosis, oxidative stress, and cardiac fibrosis. Diabetes 2008, 57, 3297–3306. [Google Scholar] [CrossRef] [PubMed]
- Bando, Y.K.; Murohara, T. Diabetes-related heart failure. Circ. J. 2014, 78, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Ziyadeh, F.N.; Sharma, K.; Ericksen, M.; Wolf, G. Stimulation of collagen gene expression and protein synthesis in murine mesangial cells by high glucose is mediated by autocrine activation of transforming growth factor-beta. J. Clin. Investig. 1994, 93, 536–542. [Google Scholar] [CrossRef]
- Singh, V.P.; Baker, K.M.; Kumar, R. Activation of the intracellular renin-angiotensin system in cardiac fibroblasts by high glucose: Role in extracellular matrix production. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1675–H1684. [Google Scholar] [CrossRef]
- Russo, I.; Frangogiannis, N.G. Diabetes-associated cardiac fibrosis: Cellular effectors, molecular mechanisms and therapeutic opportunities. J. Mol. Cell. Cardiol. 2016, 90, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Tuleta, I.; Frangogiannis, N.G. Diabetic fibrosis. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166044. [Google Scholar] [CrossRef] [PubMed]
- Boudina, S.; Abel, E.D. Diabetic cardiomyopathy, causes and effects. Rev. Endocr. Metab. Disord. 2010, 11, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Fiordaliso, F.; Li, B.; Latini, R.; Sonnenblick, E.H.; Anversa, P.; Leri, A.; Kajstura, J. Myocyte death in streptozotocin-induced diabetes in rats in angiotensin II—Dependent. Lab. Investig. 2000, 80, 513–527. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Zhao, F.; Kobayashi, T.; Hagiwara, M.; Kaminaris, A.; Li, C.; Cai, F.; Huang, Y.; Liang, Q. Hyperglycemia-induced cardiomyocyte death is mediated by lysosomal membrane injury and aberrant expression of cathepsin D. Biochem. Biophys. Res. Commun. 2020, 523, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Shi, X.; Zhang, B.; Liu, H.; Zhang, L.; Ding, W.; Zhao, Y. The imbalance of Th17/Th1/Tregs in patients with type 2 diabetes: Relationship with metabolic factors and complications. J. Mol. Med. 2012, 90, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Filardi, T.; Ghinassi, B.; Di Baldassarre, A.; Tanzilli, G.; Morano, S.; Lenzi, A.; Basili, S.; Crescioli, C. Cardiomyopathy Associated with Diabetes: The Central Role of the Cardiomyocyte. Int. J. Mol. Sci. 2019, 20, 3299. [Google Scholar] [CrossRef]
- Lappas, M.; Permezel, M.; Rice, G.E. Advanced glycation endproducts mediate pro-inflammatory actions in human gestational tissues via nuclear factor-kappaB and extracellular signal-regulated kinase 1/2. J. Endocrinol. 2007, 193, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Piek, A.; de Boer, R.A.; Sillje, H.H. The fibrosis-cell death axis in heart failure. Heart Fail. Rev. 2016, 21, 199–211. [Google Scholar] [CrossRef]
