


Pericardial Fluid Accumulates microRNAs That Regulate Heart Fibrosis after Myocardial Infarction
 , ,
, ,  , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
2.1. Pericardial Fluid from STEMI Patients Accumulates Biomarkers of Cardiac Fibrosis
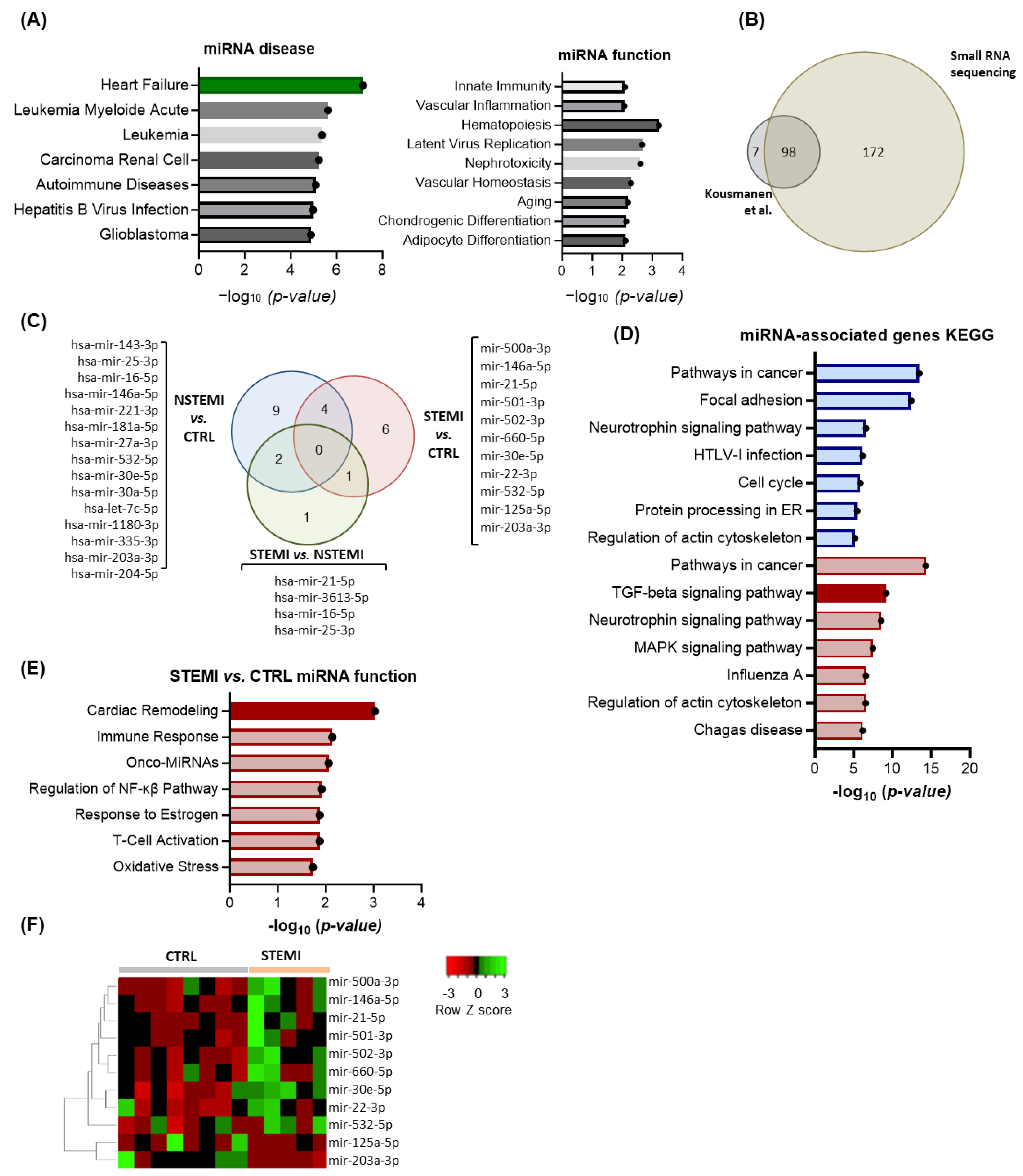
2.2. RNA Sequencing Reveals Enrichment in miRs Associated with Extracellular Matrix (ECM) Remodeling in STEMI Patients
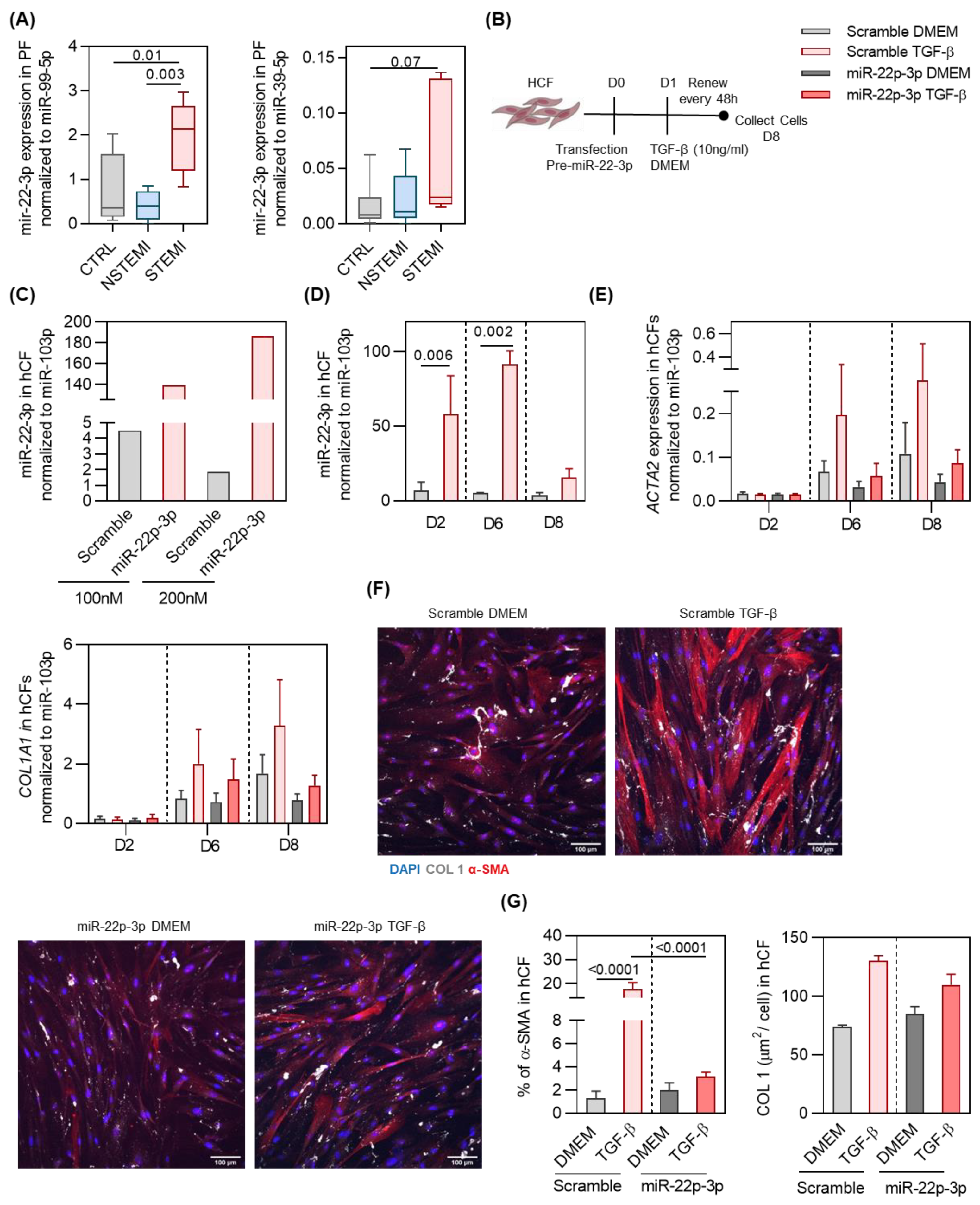
2.3. miR-22-3p Overexpression Reduces hCF Activation by TGF-β
3. Discussion
4. Materials and Methods
4.1. Study Participant Details
4.2. Sample Collection and Processing
4.3. Cardiac Fibrosis Markers Quantification
4.4. Cell Culture
4.5. Human Embryonic Stem Cells Differentiation into Ventricular Cardiomyocytes
4.6. Human Cardiac Fibroblasts Culture with PF from Patients
4.7. Transfection of Human Cardiac Fibroblasts
4.8. RNA Extraction and Reverse Transcription
4.8.1. miR Extraction from PF and Plasma and Reverse Transcription
4.8.2. RNA Extraction and Reverse Transcription from Cultured Cells
4.9. Real-Time PCR (RT-PCR)
4.10. RNA Sequencing (RNA-Seq) Analysis
4.11. Immunofluorescence Assay
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Khan, M.A.; Hashim, M.J.; Mustafa, H.; Baniyas, M.Y.; Al Suwaidi, S.; AlKatheeri, R.; Alblooshi, F.M.K.; Almatrooshi, M.; Alzaabi, M.E.H.; Al Darmaki, R.S.; et al. Global Epidemiology of Ischemic Heart Disease: Results from the Global Burden of Disease Study. Cureus 2020, 12, e9349. [Google Scholar] [CrossRef] [PubMed]
- Blom, J.N.; Feng, Q. Cardiac repair by epicardial EMT: Current targets and a potential role for the primary cilium. Pharmacol. Ther. 2018, 186, 114–129. [Google Scholar] [CrossRef] [PubMed]
