
JAK Inhibitors in Rheumatoid Arthritis: Immunomodulatory Properties and Clinical Efficacy
 , ,
, ,
Abstract
:1. Introduction
2. Brief Overview of the JAK/STAT Pathway
3. Immunomodulatory Properties of JAK Inhibitors and Preclinical Efficacy
3.1. Fibroblast-Like Synoviocytes
3.2. Macrophages
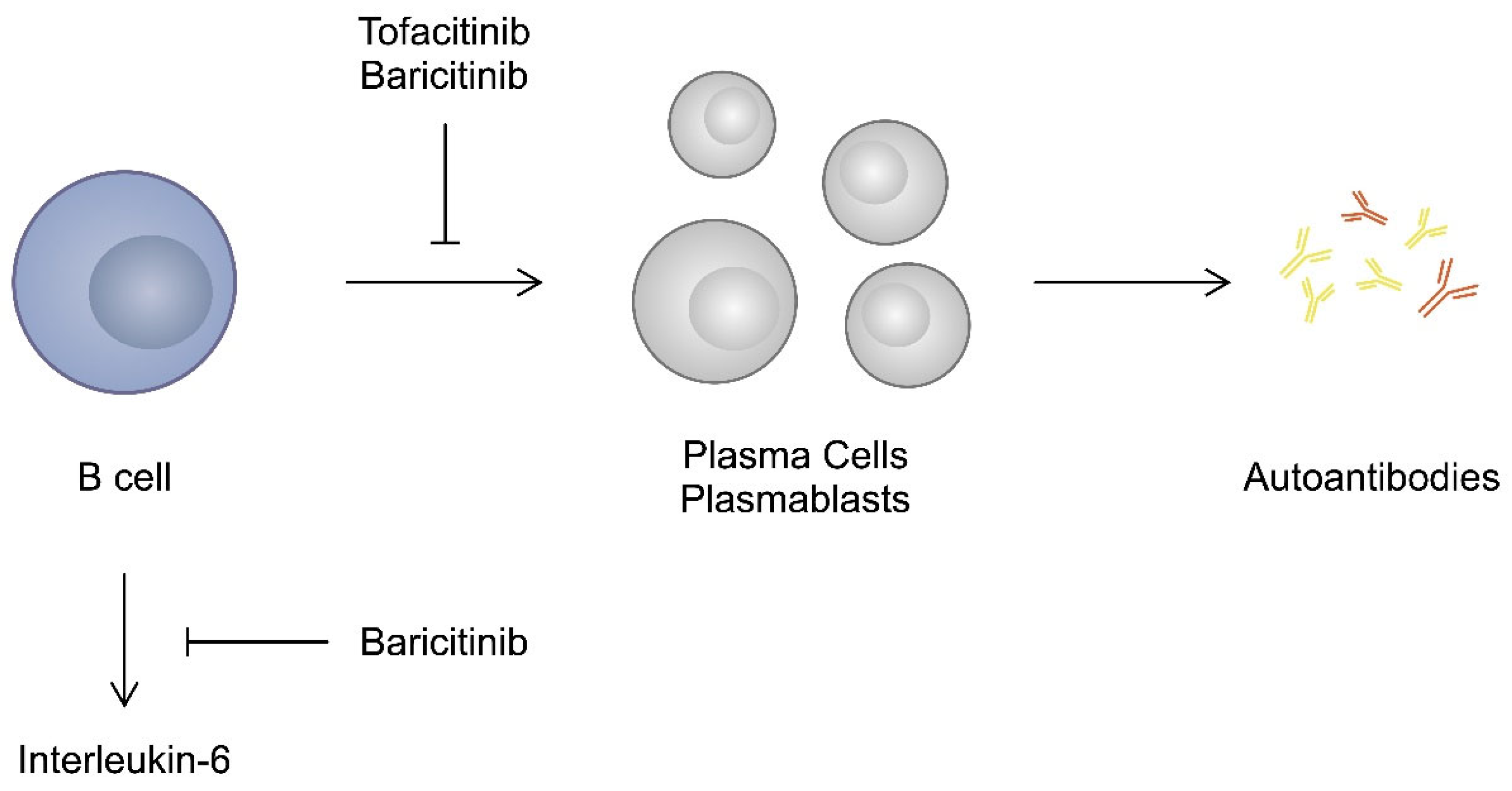
3.3. T and B Cells
3.4. Animal Studies
4. Clinical Studies
Tofacitinib
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gao, Y.; Zhang, Y.; Liu, X. Rheumatoid arthritis: Pathogenesis and therapeutic advances. MedComm 2024, 5, e509. [Google Scholar] [CrossRef] [PubMed]
- Collaborators GRA. Global, regional, and national burden of rheumatoid arthritis, 1990–2020, and projections to 2050: A systematic analysis of the Global Burden of Disease Study 2021. Lancet Rheumatol. 2023, 5, e594–e610. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.Y.; Yang, H.Y.; Luo, S.F.; Lai, J.H. From Rheumatoid Factor to Anti-Citrullinated Protein Antibodies and Anti-Carbamylated Protein Antibodies for Diagnosis and Prognosis Prediction in Patients with Rheumatoid Arthritis. Int. J. Mol. Sci. 2021, 22, 686. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wu, B.; Mo, L.; Chen, H.; Zhao, Y.; Tan, T.; Chen, L.; Li, Y.; Yao, P.; Tang, Y. Associations between biological ageing and the risk of, genetic susceptibility to, and life expectancy associated with rheumatoid arthritis: A secondary analysis of two observational studies. Lancet Healthy Longev. 2024, 5, e45–e55. [Google Scholar] [CrossRef] [PubMed]
- Imanuel, C.A.; Sivatheesan, S.; Koyanagi, A.; Smith, L.; Konrad, M.; Kostev, K. Associations between Rheumatoid Arthritis and Various Comorbid Conditions in Germany—A Retrospective Cohort Study. J. Clin. Med. 2023, 12, 7265. [Google Scholar] [CrossRef]
- Patel, A.; Varsha, B.; Manaswini, E. A Study of Haematological Profile in Newly Diagnosed Rheumatoid Arthritis and its Correlation with Disease Activity. J. Assoc. Physicians India 2022, 70, 11–12. [Google Scholar] [PubMed]
- Dar, W.R.; Mir, I.A.; Siddiq, S.; Nadeem, M.; Singh, G. The Assessment of Fatigue in Rheumatoid Arthritis Patients and Its Impact on Their Quality of Life. Clin. Pract. 2022, 12, 591–598. [Google Scholar] [CrossRef] [PubMed]
- McBeth, J.; Dixon, W.G.; Moore, S.M.; Hellman, B.; James, B.; Kyle, S.D.; Lunt, M.; Cordingley, L.; Yimer, B.B.; Druce, K.L. Sleep Disturbance and Quality of Life in Rheumatoid Arthritis: Prospective mHealth Study. J. Med. Internet Res. 2022, 24, e32825. [Google Scholar] [CrossRef] [PubMed]
- Kesharwani, D.; Paliwal, R.; Satapathy, T.; Das Paul, S. Rheumatiod Arthritis: An Updated Overview of Latest Therapy and Drug Delivery. J. Pharmacopunct. 2019, 22, 210–224. [Google Scholar] [CrossRef]
- Lane, J.C.E.; Weaver, J.; Kostka, K.; Duarte-Salles, T.; Abrahao, M.T.F.; Alghoul, H.; Alser, O.; Alshammari, T.M.; Biedermann, P.; Banda, J.M.; et al. Risk of hydroxychloroquine alone and in combination with azithromycin in the treatment of rheumatoid arthritis: A multinational, retrospective study. Lancet Rheumatol. 2020, 2, e698–e711. [Google Scholar] [CrossRef]
- Ozen, G.; Pedro, S.; Michaud, K. The Risk of Cardiovascular Events Associated with Disease-modifying Antirheumatic Drugs in Rheumatoid Arthritis. J. Rheumatol. 2021, 48, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Bergmans, B.; Jessurun, N.; van Lint, J.; Murk, J.L.; van Puijenbroek, E.; de Vries, E. Burden of non-serious infections during biological use for rheumatoid arthritis. PLoS ONE 2024, 19, e0296821. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Yang, X.; Huang, H.; Gao, D.; Ji, L.; Zhang, Z. Risk of malignancy with non-TNFi biologic or tofacitinib therapy in rheumatoid arthritis: A meta-analysis of observational studies. Semin. Arthritis Rheum. 2020, 50, 930–937. [Google Scholar] [CrossRef] [PubMed]
- Ben Mrid, R.; Bouchmaa, N.; Ainani, H.; El Fatimy, R.; Malka, G.; Mazini, L. Anti-rheumatoid drugs advancements: New insights into the molecular treatment of rheumatoid arthritis. Biomed. Pharmacother. 2022, 151, 113126. [Google Scholar] [CrossRef] [PubMed]
- Bonelli, M.; Kerschbaumer, A.; Kastrati, K.; Ghoreschi, K.; Gadina, M.; Heinz, L.X.; Smolen, J.S.; Aletaha, D.; O’Shea, J.; Laurence, A. Selectivity, efficacy and safety of JAKinibs: New evidence for a still evolving story. Ann. Rheum. Dis. 2024, 83, 139–160. [Google Scholar] [CrossRef]
- Vyas, D.; O’Dell, K.M.; Bandy, J.L.; Boyce, E.G. Tofacitinib: The First Janus Kinase (JAK) inhibitor for the treatment of rheumatoid arthritis. Ann. Pharmacother. 2013, 47, 1524–1531. [Google Scholar] [CrossRef] [PubMed]
- Urits, I.; Israel, J.; Hakobyan, H.; Yusin, G.; Lassiter, G.; Fackler, N.; Berger, A.A.; Kassem, H.; Kaye, A.; Viswanath, O. Baricitinib for the treatment of rheumatoid arthritis. Rheumatology 2020, 58, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Sanmartí, R.; Corominas, H. Upadacitinib for Patients with Rheumatoid Arthritis: A Comprehensive Review. J. Clin. Med. 2023, 12, 1734. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Molina, C.; Feliu, A.; Park, H.S.; Juanes, A.; Diaz-Torne, C.; Vidal, S.; Corominas, H. Are There Sex-Related Differences in the Effectiveness of Janus Kinase Inhibitors in Rheumatoid Arthritis Patients? J. Clin. Med. 2024, 13, 2355. [Google Scholar] [CrossRef]
- Sugahara, S.; Hanaoka, K.; Emori, T.; Takeshita, N.; Fujii, Y.; Nakano, M.; Suzuki, T.; Takahashi, J.; Nakamura, Y. Peficitinib improves bone fragility by recovering bone turnover imbalance in arthritic mice. J. Pharmacol. Sci. 2022, 148, 134–141. [Google Scholar] [CrossRef]
- Hu, Q.; Bian, Q.; Rong, D.; Wang, L.; Song, J.; Huang, H.-S.; Zeng, J.; Mei, J.; Wang, P.-Y. JAK/STAT pathway: Extracellular signals, diseases, immunity, and therapeutic regimens. Front. Bioeng. Biotechnol. 2023, 11, 1110765. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef]
- Sarapultsev, A.; Gusev, E.; Komelkova, M.; Utepova, I.; Luo, S.; Hu, D. JAK-STAT signaling in inflammation and stress-related diseases: Implications for therapeutic interventions. Mol. Biomed. 2023, 4, 40. [Google Scholar] [CrossRef]
- Barcia Durán, J.G.; Lu, T.; Houghton, S.; Geng, F.; Schreiner, R.; Xiang, J.; Rafii, S.; Redmond, D.; Lis, R. Endothelial Jak3 expression enhances pro-hematopoietic angiocrine function in mice. Commun. Biol. 2021, 4, 406. [Google Scholar] [CrossRef] [PubMed]