- Luo, M.; Anderson, M.E. Mechanisms of altered Ca2⁺ handling in heart failure. Circ. Res. 2013, 113, 690–708. [Google Scholar] [CrossRef]
- Dattani, A.; Singh, A.; McCann, G.P.; Gulsin, G.S. Myocardial Calcium Handling in Type 2 Diabetes: A Novel Therapeutic Target. J. Cardiovasc. Dev. Dis. 2023, 11, 12. [Google Scholar] [CrossRef] [PubMed]
- Stølen, T.O.; Høydal, M.A.; Kemi, O.J.; Catalucci, D.; Ceci, M.; Aasum, E.; Larsen, T.; Rolim, N.; Condorelli, G.; Smith, G.L.; et al. Interval training normalizes cardiomyocyte function, diastolic Ca2+ control, and SR Ca2+ release synchronicity in a mouse model of diabetic cardiomyopathy. Circ. Res. 2009, 105, 527–536. [Google Scholar] [CrossRef]
- Hegab, Z.; Mohamed, T.M.A.; Stafford, N.; Mamas, M.; Cartwright, E.J.; Oceandy, D. Advanced glycation end products reduce the calcium transient in cardiomyocytes by increasing production of reactive oxygen species and nitric oxide. FEBS Open Bio 2017, 7, 1672–1685. [Google Scholar] [CrossRef]
- Tang, L.; Wang, H.; Dai, B.; Wang, X.; Zhou, D.; Shen, J.; Guo, F.; Wang, J.; Zhou, J.; Wang, H.; et al. Human induced pluripotent stem cell-derived cardiomyocytes reveal abnormal TGFβ signaling in type 2 diabetes mellitus. J. Mol. Cell. Cardiol. 2020, 142, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Al Kury, L.T.; Sydorenko, V.; Smail, M.M.; Qureshi, M.A.; Shmygol, A.; Papandreou, D.; Singh, J.; Howarth, F.C. Calcium signaling in endocardial and epicardial ventricular myocytes from streptozotocin-induced diabetic rats. J. Diabetes Investig. 2021, 12, 493–500. [Google Scholar] [CrossRef]
- Ashrafi, R.; Modi, P.; Oo, A.Y.; Pullan, D.M.; Jian, K.; Zhang, H.; Gerges, J.Y.; Hart, G.; Boyett, M.R.; Davis, G.K.; et al. Arrhythmogenic gene remodelling in elderly patients with type 2 diabetes with aortic stenosis and normal left ventricular ejection fraction. Exp. Physiol. 2017, 102, 1424–1434. [Google Scholar] [CrossRef]
- Walker, C.A.; Spinale, F.G. The structure and function of the cardiac myocyte: A review of fundamental concepts. J. Thorac. Cardiovasc. Surg. 1999, 118, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Bidasee, K.R.; Nallani, K.; Yu, Y.; Cocklin, R.R.; Zhang, Y.; Wang, M.; Dincer, U.D.; Besch, H.R., Jr. Chronic diabetes increases advanced glycation end products on cardiac ryanodine receptors/calcium-release channels. Diabetes 2003, 52, 1825–1836. [Google Scholar] [CrossRef] [PubMed]
- Toyofuku, T.; Curotto Kurzydlowski, K.; Narayanan, N.; MacLennan, D.H. Identification of Ser38 as the site in cardiac sarcoplasmic reticulum Ca(2+)-ATPase that is phosphorylated by Ca2+/calmodulin-dependent protein kinase. J. Biol. Chem. 1994, 269, 26492–26496. [Google Scholar] [CrossRef]