- Abbott, J.D.; Ahmed, H.N.; Vlachos, H.A.; Selzer, F.; Williams, D.O. Comparison of Outcome in Patients With ST-Elevation Versus Non–ST-Elevation Acute Myocardial Infarction Treated With Percutaneous Coronary Intervention (from the National Heart, Lung, and Blood Institute Dynamic Registry). Am. J. Cardiol. 2007, 100, 190–195. [Google Scholar] [CrossRef] [PubMed]
- García-García, C.; Subirana, I.; Sala, J.; Bruguera, J.; Sanz, G.; Valle, V.; Arós, F.; Fiol, M.; Molina, L.; Serra, J.; et al. Long-Term Prognosis of First Myocardial Infarction According to the Electrocardiographic Pattern (ST Elevation Myocardial Infarction, Non-ST Elevation Myocardial Infarction and Non-Classified Myocardial Infarction) and Revascularization Procedures. Am. J. Cardiol. 2011, 108, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Thygesen, K.; Alpert, J.S.; Jaffe, A.S.; Simoons, M.L.; Chaitman, B.R.; White, H.D.; The Writing Group on behalf of the Joint ESC/ACCF/AHA/WHF Task Force for the Universal Definition of Myocardial Infarction. Third universal definition of myocardial infarction. Nat. Rev. Cardiol. 2012, 9, 620–633. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Sun, P.; Zhang, J.C.; Zhang, Q.; Yao, S.L. Proinflammatory effects of S100A8/A9 via TLR4 and RAGE signaling pathways in BV-2 microglial cells. Int. J. Mol. Med. 2017, 40, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. The mechanistic basis of infarct healing. Antioxid. Redox Signal. 2006, 8, 1907–1939. [Google Scholar] [CrossRef]
- Bergmann, O.; Zdunek, S.; Felker, A.; Salehpour, M.; Alkass, K.; Bernard, S.; Sjostrom, S.L.; Szewczykowska, M.; Jackowska, T.; Dos Remedios, C.; et al. Dynamics of Cell Generation and Turnover in the Human Heart. Cell 2015, 161, 1566–1575. [Google Scholar] [CrossRef]
- Bergmann, O.; Bhardwaj, R.D.; Bernard, S.; Zdunek, S.; Barnabé-Heider, F.; Walsh, S.; Zupicich, J.; Alkass, K.; Buchholz, B.A.; Druid, H.; et al. Evidence for Cardiomyocyte Renewal in Humans. Science 2009, 324, 98–102. [Google Scholar] [CrossRef]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac Fibrosis: The Fibroblast Awakens. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef]