- Rusiñol, L.; Puig, L. Tyk2 Targeting in Immune-Mediated Inflammatory Diseases. Int. J. Mol. Sci. 2023, 24, 3391. [Google Scholar] [CrossRef] [PubMed]
- Valle-Mendiola, A.; Gutiérrez-Hoya, A.; Soto-Cruz, I. JAK/STAT Signaling and Cervical Cancer: From the Cell Surface to the Nucleus. Genes 2023, 14, 1141. [Google Scholar] [CrossRef]
- Rengachari, S.; Groiss, S.; Devos, J.M.; Caron, E.; Grandvaux, N.; Panne, D. Structural basis of STAT2 recognition by IRF9 reveals molecular insights into ISGF3 function. Proc. Natl. Acad. Sci. USA 2018, 115, E601–E609. [Google Scholar] [CrossRef]
- Emori, T.; Kasahara, M.; Sugahara, S.; Hashimoto, M.; Ito, H.; Narumiya, S.; Higashi, Y.; Fujii, Y. Role of JAK-STAT signaling in the pathogenic behavior of fibroblast-like synoviocytes in rheumatoid arthritis: Effect of the novel JAK inhibitor peficitinib. Eur. J. Pharmacol. 2020, 882, 173238. [Google Scholar] [CrossRef]
- Tóthová, Z.; Tomc, J.; Debeljak, N.; Solár, P. STAT5 as a Key Protein of Erythropoietin Signalization. Int. J. Mol. Sci. 2021, 22, 7109. [Google Scholar] [CrossRef]
- Able, A.A.; Burrell, J.A.; Stephens, J.M. STAT5-Interacting Proteins: A Synopsis of Proteins that Regulate STAT5 Activity. Biology 2017, 6, 20. [Google Scholar] [CrossRef]
- Morris, R.; Kershaw, N.J.; Babon, J.J. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 2018, 27, 1984–2009. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Yao, Q.; Gu, X.; Shi, Q.; Yuan, X.; Chu, Q.; Bao, Z.; Lu, J.; Li, L. Evolving cognition of the JAK-STAT signaling pathway: Autoimmune disorders and cancer. Signal Transduct. Target. Ther. 2023, 8, 204. [Google Scholar] [CrossRef] [PubMed]
- Seif, F.; Khoshmirsafa, M.; Aazami, H.; Mohsenzadegan, M.; Sedighi, G.; Bahar, M. The role of JAK-STAT signaling pathway and its regulators in the fate of T helper cells. Cell Commun. Signal. 2017, 15, 23. [Google Scholar] [CrossRef] [PubMed]
- Migita, K.; Komori, A.; Torigoshi, T.; Maeda, Y.; Izumi, Y.; Jiuchi, Y.; Miyashita, T.; Nakamura, M.; Motokawa, S.; Ishibashi, H. CP690,550 inhibits oncostatin M-induced JAK/STAT signaling pathway in rheumatoid synoviocytes. Arthritis Res. Ther. 2011, 13, R72. [Google Scholar] [CrossRef] [PubMed]
- Dreo, B.; Muralikrishnan, A.S.; Husic, R.; Lackner, A.; Brügmann, T.; Haudum, P.; Bosch, P.; Thiel, J.; Fessler, J.; Stradner, M. JAK/STAT signaling in rheumatoid arthritis leukocytes is uncoupled from serum cytokines in a subset of patients. Clin. Immunol. 2024, 264, 110238. [Google Scholar] [CrossRef] [PubMed]
- Pizzo, M.L.; La Barbera, L.; Rizzo, C.; Mohammadnezhad, L.; Camarda, F.; Ciccia, F.; Guggino, G. JAK/STAT inhibition modifies the ILC1 immune response in patients with rheumatoid arthritis. Clin. Exp. Rheumatol. 2024, 42, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Jose, A.M.; Samarpita, S.; Panchal, N.K.; Sabina, E.P.; Rasool, M. Selective blockade of IL-21 by myricetin impedes T follicular helper cell differentiation by negatively regulating the JAK/STAT/Bcl-6 pathway in a rheumatoid arthritis animal model. 3 Biotech 2024, 14, 25. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Wang, D.; Wu, J.; Zhang, J. JAK2 inhibitors improve RA combined with pulmonary fibrosis in rats by downregulating SMAD3 phosphorylation. Int. J. Rheum. Dis. 2024, 27, e15164. [Google Scholar] [CrossRef] [PubMed]
- Golumba-Nagy, V.; Yan, S.; Steinbach-Knödgen, E.; Thiele, J.; Esser, R.L.; Haak, T.H.; Nikiforov, A.; Meyer, A.; Seeger-Nukpezah, T.; Kofler, D.M. Treatment of rheumatoid arthritis with baricitinib or upadacitinib is associated with reduced scaffold protein NEDD9 levels in CD4+ T cells. Physiol. Rep. 2023, 11, e15829. [Google Scholar] [CrossRef]
- Tawfeik, A.M.; El-Dydamoni, O.A.; Maghraby, H.M.; Elshohat, E.; Seliem, N.; Kassem, E.A.; Anani, H.A. The Relationship between FoxP3 and SOCs3 Gene Expressions and Disease Activity in Rheumatoid Arthritis. Curr. Rheumatol. Rev. 2023, 19, 76–82. [Google Scholar]
- Di Benedetto, P.; Ruscitti, P.; Berardicurti, O.; Panzera, N.; Grazia, N.; Nolfi, M.D.V.; Di Francesco, B.; Navarini, L.; Maurizi, A.; Rucci, N.; et al. Blocking Jak/STAT signalling using tofacitinib inhibits angiogenesis in experimental arthritis. Arthritis Res. Ther. 2021, 23, 213. [Google Scholar] [CrossRef] [PubMed]
- You, S.; Koh, J.H.; Leng, L.; Kim, W.; Bucala, R. Review: The Tumor-Like Phenotype of Rheumatoid Synovium: Molecular Profiling and Prospects for Precision Medicine. Arthritis Rheumatol. 2018, 70, 637–652. [Google Scholar] [CrossRef] [PubMed]
- Najm, A.; Masson, F.M.; Preuss, P.; Georges, S.; Ory, B.; Quillard, T.; Sood, S.; Goodyear, C.S.; Veale, D.J.; Fearon, U.; et al. MicroRNA-17-5p Reduces Inflammation and Bone Erosions in Mice with Collagen-Induced Arthritis and Directly Targets the JAK/STAT Pathway in Rheumatoid Arthritis Fibroblast-like Synoviocytes. Arthritis Rheumatol. 2020, 72, 2030–2039. [Google Scholar] [CrossRef]
- Yamaoka, K.; Tanaka, Y. Targeting the Janus kinases in rheumatoid arthritis: Focus on tofacitinib. Expert Opin. Pharmacother. 2014, 15, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Yan, J.; Li, T.; Lin, W.; Huang, Y.; Shen, P.; Ba, X.; Huang, Y.; Qin, K.; Geng, Y.; et al. Multifaceted oncostatin M: Novel roles and therapeutic potential of the oncostatin M signaling in rheumatoid arthritis. Front. Immunol. 2023, 14, 1258765. [Google Scholar] [CrossRef] [PubMed]
- Rosengren, S.; Corr, M.; Firestein, G.S.; Boyle, D.L. The JAK inhibitor CP-690,550 (tofacitinib) inhibits TNF-induced chemokine expression in fibroblast-like synoviocytes: Autocrine role of type I interferon. Ann. Rheum. Dis. 2012, 71, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.-J.; Han, S.-H.; Kim, D.-S.; Lee, G.-H.; Yoo, W.-H.; Kang, Y.-M.; Choi, J.-Y.; Lee, Y.C.; Park, S.J.; Jeong, S.-K.; et al. Autophagy induction and CHOP under-expression promotes survival of fibroblasts from rheumatoid arthritis patients under endoplasmic reticulum stress. Arthritis Res. Ther. 2010, 12, R19. [Google Scholar] [CrossRef] [PubMed]
- Vomero, M.; Caliste, M.; Barbati, C.; Speziali, M.; Celia, A.I.; Ucci, F.; Ciancarella, C.; Putro, E.; Colasanti, T.; Buoncuore, G.; et al. Tofacitinib Decreases Autophagy of Fibroblast-like Synoviocytes from Rheumatoid Arthritis Patients. Front. Pharmacol. 2022, 13, 852802. [Google Scholar] [CrossRef] [PubMed]