- Bidasee, K.R.; Zhang, Y.; Shao, C.H.; Wang, M.; Patel, K.P.; Dincer, U.D.; Besch, H.R., Jr. Diabetes increases formation of advanced glycation end products on Sarco(endo)plasmic reticulum Ca2+-ATPase. Diabetes 2004, 53, 463–473. [Google Scholar] [CrossRef]
- Kwak, S.H.; Park, K.S. Recent progress in genetic and epigenetic research on type 2 diabetes. Exp. Mol. Med. 2016, 48, e220. [Google Scholar] [CrossRef] [PubMed]
- Ling, C.; Groop, L. Epigenetics: A molecular link between environmental factors and type 2 diabetes. Diabetes 2009, 58, 2718–2725. [Google Scholar] [CrossRef] [PubMed]
- Khullar, M.; Cheema, B.S.; Raut, S.K. Emerging Evidence of Epigenetic Modifications in Vascular Complication of Diabetes. Front. Endocrinol. 2017, 8, 237. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.B.; Khanna, S.; Raut, S.K.; Sharma, S.; Sharma, R.; Khullar, M. DUSP-1 gene expression is not regulated by promoter methylation in diabetes-associated cardiac hypertrophy. Ther. Adv. Cardiovasc. Dis. 2017, 11, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Suo, W.; Zhang, X.; Yang, Y.; Zhao, W.; Li, H.; Ni, Q. Targeting epigenetics in diabetic cardiomyopathy: Therapeutic potential of flavonoids. Biomed. Pharmacother. 2023, 157, 114025. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Cao, Y.; Qin, J.; Song, X.; Zhang, Q.; Shi, Y.; Cao, L. DNA methylation, its mediators and genome integrity. Int. J. Biol. Sci. 2015, 11, 604–617. [Google Scholar] [CrossRef] [PubMed]
- Tabaei, S.; Tabaee, S.S. DNA methylation abnormalities in atherosclerosis. Artif. Cells Nanomed. Biotechnol. 2019, 47, 2031–2041. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.L.; Krause, B.J.; Cataldo, L.R.; Vega, J.; Salas-Pérez, F.; Mennickent, P.; Gallegos, R.; Milagro, F.I.; Prieto-Hontoria, P.; Riezu-Boj, J.I.; et al. PPARGC1A Gene Promoter Methylation as a Biomarker of Insulin Secretion and Sensitivity in Response to Glucose Challenges. Nutrients 2020, 12, 2790. [Google Scholar] [CrossRef] [PubMed]
- Kao, Y.H.; Chen, Y.C.; Cheng, C.C.; Lee, T.I.; Chen, Y.J.; Chen, S.A. Tumor necrosis factor-alpha decreases sarcoplasmic reticulum Ca2+-ATPase expressions via the promoter methylation in cardiomyocytes. Crit. Care Med. 2010, 38, 217–222. [Google Scholar] [CrossRef]
- Bogdarina, I.; Welham, S.; King, P.J.; Burns, S.P.; Clark, A.J. Epigenetic modification of the renin-angiotensin system in the fetal programming of hypertension. Circ. Res. 2007, 100, 520–526. [Google Scholar] [CrossRef]
- Stillman, B. Histone Modifications: Insights into Their Influence on Gene Expression. Cell 2018, 175, 6–9. [Google Scholar] [CrossRef]
- Weeks, K.L. HDAC inhibitors and cardioprotection: Homing in on a mechanism of action. eBioMedicine 2019, 40, 21–22. [Google Scholar] [CrossRef]