- Hinderer, S.; Schenke-Layland, K. Cardiac fibrosis—A short review of causes and therapeutic strategies. Adv. Drug Deliv. Rev. 2019, 146, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Spach, M.S.; Boineau, J.P. Microfibrosis produces electrical load variations due to loss of side-to-side cell connections: A major mechanism of structural heart disease arrhythmias. Pacing Clin. Electrophysiol. PACE 1997, 20, 397–413. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.C.; Pereira, C.; Fonseca, A.; Pinto-do-Ó, P.; Nascimento, D.S. Bearing My Heart: The Role of Extracellular Matrix on Cardiac Development, Homeostasis, and Injury Response. Front. Cell Dev. Biol. 2020, 8, 621644. [Google Scholar] [CrossRef] [PubMed]
- Perestrelo, A.R.; Silva, A.C.; Cruz, J.O.-D.L.; Martino, F.; Horváth, V.; Caluori, G.; Polanský, O.; Vinarský, V.; Azzato, G.; Marco, G.d.; et al. Multiscale Analysis of Extracellular Matrix Remodeling in the Failing Heart. Circ. Res. 2021, 128, 24–38. [Google Scholar] [CrossRef] [PubMed]
- van den Borne, S.W.M.; Diez, J.; Blankesteijn, W.M.; Verjans, J.; Hofstra, L.; Narula, J. Myocardial remodeling after infarction: The role of myofibroblasts. Nat. Reviews. Cardiol. 2010, 7, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Limana, F.; Bertolami, C.; Mangoni, A.; Di Carlo, A.; Avitabile, D.; Mocini, D.; Iannelli, P.; De Mori, R.; Marchetti, C.; Pozzoli, O.; et al. Myocardial infarction induces embryonic reprogramming of epicardial c-kit+ cells: Role of the pericardial fluid. J. Mol. Cell. Cardiol. 2010, 48, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Trindade, F.; Vitorino, R.; Leite-Moreira, A.; Falcão-Pires, I. Pericardial fluid: An underrated molecular library of heart conditions and a potential vehicle for cardiac therapy. Basic Res. Cardiol. 2019, 114, 10. [Google Scholar] [CrossRef] [PubMed]