- Silvagni, E.; Missiroli, S.; Patergnani, S.; Boncompagni, C.; D’Ugo, C.; Garaffoni, C.; Ciliento, M.S.; Lanza, G.; Bonora, M.; Gafà, R.; et al. Tofacitinib restores psoriatic arthritis fibroblast-like synoviocytes function via autophagy and mitochondrial quality control modulation. J. Autoimmun. 2024, 143, 103159. [Google Scholar] [CrossRef]
- Ruscitti, P.; Liakouli, V.; Panzera, N.; Angelucci, A.; Berardicurti, O.; Di Nino, E.; Navarini, L.; Vomero, M.; Ursini, F.; Mauro, D.; et al. Tofacitinib May Inhibit Myofibroblast Differentiation from Rheumatoid-Fibroblast-like Synoviocytes Induced by TGF-β and IL-6. Pharmaceuticals 2022, 15, 622. [Google Scholar] [CrossRef]
- Chen, J.; Che, Q.; Kou, Y.; Rong, X.; Zhang, X.; Li, M.; Shu, Q. A novel drug combination of Tofacitinib and Iguratimod alleviates rheumatoid arthritis and secondary osteoporosis. Int. Immunopharmacol. 2023, 124 Pt B, 110913. [Google Scholar] [CrossRef]
- Hammaker, D.; Nygaard, G.; Kuhs, A.; Ai, R.; Boyle, D.L.; Wang, W.; Firestein, G.S. Joint Location-Specific JAK-STAT Signaling in Rheumatoid Arthritis Fibroblast-like Synoviocytes. ACR Open Rheumatol. 2019, 1, 640–648. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, L.; Malvaso, D.; Chiricozzi, A.; Tambone, S.; D’Urso, D.F.; Guerriero, C.; Peris, K. Baricitinib: Therapeutic potential for moderate to severe atopic dermatitis. Expert Opin. Investig. Drugs 2020, 29, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Weston, S.; Macdonald, J.L.; Williams, L.M.; Roussou, E.; Kang, N.V.; Kiriakidis, S.; Taylor, P.C. The JAK inhibitor baricitinib inhibits oncostatin M induction of proinflammatory mediators in ex-vivo synovial derived cells. Clin. Exp. Rheumatol. 2022, 40, 1620–1628. [Google Scholar] [CrossRef] [PubMed]
- Joyo, Y.; Kawaguchi, Y.; Yonezu, H.; Senda, H.; Yasuma, S.; Shiraga, H.; Nozaki, M.; Aoyama, M.; Asai, K.; Murakami, H.; et al. The Janus kinase inhibitor (baricitinib) suppresses the rheumatoid arthritis active marker gliostatin/thymidine phosphorylase in human fibroblast-like synoviocytes. Immunol. Res. 2022, 70, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Karonitsch, T.; Beckmann, D.; Dalwigk, K.; Niederreiter, B.; Studenic, P.; Byrne, R.A.; Holinka, J.; Sevelda, F.; Korb-Pap, A.; Steiner, G.; et al. Targeted inhibition of Janus kinases abates interfon gamma-induced invasive behaviour of fibroblast-like synoviocytes. Rheumatology 2018, 57, 572–577. [Google Scholar] [CrossRef] [PubMed]
- Bonelli, M.; Dalwigk, K.; Platzer, A.; Olmos Calvo, I.; Hayer, S.; Niederreiter, B.; Holinka, J.; Sevelda, F.; Pap, T.; Steiner, G.; et al. IRF1 is critical for the TNF-driven interferon response in rheumatoid fibroblast-like synoviocytes: JAKinibs suppress the interferon response in RA-FLSs. Exp. Mol. Med. 2019, 51, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Izutsu, H. Peficitinib for the treatment of rheumatoid arthritis: An overview from clinical trials. Expert Opin. Pharmacother. 2020, 21, 1015–1025. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.-H.; Lu, Y.-H.; Wu, L.-F.; Lu, X.; Guo, W.; Deng, F.-Y.; Lei, S.-F. Evaluation of plasma cytokine protein array profile: The highlighted PDGF-BB in rheumatoid arthritis. Clin. Rheumatol. 2020, 39, 3323–3330. [Google Scholar] [CrossRef]
- Ishikawa, G.; Kwon, C.; Fujii, Y. Peficitinib inhibits fibroblast-like synoviocyte activation and angiogenic vascular endothelial tube formation via inhibitory effects on PDGF and VEGF signaling in addition to JAK. J. Pharmacol. Sci. 2022, 150, 74–80. [Google Scholar] [CrossRef]
- Marrelli, A.; Cipriani, P.; Liakouli, V.; Carubbi, F.; Perricone, C.; Perricone, R.; Giacomelli, R. Angiogenesis in rheumatoid arthritis: A disease specific process or a common response to chronic inflammation? Autoimmun. Rev. 2011, 10, 595–598. [Google Scholar] [CrossRef] [PubMed]
- Ikari, Y.; Isozaki, T.; Tsubokura, Y.; Kasama, T. Peficitinib Inhibits the Chemotactic Activity of Monocytes via Proinflammatory Cytokine Production in Rheumatoid Arthritis Fibroblast-Like Synoviocytes. Cells 2019, 8, 561. [Google Scholar] [CrossRef] [PubMed]
- Diller, M.; Hasseli, R.; Hülser, M.L.; Aykara, I.; Frommer, K.; Rehart, S.; Müller-Ladner, U.; Neumann, E. Targeting Activated Synovial Fibroblasts in Rheumatoid Arthritis by Peficitinib. Front. Immunol. 2019, 10, 541. [Google Scholar] [CrossRef]
- Srivastava, S.; Samarpita, S.; Ganesan, R.; Rasool, M. CYT387 Inhibits the Hyperproliferative Potential of Fibroblast-like Synoviocytes via Modulation of IL-6/JAK1/STAT3 Signaling in Rheumatoid Arthritis. Immunol. Investig. 2022, 51, 1582–1597. [Google Scholar] [CrossRef]
- Song, Y.; Gao, N.; Yang, Z.; Zhang, L.; Wang, Y.; Zhang, S.; Fan, T. Characteristics, polarization and targeted therapy of mononuclear macrophages in rheumatoid arthritis. Am. J. Transl. Res. 2023, 15, 2109–2121. [Google Scholar]
- Cutolo, M.; Campitiello, R.; Gotelli, E.; Soldano, S. The Role of M1/M2 Macrophage Polarization in Rheumatoid Arthritis Synovitis. Front. Immunol. 2022, 13, 867260. [Google Scholar] [CrossRef]
- Roszkowski, L.; Ciechomska, M. Tuning Monocytes and Macrophages for Personalized Therapy and Diagnostic Challenge in Rheumatoid Arthritis. Cells 2021, 10, 1860. [Google Scholar] [CrossRef]
- Smiljanovic, B.; Radzikowska, A.; Kuca-Warnawin, E.; Kurowska, W.; Grün, J.R.; Stuhlmüller, B.; Bonin, M.; Schulte-Wrede, U.; Sörensen, T.; Kyogoku, C.; et al. Monocyte alterations in rheumatoid arthritis are dominated by preterm release from bone marrow and prominent triggering in the joint. Ann. Rheum. Dis. 2018, 77, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Rückert, R.; Brandt, K.; Ernst, M.; Marienfeld, K.; Csernok, E.; Metzler, C.; Budagian, V.; Bulanova, E.; Paus, R.; Bulfone-Paus, S. Interleukin-15 stimulates macrophages to activate CD4+ T cells: A role in the pathogenesis of rheumatoid arthritis? Immunology 2009, 126, 63–73. [Google Scholar] [CrossRef]
- Degboé, Y.; Poupot, R.; Poupot, M. Repolarization of Unbalanced Macrophages: Unmet Medical Need in Chronic Inflammation and Cancer. Int. J. Mol. Sci. 2022, 23, 1496. [Google Scholar] [CrossRef]
- Siouti, E.; Andreakos, E. The many facets of macrophages in rheumatoid arthritis. Biochem. Pharmacol. 2019, 165, 152–169. [Google Scholar] [CrossRef] [PubMed]