- Reddy, M.A.; Sahar, S.; Villeneuve, L.M.; Lanting, L.; Natarajan, R. Role of Src tyrosine kinase in the atherogenic effects of the 12/15-lipoxygenase pathway in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 387–393. [Google Scholar] [CrossRef]
- Feng, B.; Chen, S.; Chiu, J.; George, B.; Chakrabarti, S. Regulation of cardiomyocyte hypertrophy in diabetes at the transcriptional level. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E1119–E1126. [Google Scholar] [CrossRef] [PubMed]
- Marzano, F.; Faienza, M.F.; Caratozzolo, M.F.; Brunetti, G.; Chiara, M.; Horner, D.S.; Annese, A.; D’Erchia, A.M.; Consiglio, A.; Pesole, G.; et al. Pilot study on circulating miRNA signature in children with obesity born small for gestational age and appropriate for gestational age. Pediatr. Obes. 2018, 13, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Costantino, S.; Paneni, F.; Lüscher, T.F.; Cosentino, F. MicroRNA profiling unveils hyperglycaemic memory in the diabetic heart. Eur. Heart J. 2016, 37, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Nair, S. Role of microRNA in diabetic cardiomyopathy: From mechanism to intervention. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2070–2077. [Google Scholar] [CrossRef]
- Yang, X.; Li, X.; Lin, Q.; Xu, Q. Up-regulation of microRNA-203 inhibits myocardial fibrosis and oxidative stress in mice with diabetic cardiomyopathy through the inhibition of PI3K/Akt signaling pathway via PIK3CA. Gene 2019, 715, 143995. [Google Scholar] [CrossRef]
- Palareti, G.; Legnani, C.; Cosmi, B.; Antonucci, E.; Erba, N.; Poli, D.; Testa, S.; Tosetto, A. Comparison between different D-Dimer cutoff values to assess the individual risk of recurrent venous thromboembolism: Analysis of results obtained in the DULCIS study. Int. J. Lab. Hematol. 2016, 38, 42–49. [Google Scholar] [CrossRef]
- Yu, M.; Liu, Y.; Zhang, B.; Shi, Y.; Cui, L.; Zhao, X. Inhibiting microRNA-144 abates oxidative stress and reduces apoptosis in hearts of streptozotocin-induced diabetic mice. Cardiovasc. Pathol. 2015, 24, 375–381. [Google Scholar] [CrossRef]
- Liang, C.; Gao, L.; Liu, Y.; Liu, Y.; Yao, R.; Li, Y.; Xiao, L.; Wu, L.; Du, B.; Huang, Z.; et al. MiR-451 antagonist protects against cardiac fibrosis in streptozotocin-induced diabetic mouse heart. Life Sci. 2019, 224, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Balducci, S.; Sacchetti, M.; Haxhi, J.; Orlando, G.; D’Errico, V.; Fallucca, S.; Menini, S.; Pugliese, G. Physical exercise as therapy for type 2 diabetes mellitus. Diabetes Metab. Res. Rev. 2014, 30 (Suppl. 1), 13–23. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Wang, H.; Zhu, X.; Huang, J.; Li, Y.; Zhou, K.; Hua, Y.; Yan, F.; Wang, D.Z.; Luo, Y. Adeno-associated virus-mediated delivery of anti-miR-199a tough decoys attenuates cardiac hypertrophy by targeting PGC-1alpha. Mol. Ther. Nucleic Acids 2021, 23, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.Y.; Song, Y.H.; Geng, Y.J.; Lin, Q.X.; Shan, Z.X.; Lin, S.G.; Li, Y. Glucose induces apoptosis of cardiomyocytes via microRNA-1 and IGF-1. Biochem. Biophys. Res. Commun. 2008, 376, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Fang, H.; Xu, Q.; Xu, C.; Yang, L.; Huang, C. LncRNA GAS5 inhibits NLRP3 inflammasome activation-mediated pyroptosis in diabetic cardiomyopathy by targeting miR-34b-3p/AHR. Cell Cycle 2020, 19, 3054–3065. [Google Scholar] [CrossRef] [PubMed]
- Raut, S.K.; Singh, G.B.; Rastogi, B.; Saikia, U.N.; Mittal, A.; Dogra, N.; Singh, S.; Prasad, R.; Khullar, M. miR-30c and miR-181a synergistically modulate p53-p21 pathway in diabetes induced cardiac hypertrophy. Mol. Cell. Biochem. 2016, 417, 191–203. [Google Scholar] [CrossRef]
- Zheng, D.; Ma, J.; Yu, Y.; Li, M.; Ni, R.; Wang, G.; Chen, R.; Li, J.; Fan, G.C.; Lacefield, J.C.; et al. Silencing of miR-195 reduces diabetic cardiomyopathy in C57BL/6 mice. Diabetologia 2015, 58, 1949–1958. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, Y.; Horie, T.; Baba, O.; Watanabe, S.; Nishiga, M.; Usami, S.; Izuhara, M.; Nakao, T.; Nishino, T.; Otsu, K.; et al. MicroRNA-451 exacerbates lipotoxicity in cardiac myocytes and high-fat diet-induced cardiac hypertrophy in mice through suppression of the LKB1/AMPK pathway. Circ. Res. 2015, 116, 279–288. [Google Scholar] [CrossRef]
- Magenta, A.; Greco, S.; Gaetano, C.; Martelli, F. Oxidative stress and microRNAs in vascular diseases. Int. J. Mol. Sci. 2013, 14, 17319–17346. [Google Scholar] [CrossRef]
- Chandrasekera, D.N.K.; Neale, J.P.H.; van Hout, I.; Rawal, S.; Coffey, S.; Jones, G.T.; Bunton, R.; Sugunesegran, R.; Parry, D.; Davis, P.; et al. Upregulation of microRNA-532 enhances cardiomyocyte apoptosis in the diabetic heart. Apoptosis 2020, 25, 388–399. [Google Scholar] [CrossRef]
- Huang, J.H.; Chen, Y.C.; Lee, T.I.; Kao, Y.H.; Chazo, T.F.; Chen, S.A.; Chen, Y.J. Glucagon-like peptide-1 regulates calcium homeostasis and electrophysiological activities of HL-1 cardiomyocytes. Peptides 2016, 78, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, M.; Abate, N.; Chandalia, M.; Rizvi, A.A.; Giglio, R.V.; Nikolic, D.; Marino Gammazza, A.; Barbagallo, I.; Isenovic, E.R.; Banach, M.; et al. Liraglutide reduces oxidative stress and restores heme oxygenase-1 and ghrelin levels in patients with type 2 diabetes: A prospective pilot study. J. Clin. Endocrinol. Metab. 2015, 100, 603–606. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.D.; Li, Y.G.; Wang, G.Y.; Bi, Y.G.; Zhao, Y.; Yan, M.L.; Liu, X.; Wei, M.; Wan, L.L.; Zhang, Q.Y. Metformin protects high glucose-cultured cardiomyocytes from oxidative stress by promoting NDUFA13 expression and mitochondrial biogenesis via the AMPK signaling pathway. Mol. Med. Rep. 2020, 22, 5262–5270. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Ye, P.; Liao, H.; Chen, M.; Yang, F. Metformin Protects H9C2 Cardiomyocytes from High-Glucose and Hypoxia/Reoxygenation Injury via Inhibition of Reactive Oxygen Species Generation and Inflammatory Responses: Role of AMPK and JNK. J. Diabetes Res. 2016, 2016, 2961954. [Google Scholar] [CrossRef] [PubMed]
- Bhansali, S.; Bhansali, A.; Dutta, P.; Walia, R.; Dhawan, V. Metformin upregulates mitophagy in patients with T2DM: A randomized placebo-controlled study. J. Cell. Mol. Med. 2020, 24, 2832–2846. [Google Scholar] [CrossRef] [PubMed]
- Dia, M.; Leon, C.; Chanon, S.; Bendridi, N.; Gomez, L.; Rieusset, J.; Thibault, H.; Paillard, M. Effect of Metformin on T2D-Induced MAM Ca(2+) Uncoupling and Contractile Dysfunction in an Early Mouse Model of Diabetic HFpEF. Int. J. Mol. Sci. 2022, 23, 3569. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Li, D.; Yu, J.; Jiang, W.; Liao, X.; Zhao, Q. Potential diabetic cardiomyopathy therapies targeting pyroptosis: A mini review. Front. Cardiovasc. Med. 2022, 9, 985020. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, M.S.; Pillai, K.K.; Hassan, M.Q.; Dhyani, N.; Ismail, M.V.; Najmi, A.K. Levosimendan reduces myocardial damage and improves cardiodynamics in streptozotocin induced diabetic cardiomyopathy via SERCA2a/NCX1 pathway. Life Sci. 2016, 153, 55–65. [Google Scholar] [CrossRef]
- Torre, E.; Arici, M.; Lodrini, A.M.; Ferrandi, M.; Barassi, P.; Hsu, S.C.; Chang, G.J.; Boz, E.; Sala, E.; Vagni, S.; et al. SERCA2a stimulation by istaroxime improves intracellular Ca2+ handling and diastolic dysfunction in a model of diabetic cardiomyopathy. Cardiovasc. Res. 2022, 118, 1020–1032. [Google Scholar] [CrossRef]
- Chen, Y.; Du, J.; Zhao, Y.T.; Zhang, L.; Lv, G.; Zhuang, S.; Qin, G.; Zhao, T.C. Histone deacetylase (HDAC) inhibition improves myocardial function and prevents cardiac remodeling in diabetic mice. Cardiovasc. Diabetol. 2015, 14, 99. [Google Scholar] [CrossRef]
- Xu, Z.; Tong, Q.; Zhang, Z.; Wang, S.; Zheng, Y.; Liu, Q.; Qian, L.B.; Chen, S.Y.; Sun, J.; Cai, L. Inhibition of HDAC3 prevents diabetic cardiomyopathy in OVE26 mice via epigenetic regulation of DUSP5-ERK1/2 pathway. Clin. Sci. 2017, 131, 1841–1857. [Google Scholar] [CrossRef] [PubMed]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Kober, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Belohlavek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Dhingra, N.K.; Butler, J.; Anker, S.D.; Ferreira, J.P.; Filippatos, G.; Januzzi, J.L.; Lam, C.S.P.; Sattar, N.; Peil, B.; et al. Empagliflozin in the treatment of heart failure with reduced ejection fraction in addition to background therapies and therapeutic combinations (EMPEROR-Reduced): A post-hoc analysis of a randomised, double-blind trial. Lancet Diabetes Endocrinol. 