- Kerkman, P.F.; Kempers, A.C.; van der Voort, E.I.; van Oosterhout, M.; Huizinga, T.W.; Toes, R.E.; Scherer, H.U. Synovial fluid mononuclear cells provide an environment for long-term survival of antibody-secreting cells and promote the spontaneous production of anti-citrullinated protein antibodies. Ann Rheum Dis. 2016, 75, 2201–2207. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Liu, R.; Zhang, L. Advance in bone destruction participated by JAK/STAT in rheumatoid arthritis and therapeutic effect of JAK/STAT inhibitors. Int. Immunopharmacol. 2022, 111, 109095. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Xiu, H.; Zhang, S.; Zhang, G. The Role of Macrophages in the Pathogenesis of ALI/ARDS. Mediat. Inflamm. 2018, 2018, 1264913. [Google Scholar] [CrossRef]
- Zheng, Y.; Wei, K.; Jiang, P.; Zhao, J.; Shan, Y.; Shi, Y.; Zhao, F.; Chang, C.; Li, Y.; Zhou, M.; et al. Macrophage polarization in rheumatoid arthritis: Signaling pathways, metabolic reprogramming, and crosstalk with synovial fibroblasts. Front. Immunol. 2024, 15, 1394108. [Google Scholar] [CrossRef] [PubMed]
- Xia, T.; Zhang, M.; Lei, W.; Yang, R.; Fu, S.; Fan, Z.; Yang, Y.; Zhang, T. Advances in the role of STAT3 in macrophage polarization. Front. Immunol. 2023, 14, 1160719. [Google Scholar] [CrossRef]
- Malemud, C.J. The role of the JAK/STAT signal pathway in rheumatoid arthritis. Ther. Adv. Musculoskelet. Dis. 2018, 10, 117–127. [Google Scholar] [CrossRef] [PubMed]
- De Vries, L.C.S.; Duarte, J.M.; De Krijger, M.; Welting, O.; Van Hamersveld, P.H.P.; Van Leeuwen-Hilbers, F.W.M.; Moerland, P.D.; Jongejan, A.; D’haens, G.R.; De Jonge, W.J.; et al. A JAK1 Selective Kinase Inhibitor and Tofacitinib Affect Macrophage Activation and Function. Inflamm. Bowel Dis. 2019, 25, 647–660. [Google Scholar] [CrossRef] [PubMed]
- Lescoat, A.; Lelong, M.; Jeljeli, M.; Piquet-Pellorce, C.; Morzadec, C.; Ballerie, A.; Jouneau, S.; Jego, P.; Vernhet, L.; Batteux, F.; et al. Combined anti-fibrotic and anti-inflammatory properties of JAK-inhibitors on macrophages in vitro and in vivo: Perspectives for scleroderma-associated interstitial lung disease. Biochem. Pharmacol. 2020, 178, 114103. [Google Scholar] [CrossRef]
- Wang, X.; Chen, J.; Shen, Y.; Zhang, H.; Xu, Y.; Zhang, J.; Cheng, L. Baricitinib protects ICIs-related myocarditis by targeting JAK1/STAT3 to regulate Macrophage polarization. Cytokine 2024, 179, 156620. [Google Scholar] [CrossRef]
- Jinnouchi, H.; Guo, L.; Sakamoto, A.; Torii, S.; Sato, Y.; Cornelissen, A.; Kuntz, S.; Paek, K.H.; Fernandez, R.; Fuller, D.; et al. Diversity of macrophage phenotypes and responses in atherosclerosis. Cell. Mol. Life Sci. 2020, 77, 1919–1932. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.; Kwon, E.-J.; Lee, J.J. Rheumatoid Arthritis: Pathogenic Roles of Diverse Immune Cells. Int. J. Mol. Sci. 2022, 23, 905. [Google Scholar] [CrossRef] [PubMed]
- Yap, H.Y.; Tee, S.Z.; Wong, M.M.; Chow, S.K.; Peh, S.C.; Teow, S.Y. Pathogenic Role of Immune Cells in Rheumatoid Arthritis: Implications in Clinical Treatment and Biomarker Development. Cells 2018, 7, 161. [Google Scholar] [CrossRef] [PubMed]
- Schulze-Koops, H.; Kalden, J.R. The balance of Th1/Th2 cytokines in rheumatoid arthritis. Best Pract. Res. Clin. Rheumatol. 2001, 15, 677–691. [Google Scholar] [CrossRef] [PubMed]
- Skapenko, A.; Leipe, J.; Lipsky, P.E.; Schulze-Koops, H. The role of the T cell in autoimmune inflammation. Arthritis Res. Ther. 2005, 7 (Suppl. S2), S4–S14. [Google Scholar] [CrossRef] [PubMed]
- Gol-Ara, M.; Jadidi-Niaragh, F.; Sadria, R.; Azizi, G.; Mirshafiey, A. The role of different subsets of regulatory T cells in immunopathogenesis of rheumatoid arthritis. Arthritis 2012, 2012, 805875. [Google Scholar] [CrossRef] [PubMed]
- Eddahri, F.; Denanglaire, S.; Bureau, F.; Spolski, R.; Leonard, W.J.; Leo, O.; Andris, F. Interleukin-6/STAT3 signaling regulates the ability of naive T cells to acquire B-cell help capacities. Blood 2009, 113, 2426–2433. [Google Scholar] [CrossRef]
- Kuuliala, K.; Kuuliala, A.; Koivuniemi, R.; Oksanen, S.; Hämäläinen, M.; Moilanen, E.; Kautiainen, H.; Leirisalo-Repo, M.; Repo, H. Constitutive STAT3 Phosphorylation in Circulating CD4+ T Lymphocytes Associates with Disease Activity and Treatment Response in Recent-Onset Rheumatoid Arthritis. PLoS ONE 2015, 10, e0137385. [Google Scholar] [CrossRef]
- Kuuliala, K.; Kuuliala, A.; Koivuniemi, R.; Kautiainen, H.; Repo, H.; Leirisalo-Repo, M. STAT6 and STAT1 Pathway Activation in Circulating Lymphocytes and Monocytes as Predictor of Treatment Response in Rheumatoid Arthritis. PLoS ONE 2016, 11, e0167975. [Google Scholar] [CrossRef]
- Sanchez-Guajardo, V.; Tanchot, C.; O’malley, J.T.; Kaplan, M.H.; Garcia, S.; Freitas, A.A. Agonist-driven development of CD4+CD25+Foxp3+ regulatory T cells requires a second signal mediated by Stat6. J. Immunol. 2007, 178, 7550–7556. [Google Scholar] [CrossRef]
- Maeshima, K.; Yamaoka, K.; Kubo, S.; Nakano, K.; Iwata, S.; Saito, K.; Ohishi, M.; Miyahara, H.; Tanaka, S.; Ishii, K.; et al. The JAK inhibitor tofacitinib regulates synovitis through inhibition of interferon-γ and interleukin-17 production by human CD4+ T cells. Arthritis Rheum. 2012, 64, 1790–1798. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, R.; Kremer, J.; Cush, J.; Schulze-Koops, H.; Connell, C.A.; Bradley, J.D.; Gruben, D.; Wallenstein, G.V.; Zwillich, S.H.; Kanik, K.S. Placebo-controlled trial of tofacitinib monotherapy in rheumatoid arthritis. N. Engl. J. Med. 2012, 367, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Paradowska-Gorycka, A.; Wajda, A.; Romanowska-Próchnicka, K.; Walczuk, E.; Kuca-Warnawin, E.; Kmiolek, T.; Stypinska, B.; Rzeszotarska, E.; Majewski, D.; Jagodzinski, P.P.; et al. Th17/Treg-Related Transcriptional Factor Expression and Cytokine Profile in Patients with Rheumatoid Arthritis. Front. Immunol. 2020, 11, 572858. [Google Scholar] [CrossRef] [PubMed]