2022, 10, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.; Butler, J.; Zannad, F.; Filippatos, G.; Ferreira, J.P.; Pocock, S.J.; Carson, P.; Anand, I.; Doehner, W.; Haass, M.; et al. Effect of Empagliflozin on Worsening Heart Failure Events in Patients with Heart Failure and Preserved Ejection Fraction: EMPEROR-Preserved Trial. Circulation 2021, 144, 1284–1294. [Google Scholar] [CrossRef] [PubMed]
- Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Bocchi, E.; Bohm, M.; Brunner-La Rocca, H.P.; Choi, D.J.; Chopra, V.; Chuquiure-Valenzuela, E.; et al. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N. Engl. J. Med. 2021, 385, 1451–1461. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhang, J.; Xue, M.; Li, X.; Han, F.; Liu, X.; Xu, L.; Lu, Y.; Cheng, Y.; Li, T.; et al. SGLT2 inhibition with empagliflozin attenuates myocardial oxidative stress and fibrosis in diabetic mice heart. Cardiovasc. Diabetol. 2019, 18, 15. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Bajaj, M.; Yang, H.C.; Perez-Polo, J.R.; Birnbaum, Y. SGLT-2 Inhibition with Dapagliflozin Reduces the Activation of the Nlrp3/ASC Inflammasome and Attenuates the Development of Diabetic Cardiomyopathy in Mice with Type 2 Diabetes. Further Augmentation of the Effects with Saxagliptin, a DPP4 Inhibitor. Cardiovasc. Drugs Ther. 2017, 31, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Tahara, A.; Kurosaki, E.; Yokono, M.; Yamajuku, D.; Kihara, R.; Hayashizaki, Y.; Takasu, T.; Imamura, M.; Li, Q.; Tomiyama, H.; et al. Effects of sodium-glucose cotransporter 2 selective inhibitor ipragliflozin on hyperglycaemia, oxidative stress, inflammation and liver injury in streptozotocin-induced type 1 diabetic rats. J. Pharm. Pharmacol. 2014, 66, 975–987. [Google Scholar] [CrossRef]
- Lee, T.I.; Chen, Y.C.; Lin, Y.K.; Chung, C.C.; Lu, Y.Y.; Kao, Y.H.; Chen, Y.J. Empagliflozin Attenuates Myocardial Sodium and Calcium Dysregulation and Reverses Cardiac Remodeling in Streptozotocin-Induced Diabetic Rats. Int. J. Mol. Sci. 2019, 20, 1680. [Google Scholar] [CrossRef]
- Cortese, F.; Scicchitano, P.; Cortese, A.M.; Meliota, G.; Andriani, A.; Truncellito, L.; Calculli, G.; Giordano, P.; Ciccone, M.M. Uric Acid in Metabolic and Cerebrovascular Disorders: A Review. Curr. Vasc. Pharmacol. 2020, 18, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Cortese, F.; Giordano, P.; Scicchitano, P.; Faienza, M.F.; De Pergola, G.; Calculli, G.; Meliota, G.; Ciccone, M.M. Uric acid: From a biological advantage to a potential danger. A focus on cardiovascular effects. Vasc. Pharmacol. 2019, 120, 106565. [Google Scholar] [CrossRef] [PubMed]