- Ghoreschi, K.; Jesson, M.I.; Li, X.; Lee, J.L.; Ghosh, S.; Alsup, J.W.; Warner, J.D.; Tanaka, M.; Steward-Tharp, S.M.; Gadina, M.; et al. Modulation of innate and adaptive immune responses by tofacitinib (CP-690,550). J. Immunol. 2011, 186, 4234–4243. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yuan, L.; Yang, J.; Lei, Y.; Zhang, H.; Xia, L.; Shen, H.; Lu, J. Changes in Serum Cytokines May Predict Therapeutic Efficacy of Tofacitinib in Rheumatoid Arthritis. Mediat. Inflamm. 2019, 2019, 5617431. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Kimura, A.; Fukaya, T.; Sekiya, T.; Morita, R.; Shichita, T.; Inoue, H.; Yoshimura, A. Low dose CP-690,550 (tofacitinib), a pan-JAK inhibitor, accelerates the onset of experimental autoimmune encephalomyelitis by potentiating Th17 differentiation. Biochem. Biophys. Res. Commun. 2012, 418, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhan, N.; Jin, Y.; Ling, H.; Xiao, C.; Xie, Z.; Zhong, H.; Yu, X.; Tang, R.; Ma, J. Tofacitinib restores the balance of gammadeltaTreg/gammadeltaT17 cells in rheumatoid arthritis by inhibiting the NLRP3 inflammasome. Theranostics 2021, 11, 1446–1457. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Furuya, M.Y.; Asano, T.; Sumichika, Y.; Sato, S.; Kobayashi, H.; Watanabe, H.; Suzuki, E.; Kozuru, H.; Yatsuhashi, H.; Koga, T.; et al. Tofacitinib inhibits granulocyte–macrophage colony-stimulating factor-induced NLRP3 inflammasome activation in human neutrophils. Arthritis Res. Ther. 2018, 20, 196. [Google Scholar] [CrossRef]
- Sun, H.-G.; Jiang, Q.; Fan, W.-J.; Shen, X.-Y.; Wang, Z.-W.; Wang, X. TAGAP activates Th17 cell differentiation by promoting RhoA and NLRP3 to accelerate rheumatoid arthritis development. Clin. Exp. Immunol. 2023, 214, 26–35. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Q.; Cao, G.; Luo, M.; Hou, H.; Yue, C. Pyroptosis by NLRP3/caspase-1/gasdermin-D pathway in synovial tissues of rheumatoid arthritis patients. J. Cell. Mol. Med. 2023, 27, 2448–2456. [Google Scholar] [CrossRef] [PubMed]
- Alamino, V.A.; Onofrio, L.I.; Acosta, C.d.V.; Ferrero, P.V.; Zacca, E.R.; Cadile, I.I.; Mussano, E.D.; Onetti, L.B.; Montes, C.L.; Gruppi, A.; et al. Tofacitinib treatment of rheumatoid arthritis increases senescent T cell frequency in patients and limits T cell function in vitro. Eur. J. Immunol. 2023, 53, e2250353. [Google Scholar] [CrossRef] [PubMed]
- Rao, D.A. Editorial: Lymphocyte Highs and Lows with Baricitinib. Arthritis Rheumatol. 2018, 70, 1897–1900. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; McInnes, I.B.; Taylor, P.C.; Byers, N.L.; Chen, L.; de Bono, S.; Issa, M.; Macias, W.L.; Rogai, V.; Rooney, T.P.; et al. Characterization and Changes of Lymphocyte Subsets in Baricitinib-Treated Patients with Rheumatoid Arthritis: An Integrated Analysis. Arthritis Rheumatol. 2018, 70, 1923–1932. [Google Scholar] [CrossRef] [PubMed]
- Katayose, T.; Iwata, S.; Oyaizu, N.; Hosono, O.; Yamada, T.; Dang, N.H.; Hatano, R.; Tanaka, H.; Ohnuma, K.; Morimoto, C. The role of Cas-L/NEDD9 as a regulator of collagen-induced arthritis in a murine model. Biochem. Biophys. Res. Commun. 2015, 460, 1069–1075. [Google Scholar] [CrossRef]
- Nyirenda, M.H.; Nijjar, J.S.; Frleta-Gilchrist, M.; Gilchrist, D.S.; Porter, D.; Siebert, S.; Goodyear, C.S.; McInnes, I.B. JAK inhibitors disrupt T cell-induced proinflammatory macrophage activation. RMD Open 2023, 9, e002671. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Gao, J.; Kang, J.; Wang, X.; Niu, Q.; Liu, J.; Zhang, L. B Cells in Rheumatoid Arthritis: Pathogenic Mechanisms and Treatment Prospects. Front. Immunol. 2021, 12, 750753. [Google Scholar] [CrossRef] [PubMed]
- Isailovic, N.; Ceribelli, A.; Cincinelli, G.; Vecellio, M.; Guidelli, G.M.; Caprioli, M.; Luciano, N.; Motta, F.; Selmi, C.; De Santis, M. Lymphocyte modulation by tofacitinib in patients with rheumatoid arthritis. Clin. Exp. Immunol. 2021, 205, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Avery, D.T.; Deenick, E.K.; Ma, C.S.; Suryani, S.; Simpson, N.; Chew, G.Y.; Chan, T.D.; Palendira, U.; Bustamante, J.; Boisson-Dupuis, S.; et al. B cell–intrinsic signaling through IL-21 receptor and STAT3 is required for establishing long-lived antibody responses in humans. J. Exp. Med. 2010, 207, 155–171. [Google Scholar] [CrossRef]
- Rizzi, M.; Lorenzetti, R.; Fischer, K.; Staniek, J.; Janowska, I.; Troilo, A.; Strohmeier, V.; Erlacher, M.; Kunze, M.; Bannert, B.; et al. Impact of tofacitinib treatment on human B-cells in vitro and in vivo. J. Autoimmun. 2017, 77, 55–66. [Google Scholar] [CrossRef]
- Martina, M.; Bajo, M.R.; Bañon-Maneus, E.; Rull, D.M.; Hierro-Garcia, N.; Revuelta, I.; Campistol, J.; Rovira, J.; Diekmann, F. Inhibition of JAK3 and PKC via Immunosuppressive Drugs Tofacitinib and Sotrastaurin Inhibits Proliferation of Human B Lymphocytes In Vitro. Transplant. Proc. 2016, 48, 3046–3052. [Google Scholar] [CrossRef] [PubMed]
- Kubo, S.; Nakayamada, S.; Sakata, K.; Kitanaga, Y.; Ma, X.; Lee, S.; Ishii, A.; Yamagata, K.; Nakano, K.; Tanaka, Y. Janus Kinase Inhibitor Baricitinib Modulates Human Innate and Adaptive Immune System. Front. Immunol. 2018, 9, 1510. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.-S.; Jeong, G.H.; Yoo, S.-A. The use of animal models in rheumatoid arthritis research. Yeungnam Univ. J. Med. 2023, 40, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Caplazi, P.; Baca, M.; Barck, K.; Carano, R.A.D.; DeVoss, J.; Lee, W.P.; Bolon, B.; Diehl, L. Mouse Models of Rheumatoid Arthritis. Vet. Pathol. 2015, 52, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Inglis, J.J.; Notley, C.A.; Essex, D.; Wilson, A.W.; Feldmann, M.; Anand, P.; Williams, R. Collagen-induced arthritis as a model of hyperalgesia: Functional and cellular analysis of the analgesic actions of tumor necrosis factor blockade. Arthritis Rheum. 2007, 56, 4015–4023. [Google Scholar] [CrossRef] [PubMed]
- Asquith, D.L.; Miller, A.M.; McInnes, I.B.; Liew, F.Y. Animal models of rheumatoid arthritis. Eur. J. Immunol. 2009, 39, 2040–2044. [Google Scholar] [CrossRef] [PubMed]
- Giant, T.T.; Mikecz, K.; Bartlett, R.R.; Deák, F.; Eugene, J.M.T.; Williams, J.M.; Mattar, T.; Kuettner, K.E.; Schleyerbach, R. Immunomodulation of proteoglycan-induced progressive polyarthritis by leflunomide. Immunopharmacology 1992, 23, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Christensen, A.D.; Haase, C.; Cook, A.D.; Hamilton, J.A. K/BxN Serum-Transfer Arthritis as a Model for Human Inflammatory Arthritis. Front. Immunol. 2016, 7, 213. [Google Scholar] [CrossRef] [PubMed]
- Hablot, J.; Ferhat, M.; Lavelle, A.; Salem, F.; Taieb, M.; Medvedovic, J.; Kindt, N.; Reboul, P.; Cailotto, F.; Jouzeau, J.; et al. Tofacitinib treatment alters mucosal immunity and gut microbiota during experimental arthritis. Clin. Transl. Med. 2020, 10, e163. [Google Scholar] [CrossRef]
- LaBranche, T.P.; Jesson, M.I.; Radi, Z.A.; Storer, C.E.; Guzova, J.A.; Bonar, S.L.; Thompson, J.M.; Happa, F.A.; Stewart, Z.S.; Zhan, Y.; et al. JAK inhibition with tofacitinib suppresses arthritic joint structural damage through decreased RANKL production. Arthritis Rheum. 2012, 64, 3531–3542. [Google Scholar] [CrossRef]
- Gertel, S.; Mahagna, H.; Karmon, G.; Watad, A.; Amital, H. Tofacitinib attenuates arthritis manifestations and reduces the pathogenic CD4 T cells in adjuvant arthritis rats. Clin. Immunol. 2017, 184, 77–81. [Google Scholar] [CrossRef]
- Makabe, K.; Okada, H.; Tachibana, N.; Ishikura, H.; Ito, N.; Tanaka, M.; Chijimatsu, R.; Terashima, A.; Yano, F.; Asaka, M.; et al. Baricitinib ameliorates inflammatory and neuropathic pain in collagen antibody-induced arthritis mice by modulating the IL-6/JAK/STAT3 pathway and CSF-1 expression in dorsal root ganglion neurons. Arthritis Res. Ther. 2024, 26, 121. [Google Scholar] [CrossRef]
- Mahajan, S.; Hogan, J.K.; Shlyakhter, D.; Oh, L.; Salituro, F.G.; Farmer, L.; Hoock, T.C. VX-509 (decernotinib) is a potent and selective janus kinase 3 inhibitor that attenuates inflammation in animal models of autoimmune disease. J. Pharmacol. Exp. Ther. 2015, 353, 405–414. [Google Scholar] [CrossRef] [PubMed]
- van Vollenhoven, R.F.; Fleischmann, R.; Cohen, S.; Lee, E.B.; Meijide, J.A.G.; Wagner, S.; Forejtova, S.; Zwillich, S.H.; Gruben, D.; Koncz, T.; et al. Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N. Engl. J. Med. 2012, 367, 508–519. [Google Scholar] [CrossRef]
- Lee, E.B.; Fleischmann, R.; Hall, S.; Wilkinson, B.; Bradley, J.D.; Gruben, D.; Koncz, T.; Krishnaswami, S.; Wallenstein, G.V.; Zang, C.; et al. Tofacitinib versus methotrexate in rheumatoid arthritis. N. Engl. J. Med. 2014, 370, 2377–2386. [Google Scholar] [CrossRef] [PubMed]
- van der Heijde, D.; Tanaka, Y.; Fleischmann, R.; Keystone, E.; Kremer, J.; Zerbini, C.; Cardiel, M.H.; Cohen, S.; Nash, P.; Song, Y.W.; et al. Tofacitinib (CP-690,550) in patients with rheumatoid arthritis receiving methotrexate: Twelve-month data from a twenty-four-month phase III randomized radiographic study. Arthritis Rheum. 2013, 65, 559–570. [Google Scholar] [CrossRef] [PubMed]
- van der Heijde, D.; Strand, V.; Tanaka, Y.; Keystone, E.; Kremer, J.; Zerbini, C.A.; Cardiel, M.H.; Cohen, S.; Nash, P.; Song, Y.W.; et al. Tofacitinib in Combination with Methotrexate in Patients with Rheumatoid Arthritis: Clinical Efficacy, Radiographic, and Safety Outcomes from a Twenty-Four-Month, Phase III Study. Arthritis Rheumatol. 2019, 71, 878–891. [Google Scholar] [CrossRef]
- Burmester, G.R.; Blanco, R.; Charles-Schoeman, C.; Wollenhaupt, J.; Zerbini, C.; Benda, B.; Gruben, D.; Wallenstein, G.; Krishnaswami, S.; Zwillich, S.H.; et al. Tofacitinib (CP-690,550) in combination with methotrexate in patients with active rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitors: A randomised phase 3 trial. Lancet 2013, 381, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, R.; Mysler, E.; Hall, S.; Kivitz, A.J.; Moots, R.J.; Luo, Z.; Demasi, R.; Soma, K.; Zhang, R.; Takiya, L.; et al. Efficacy and safety of tofacitinib monotherapy, tofacitinib with methotrexate, and adalimumab with methotrexate in patients with rheumatoid arthritis (ORAL Strategy): A phase 3b/4, double-blind, head-to-head, randomised controlled trial. Lancet 2017, 390, 457–468. [Google Scholar] [CrossRef]
- Wollenhaupt, J.; Lee, E.B.; Curtis, J.R.; Silverfield, J.; Terry, K.; Soma, K.; Mojcik, C.; DeMasi, R.; Strengholt, S.; Kwok, K.; et al. Safety and efficacy of tofacitinib for up to 9.5 years in the treatment of rheumatoid arthritis: Final results of a global, open-label, long-term extension study. Arthritis Res. Ther. 2019, 21, 89. [Google Scholar] [CrossRef]
- Ytterberg, S.R.; Bhatt, D.L.; Mikuls, T.R.; Koch, G.G.; Fleischmann, R.; Rivas, J.L.; Germino, R.; Menon, S.; Sun, Y.; Wang, C.; et al. Cardiovascular and Cancer Risk with Tofacitinib in Rheumatoid Arthritis. N. Engl. J. Med. 2022, 386, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Buch, M.H.; Bhatt, D.L.; Charles-Schoeman, C.; Giles, J.T.; Mikuls, T.; Koch, G.G.; Ytterberg, S.; Nagy, E.; Jo, H.; Kwok, K.; et al. Risk of extended major adverse cardiovascular event endpoints with tofacitinib versus TNF inhibitors in patients with rheumatoid arthritis: A post hoc analysis of a phase 3b/4 randomised safety study. RMD Open 2024, 10, e003912. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, L.E.; Danese, S.; Yndestad, A.; Wang, C.; Nagy, E.; Modesto, I.; Rivas, J.; Benda, B. Identification of two tofacitinib subpopulations with different relative risk versus TNF inhibitors: An analysis of the open label, randomised controlled study ORAL Surveillance. Ann. Rheum. Dis. 2023, 82, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Khosrow-Khavar, F.; Kim, S.C.; Lee, H.; Lee, S.B.; Desai, R.J. Tofacitinib and risk of cardiovascular outcomes: Results from the Safety of TofAcitinib in Routine care patients with Rheumatoid Arthritis (STAR-RA) study. Ann. Rheum. Dis. 2022, 81, 798–804. [Google Scholar] [CrossRef] [PubMed]
- Balanescu, A.R.; Citera, G.; Pascual-Ramos, V.; Bhatt, D.L.; Connell, C.A.; Gold, D.; Chen, A.S.; Sawyerr, G.; Shapiro, A.B.; Pope, J.E.; et al. Infections in patients with rheumatoid arthritis receiving tofacitinib versus tumour necrosis factor inhibitors: Results from the open-label, randomised controlled ORAL Surveillance trial. Ann. Rheum. Dis. 2022, 81, 1491–1503. [Google Scholar] [CrossRef] [PubMed]
- Kubo, S.; Miyazaki, Y.; Amano, K.; Matsui, K.; Kameda, H.; Inoue, Y.; Nakayamada, S.; Ogura, T.; Kaneko, Y.; Yamaoka, K.; et al. Sustained remission following the discontinuation of tofacitinib in patients with rheumatoid arthritis (XANADU study): An open-label randomised study. RMD Open 2023, 9, e003029. [Google Scholar] [CrossRef]
- Genovese, M.C.; Kremer, J.; Zamani, O.; Ludivico, C.; Krogulec, M.; Xie, L.; Beattie, S.D.; Koch, A.E.; Cardillo, T.E.; Rooney, T.P.; et al. Baricitinib in Patients with Refractory Rheumatoid Arthritis. N. Engl. J. Med. 2016, 374, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.C.; Keystone, E.C.; van der Heijde, D.; Weinblatt, M.E.; Del Carmen Morales, L.; Gonzaga, J.R.; Yakushin, S.; Ishii, T.; Emoto, K.; Beattie, S.; et al. Baricitinib versus Placebo or Adalimumab in Rheumatoid Arthritis. N. Engl. J. Med. 2017, 376, 652–662. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, R.; Schiff, M.; van der Heijde, D.; Ramos-Remus, C.; Spindler, A.; Stanislav, M.; Zerbini, C.A.; Gurbuz, S.; Dickson, C.; de Bono, S.; et al. Baricitinib, Methotrexate, or Combination in Patients with Rheumatoid Arthritis and No or Limited Prior Disease-Modifying Antirheumatic Drug Treatment. Arthritis Rheumatol. 2017, 69, 506–517. [Google Scholar] [CrossRef]