- Hatem, S.N.; Sanders, P. Epicardial adipose tissue and atrial fibrillation. Cardiovasc. Res. 2014, 102, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Dhingra, N.K.; Mistry, N.; Puar, P.; Verma, R.; Anker, S.; Mazer, C.D.; Verma, S. SGLT2 inhibitors and cardiac remodelling: A systematic review and meta-analysis of randomized cardiac magnetic resonance imaging trials. ESC Heart Fail. 2021, 8, 4693–4700. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.M.; Chang, N.C.; Lin, S.Z. Dapagliflozin, a selective SGLT2 Inhibitor, attenuated cardiac fibrosis by regulating the macrophage polarization via STAT3 signaling in infarcted rat hearts. Free Radic. Biol. Med. 2017, 104, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Quagliariello, V.; De Laurentiis, M.; Rea, D.; Barbieri, A.; Monti, M.G.; Carbone, A.; Paccone, A.; Altucci, L.; Conte, M.; Canale, M.L.; et al. The SGLT-2 inhibitor empagliflozin improves myocardial strain, reduces cardiac fibrosis and pro-inflammatory cytokines in non-diabetic mice treated with doxorubicin. Cardiovasc. Diabetol. 2021, 20, 150. [Google Scholar] [CrossRef]
- Kang, S.; Verma, S.; Hassanabad, A.F.; Teng, G.; Belke, D.D.; Dundas, J.A.; Guzzardi, D.G.; Svystonyuk, D.A.; Pattar, S.S.; Park, D.S.J.; et al. Direct Effects of Empagliflozin on Extracellular Matrix Remodelling in Human Cardiac Myofibroblasts: Novel Translational Clues to Explain EMPA-REG OUTCOME Results. Can. J. Cardiol. 2020, 36, 543–553. [Google Scholar] [CrossRef]
- Ferrannini, E.; Mark, M.; Mayoux, E. CV Protection in the EMPA-REG OUTCOME Trial: A “Thrifty Substrate” Hypothesis. Diabetes Care 2016, 39, 1108–1114. [Google Scholar] [CrossRef]
- Janardhan, A.; Chen, J.; Crawford, P.A. Altered systemic ketone body metabolism in advanced heart failure. Tex. Heart Inst. J. 2011, 38, 533–538. [Google Scholar]
- Mudaliar, S.; Alloju, S.; Henry, R.R. Can a Shift in Fuel Energetics Explain the Beneficial Cardiorenal Outcomes in the EMPA-REG OUTCOME Study? A Unifying Hypothesis. Diabetes Care 2016, 39, 1115–1122. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Verma, S. Empagliflozin’s Fuel Hypothesis: Not so Soon. Cell Metab. 2016, 24, 200–202. [Google Scholar] [CrossRef] [PubMed]
- Ucar, A.; Gupta, S.K.; Fiedler, J.; Erikci, E.; Kardasinski, M.; Batkai, S.; Dangwal, S.; Kumarswamy, R.; Bang, C.; Holzmann, A.; et al. The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat. Commun. 2012, 3, 1078. [Google Scholar] [CrossRef] [PubMed]
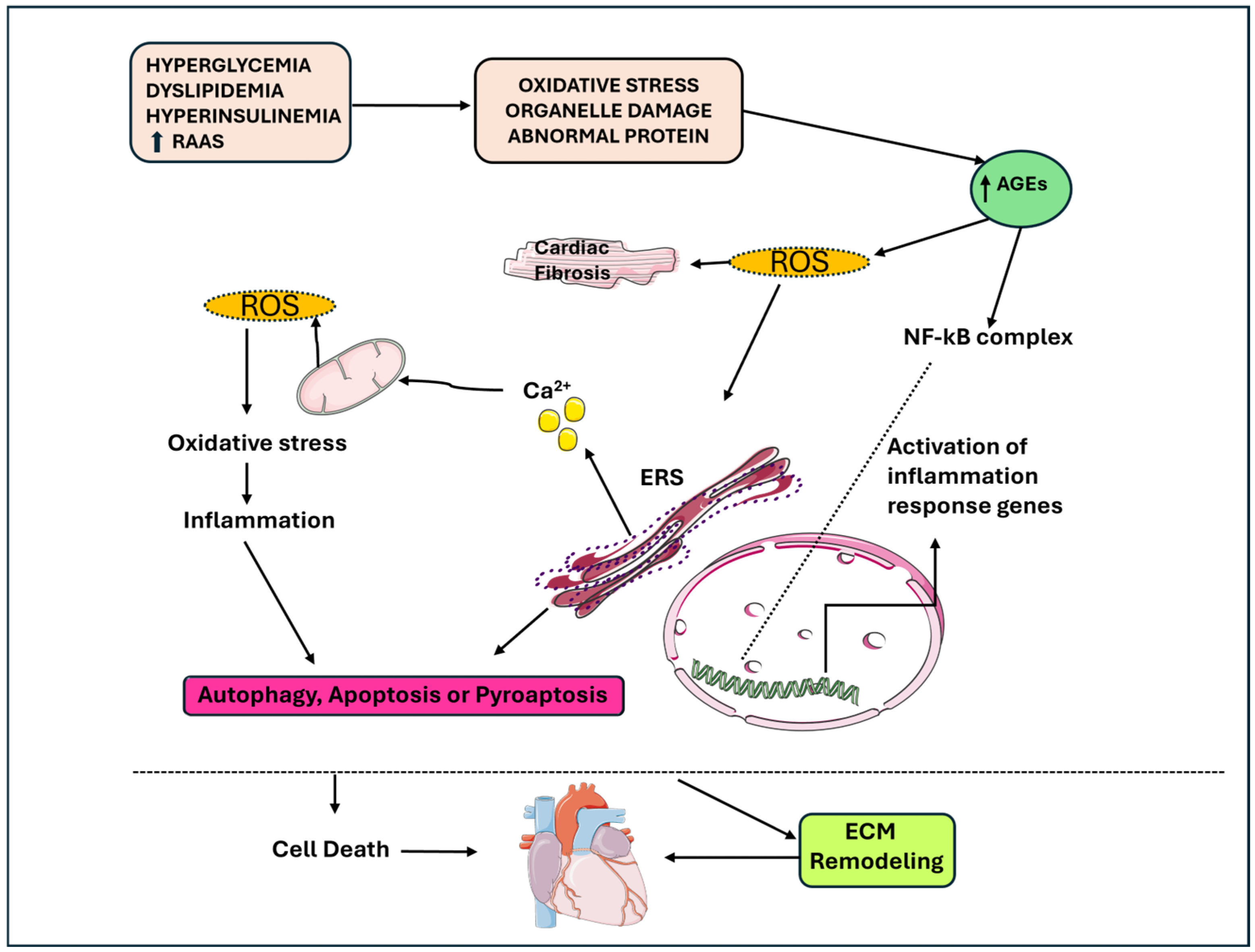
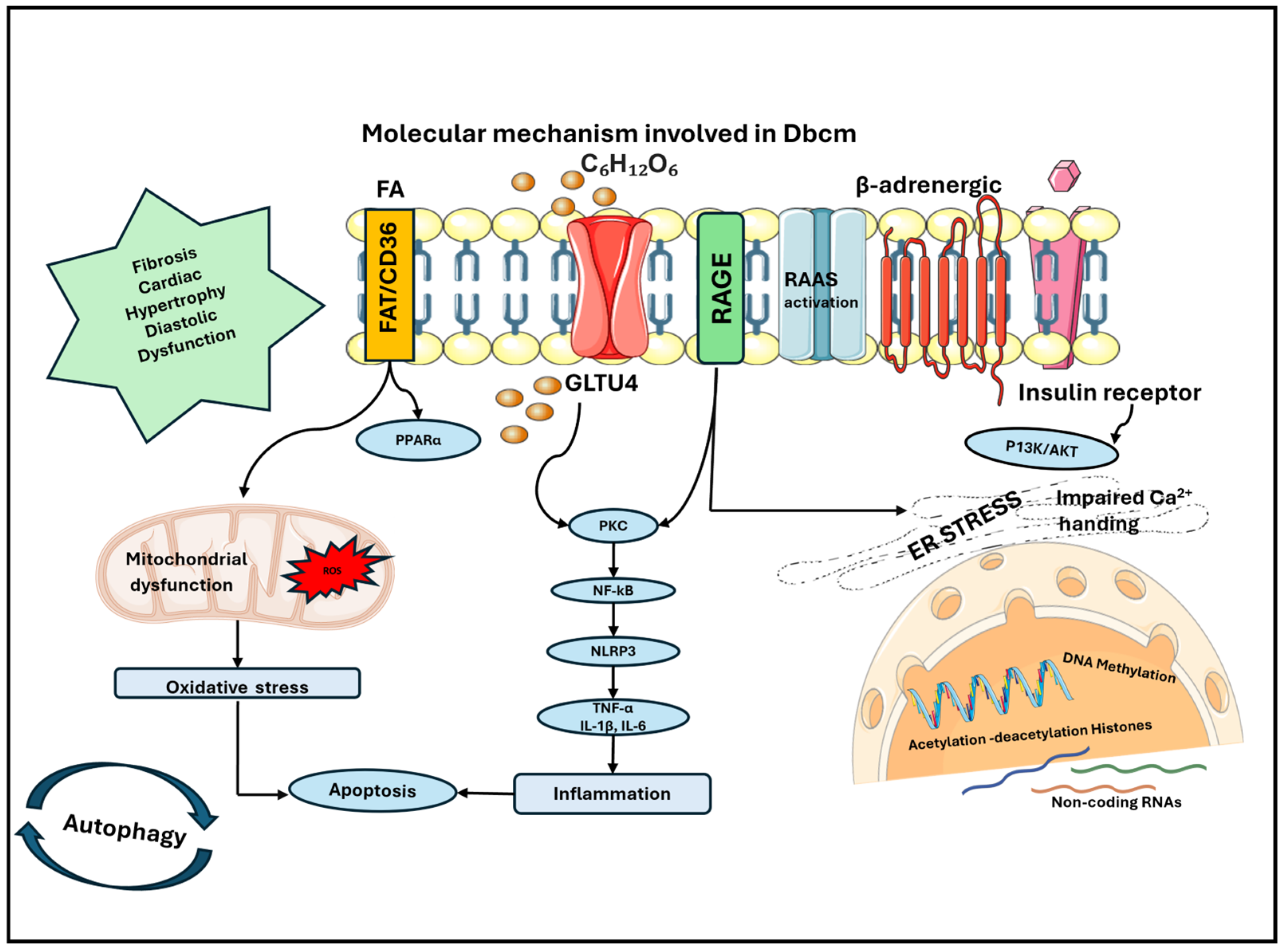
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).