- Simon, D.; Minopoulou, I.; Kemenes, S.; Bayat, S.; Tascilar, K.; Mutlu, M.Y.; Valor-Méndez, L.; Krönke, G.; Hueber, A.J.; Schett, G.; et al. Baricitinib Improves Bone Properties and Biomechanics in Patients with Rheumatoid Arthritis: Results of the Prospective Interventional BARE BONE Trial. Arthritis Rheumatol. 2023, 75, 1923–1934. [Google Scholar] [CrossRef]
- Taylor, P.C.; Weinblatt, M.E.; Burmester, G.R.; Rooney, T.P.; Witt, S.; Walls, C.D.; Issa, M.; Salinas, C.A.; Saifan, C.; Zhang, X.; et al. Cardiovascular Safety during Treatment with Baricitinib in Rheumatoid Arthritis. Arthritis Rheumatol. 2019, 71, 1042–1055. [Google Scholar] [CrossRef]
- Taylor, P.C.; Takeuchi, T.; Burmester, G.R.; Durez, P.; Smolen, J.S.; Deberdt, W.; Issa, M.; Terres, J.R.; Bello, N.; Winthrop, K.L. Safety of baricitinib for the treatment of rheumatoid arthritis over a median of 4.6 and up to 9.3 years of treatment: Final results from long-term extension study and integrated database. Ann. Rheum. Dis. 2022, 81, 335–343. [Google Scholar] [CrossRef]
- Liu, H.; Yang, Y.; Zhang, J.; Li, X. Baricitinib improves pulmonary fibrosis in mice with rheumatoid arthritis-associated interstitial lung disease by inhibiting the Jak2/Stat3 signaling pathway. Adv. Rheumatol. 2023, 63, 45. [Google Scholar] [CrossRef]
- Tucci, G.; Garufi, C.; Pacella, I.; Zagaglioni, M.; Pinzon Grimaldos, A.; Ceccarelli, F.; Conti, F.; Spinelli, F.R.; Piconese, S. Baricitinib therapy response in rheumatoid arthritis patients associates to STAT1 phosphorylation in monocytes. Front. Immunol. 2022, 13, 932240. [Google Scholar] [CrossRef]
- Smolen, J.S.; Pangan, A.L.; Emery, P.; Rigby, W.; Tanaka, Y.; Vargas, J.I.; Zhang, Y.; Damjanov, N.; Friedman, A.; Othman, A.A.; et al. Upadacitinib as monotherapy in patients with active rheumatoid arthritis and inadequate response to methotrexate (SELECT-MONOTHERAPY): A randomised, placebo-controlled, double-blind phase 3 study. Lancet 2019, 393, 2303–2311. [Google Scholar] [CrossRef]
- van Vollenhoven, R.; Takeuchi, T.; Pangan, A.L.; Friedman, A.; Mohamed, M.E.F.; Chen, S.; Rischmueller, M.; Blanco, R.; Xavier, R.M.; Strand, V. Efficacy and Safety of Upadacitinib Monotherapy in Methotrexate-Naive Patients with Moderately-to-Severely Active Rheumatoid Arthritis (SELECT-EARLY): A Multicenter, Multi-Country, Randomized, Double-Blind, Active Comparator-Controlled Trial. Arthritis Rheumatol. 2020, 72, 1607–1620. [Google Scholar] [CrossRef]
- Strand, V.; Tundia, N.; Wells, A.; Buch, M.H.; Radominski, S.C.; Camp, H.S.; Friedman, A.; Suboticki, J.L.; Dunlap, K.; Goldschmidt, D.; et al. Upadacitinib monotherapy improves patient-reported outcomes in rheumatoid arthritis: Results from SELECT-EARLY and SELECT-MONOTHERAPY. Rheumatology 2021, 60, 3209–3221. [Google Scholar] [CrossRef]
- Fleischmann, R.; Pangan, A.L.; Song, I.; Mysler, E.; Bessette, L.; Peterfy, C.; Durez, P.; Ostor, A.J.; Li, Y.; Zhou, Y.; et al. Upadacitinib Versus Placebo or Adalimumab in Patients with Rheumatoid Arthritis and an Inadequate Response to Methotrexate: Results of a Phase III, Double-Blind, Randomized Controlled Trial. Arthritis Rheumatol. 2019, 71, 1788–1800. [Google Scholar] [CrossRef]
- Peterfy, C.G.; Strand, V.; Friedman, A.; Hall, S.; Mysler, E.; Durez, P.; Baraliakos, X.; Enejosa, J.V.; Shaw, T.; Li, Y.; et al. Inhibition of structural joint damage progression with upadacitinib in rheumatoid arthritis: 1-year outcomes from the SELECT phase 3 program. Rheumatology 2022, 61, 3246–3256. [Google Scholar] [CrossRef]
- Genovese, M.C.; Fleischmann, R.; Combe, B.; Hall, S.; Rubbert-Roth, A.; Zhang, Y.; Zhou, Y.; Mohamed, M.E.F.; Meerwein, S.; Pangan, A.L. Safety and efficacy of upadacitinib in patients with active rheumatoid arthritis refractory to biologic disease-modifying anti-rheumatic drugs (SELECT-BEYOND): A double-blind, randomised controlled phase 3 trial. Lancet 2018, 391, 2513–2524. [Google Scholar] [CrossRef]
- Rubbert-Roth, A.; Enejosa, J.; Pangan, A.L.; Haraoui, B.; Rischmueller, M.; Khan, N.; Zhang, Y.; Martin, N.; Xavier, R.M. Trial of Upadacitinib or Abatacept in Rheumatoid Arthritis. N. Engl. J. Med. 2020, 383, 1511–1521. [Google Scholar] [CrossRef] [PubMed]
- Combe, B.; Kivitz, A.; Tanaka, Y.; van der Heijde, D.; Simon, J.A.; Baraf, H.S.B.; Kumar, U.; Matzkies, F.; Bartok, B.; Ye, L.; et al. Filgotinib versus placebo or adalimumab in patients with rheumatoid arthritis and inadequate response to methotrexate: A phase III randomised clinical trial. Ann. Rheum. Dis. 2021, 80, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Genovese, M.C.; Kalunian, K.; Gottenberg, J.E.; Mozaffarian, N.; Bartok, B.; Matzkies, F.; Gao, J.; Guo, Y.; Tasset, C.; Sundy, J.S.; et al. Effect of Filgotinib vs. Placebo on Clinical Response in Patients with Moderate to Severe Rheumatoid Arthritis Refractory to Disease-Modifying Antirheumatic Drug Therapy: The FINCH 2 Randomized Clinical Trial. JAMA 2019, 322, 315–325. [Google Scholar] [CrossRef]
- Westhovens, R.; Rigby, W.F.C.; van der Heijde, D.; Ching, D.W.T.; Stohl, W.; Kay, J.; Chopra, A.; Bartok, B.; Matzkies, F.; Yin, Z.; et al. Filgotinib in combination with methotrexate or as monotherapy versus methotrexate monotherapy in patients with active rheumatoid arthritis and limited or no prior exposure to methotrexate: The phase 3, randomised controlled FINCH 3 trial. Ann. Rheum. Dis. 2021, 80, 727–738. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
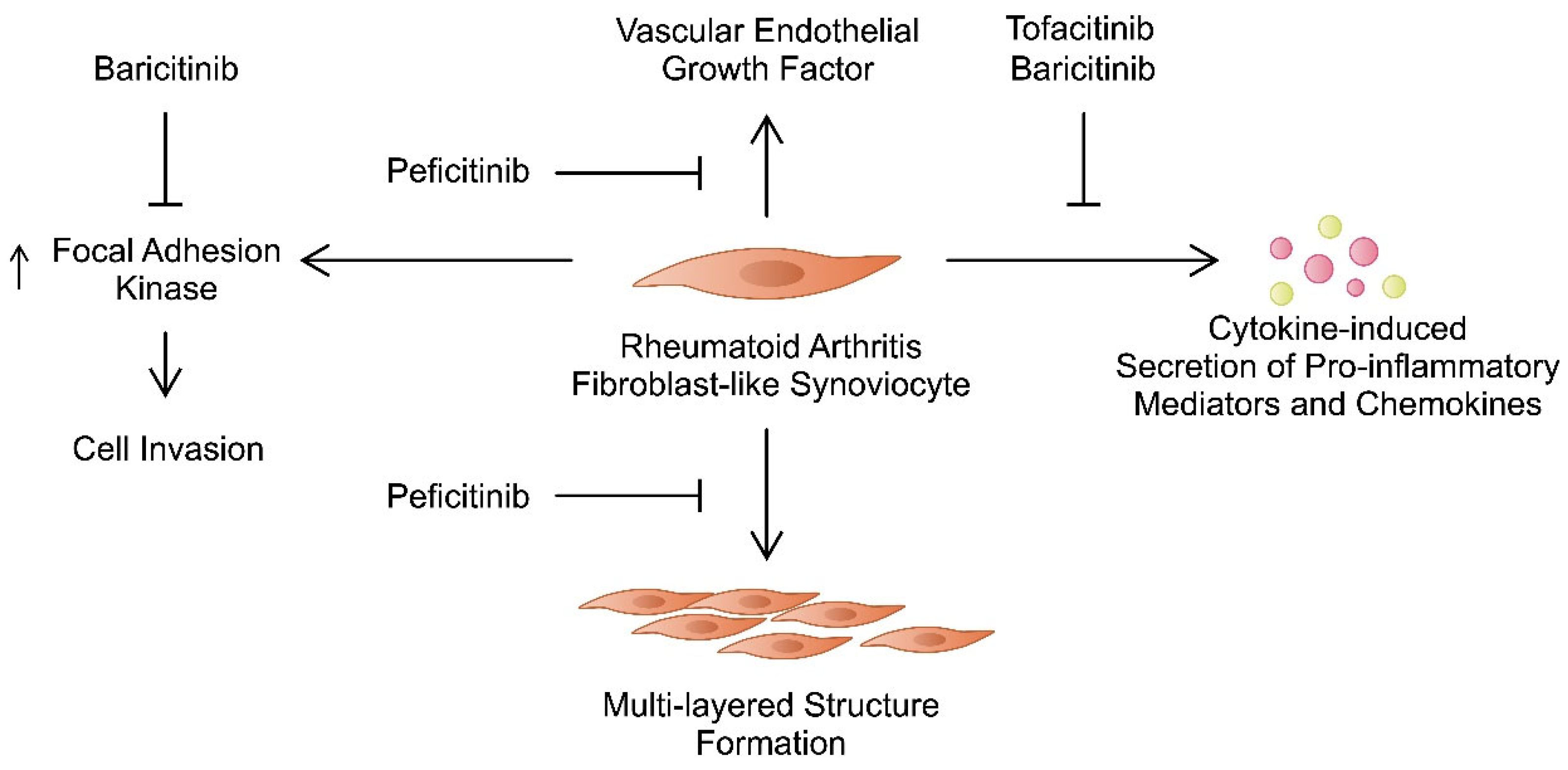
| JAK Inhibitor | Targets | Impact on Fibroblast-like Synoviocytes | References |
|---|---|---|---|
| Tofacitinib | JAK1, JAK2, JAK3 | Tofacitinib suppresses the activity of JAK/STAT pathway induced by oncostatin M, decreases the expression of chemokines, regulates apoptosis and myofibroblast differentiation of FLSs. | [34,44,46,48,50] |
| Baricitinib | JAK1, JAK2 | Baricitinib inhibits inflammatory responses stimulated by OSM, together with IFNγ-induced expression of gliostatin/thymidine phosphorylase. Baricitinib negatively regulates IFN signalling I FLSs. | [53,54,55,56] |
| Peficitinib | Pan-JAK inhibitor | Peficitinib can suppress the formation of multi-layered structures by RA-FLSs, as well as the production of MMPs, IL-6, and VEGF from these structures. The drug inhibits the secretion of MMP-3, IL-6, and VEGF from PDGF-BB-stimulated FLSs. Moreover, it suppresses FLS proliferation and chemotactic properties. | [28,58,60,62] |
| Clinical Trial | Arms | Number of Participants | Efficacy | Toxicities | Reference |
|---|---|---|---|---|---|
| ORAL Solo (NCT00814307) | Placebo | 122 | ACR20: 26.7% | Serious AEs at 3 months: 4.9% | [92] |
| Tofacitinib 5 mg | 243 | ACR20: 59.8% | Serious AEs at 3 months: 0.4% | ||
| Tofacitinib 10 mg | 245 | ACR20: 65.7% | Serious AEs at 3 months: 2% | ||
| ORAL Standard (NCT00853385) | Placebo followed by tofacitinib 5 mg | 56 | ACR20: 28.3% | Serious AEs at 6 months: 0% | [124] |
| Placebo followed by tofacitinib 10 mg | 52 | Serious AEs at 6 months: 0% | |||
| Tofacitinib 5 mg | 204 | ACR20: 51.5% | Serious AEs at 3 months: 5.9% | ||
| Tofacitinib 10 mg | 201 | ACR20: 52.6% | Serious AEs at 3 months: 5% | ||
| Adalimumab 40 mg | 204 | ACR20: 47.2% | Serious AEs at 3 months: 2.5% | ||
| ORAL Start (NCT01039688) | Tofacitinib 5 mg | 373 | ACR70: 25.5 ± 2.3% | Serious AEs at 24 months: 10.7% | [125] |
| Tofacitinib 10 mg | 397 | ACR70: 37.7 ± 2.4% | Serious AEs at 24 months: 10.8% | ||
| Methotrexate | 186 | ACR70: 12.0 ± 2.4% | Serious AEs at 24 months: 11.8% | ||
| ORAL Scan (NCT00847613) | Tofacitinib 5 mg + methotrexate | 321 | ACR20: 51.5% | Serious AEs at 6 months: 5.3% | [126] |
| Tofacitinib 10 mg + MTX | 316 | ACR20: 61.8% | Serious AEs at 6 months: 2.2% | ||
| Placebo to tofacitinib 5 mg + MTX | 81 | ACR20: 25.3% | Serious AEs at 6 months: 2.4% | ||
| Placebo to tofacitinib 10 mg + MTX | 79 | Serious AEs at 6 months: 2.7% | |||
| ORAL Step (NCT00960440) | Placebo + MTX | 132 | ACR20 at 3 months: 41.7% | - | [128] |
| Tofacitinib 5 mg + MTX | 133 | ACR20 at 3 months: 48.1% | Serious AEs at 6 months: 3.8% | ||
| Tofacitinib 10 mg + MTX | 134 | ACR20 at 3 months: 24.4% | Serious AEs at 6 months: 4.5% | ||
| ORAL Strategy (NCT02187055) | Tofacitinib 5 mg | 384 | ACR50 at 6 months: 38% | Serious AEs at 6 months: 9% | [129] |
| Tofacitinib 5 mg + MTX | 376 | ACR50 at 6 months: 46% | Serious AEs at 6 months: 7% | ||
| Adalimumab 40 mg + MTX | 386 | ACR50 at 6 months: 44% | Serious AEs at 6 months: 6% | ||
| RA-BEACON (NCT01721044) | Placebo | 176 | ACR20 at 12 weeks: 27% | Serious AEs at 24 weeks: 7% | [137] |
| Baricitinib 2 mg | 174 | - | Serious AEs at 24 weeks: 4% | ||
| Baricitinib 4 mg | 177 | ACR20 at 12 weeks: 55% | Serious AEs at 24 weeks: 10% | ||
| RA-BEAM (NCT01710358) | Placebo | 488 | ACR20 at 12 weeks: 40% | Serious AEs at 24 weeks: 5% | [138] |
| Baricitinib | 487 | ACR20 at 12 weeks: 70% | Serious AEs at 24 weeks: 5% | ||
| Adalimumab | 330 | ACR20 at 12 weeks: 61% | Serious AEs at 24 weeks: 2% | ||
| RA-BEGIN (NCT01711359) | MTX | 210 | ACR20 at 24 weeks: 62% | Serious AEs at 52 weeks: 10% | [139] |
| Baricitinib | 159 | ACR20 at 24 weeks: 77% | Serious AEs at 52 weeks: 8% | ||
| MTX + baricitinib | 215 | ACR20 at 24 weeks: 78% | Serious AEs at 52 weeks: 8% | ||
| SELECT- MONOTHERAPY (NCT02706951) | MTX | 216 | ACR20 at 14 weeks: 41% | Serious AEs at 14 weeks: 3% | [145] |
| Upadacitinib 15 mg | 217 | ACR20 at 14 weeks: 68% | Serious AEs at 14 weeks: 4% | ||
| Upadacitinib 30 mg | 215 | ACR20 at 14 weeks: 71% | Serious AEs at 14 weeks: 3% | ||
| SELECT-EARLY (NCT02706873) | MTX | 314 | ACR50 at 12 weeks: 28% | Serious AEs at 24 weeks: 4.1% | [146] |
| Upadacitinib 15 mg | 317 | ACR50 at 12 weeks: 52% | Serious AEs at 24 weeks: 4.7% | ||
| Upadacitinib 30 mg | 314 | ACR50 at 12 weeks: 56% | Serious AEs at 24 weeks: 6.4% | ||
| SELECT- COMPARE (NCT02629159) | Placebo + MTX | 651 | ACR20 at 12 weeks: 36% | Serious AEs at 26 weeks: 2.9% | [148] |
| Upadacitinib 15 mg + MTX | 651 | ACR20 at 12 weeks: 71% | Serious AEs at 26 weeks: 3.7% | ||
| Adalimumab + MTX | 327 | ACR20 at 12 weeks: 63% | Serious AEs at 26 weeks: 4.3% | ||
| SELECT-BEYOND (NCT02706847) | Placebo | 169 | ACR20 at 12 weeks: 28% | Serious AEs at 12 weeks: 0% | [150] |
| Upadacitinib 15 mg | 164 | ACR20 at 12 weeks: 65% | Serious AEs at 12 weeks: 5% | ||
| Upadacitinib 30 mg | 165 | ACR20 at 12 weeks: 56% | Serious AEs at 12 weeks: 7% | ||
| SELECT-CHOICE (NCT03086343) | Upadacitinib | 303 | DAS28-CRP at 12 weeks: −2.52 DAS28-CRP < 2.6: 30% | Serious AEs at 24 weeks: 3.3% | [151] |
| Abatacept | 309 | DAS28-CRP at 12 weeks: −2.00 DAS28-CRP < 2.6: 13.3% | Serious AEs at 24 weeks: 1.6% | ||
| FINCH 1 (NCT02889796) | Placebo + MTX | 475 | ACR20 at 12 weeks: 49.9% | Serious AEs at 24 weeks: 4.2% | [152] |
| Adalimumab + MTX | 325 | ACR20 at 12 weeks: 70.5% | Serious AEs at 24 weeks: 4.3% | ||
| Filgotinib 100 mg + MTX | 480 | ACR20 at 12 weeks: 69.8% | Serious AEs at 24 weeks: 5% | ||
| Filgotinib 200 mg + MTX | 475 | ACR20 at 12 weeks: 76.6% | Serious AEs at 24 weeks: 4.4% | ||
| FINCH 2 (NCT02873936) | Placebo | 148 | ACR20 at 12 weeks: 31.1% DAS28-CRP < 3.2: 15.5% | Serious AEs at 24 weeks: 3.4% | [153] |
| Filgotinib 100 mg | 153 | ACR20 at 12 weeks: 57.5% DAS28-CRP < 3.2: 37.3% | Serious AEs at 24 weeks: 5.2% | ||
| Filgotinib 200 mg | 147 | ACR20 at 12 weeks: 66% DAS28-CRP < 3.2: 40.8% | Serious AEs at 24 weeks: 4.1% | ||
| FINCH 3 (NCT02886728) | MTX | 416 | ACR20 at 24 weeks: 71% | Serious AEs up to 52 weeks: 7% | [154] |
| Filgotinib 200 mg | 210 | ACR20 at 24 weeks: 78.1% | Serious AEs up to 52 weeks: 8% | ||
| Filgotinib 100 mg + MTX | 207 | ACR20 at 24 weeks: 80% | Serious AEs up to 52 weeks: 6% | ||
| Filgotinib 200 mg + MTX | 416 | ACR20 at 24 weeks: 81% | Serious AEs up to 52 weeks: 6% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiełbowski, K.; Plewa, P.; Bratborska, A.W.; Bakinowska, E.; Pawlik, A. JAK Inhibitors in Rheumatoid Arthritis: Immunomodulatory Properties and Clinical Efficacy. Int. J. Mol. Sci. 2024, 25, 8327. https://doi.org/10.3390/ijms25158327
Kiełbowski K, Plewa P, Bratborska AW, Bakinowska E, Pawlik A. JAK Inhibitors in Rheumatoid Arthritis: Immunomodulatory Properties and Clinical Efficacy. International Journal of Molecular Sciences. 2024; 25(15):8327. https://doi.org/10.3390/ijms25158327
Chicago/Turabian StyleKiełbowski, Kajetan, Paulina Plewa, Aleksandra Wiktoria Bratborska, Estera Bakinowska, and Andrzej Pawlik. 2024. "JAK Inhibitors in Rheumatoid Arthritis: Immunomodulatory Properties and Clinical Efficacy" International Journal of Molecular Sciences 25, no. 15: 8327. https://doi.org/10.3390/ijms25158327