- Beltrami, C.; Besnier, M.; Shantikumar, S.; Shearn, A.I.; Rajakaruna, C.; Laftah, A.; Sessa, F.; Spinetti, G.; Petretto, E.; Angelini, G.D.; et al. Human Pericardial Fluid Contains Exosomes Enriched with Cardiovascular-Expressed MicroRNAs and Promotes Therapeutic Angiogenesis. Mol. Ther. 2017, 25, 679–693. [Google Scholar] [CrossRef] [PubMed]
- Foglio, E.; Puddighinu, G.; Fasanaro, P.; D’Arcangelo, D.; Perrone, G.A.; Mocini, D.; Campanella, C.; Coppola, L.; Logozzi, M.; Azzarito, T.; et al. Exosomal clusterin, identified in the pericardial fluid, improves myocardial performance following MI through epicardial activation, enhanced arteriogenesis and reduced apoptosis. Int. J. Cardiol. 2015, 197, 333–347. [Google Scholar] [CrossRef] [PubMed]
- Catalanotto, C.; Cogoni, C.; Zardo, G. MicroRNA in Control of Gene Expression: An Overview of Nuclear Functions. Int. J. Mol. Sci. 2016, 17, 1712. [Google Scholar] [CrossRef]
- Varzideh, F.; Kansakar, U.; Donkor, K.; Wilson, S.; Jankauskas, S.S.; Mone, P.; Wang, X.; Lombardi, A.; Santulli, G. Cardiac Remodeling After Myocardial Infarction: Functional Contribution of microRNAs to Inflammation and Fibrosis. Front. Cardiovasc. Med. 2022, 9, 863238. [Google Scholar] [CrossRef] [PubMed]
- Kuosmanen, S.M.; Hartikainen, J.; Hippeläinen, M.; Kokki, H.; Levonen, A.L.; Tavi, P. MicroRNA profiling of pericardial fluid samples from patients with heart failure. PLoS ONE 2015, 10, e0119646. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Chen, Y.; Shu, J.; Tang, C.-E.; Jiang, Y.; Luo, F. Identification of microRNAs enriched in exosomes in human pericardial fluid of patients with atrial fibrillation based on bioinformatic analysis. J. Thorac. Dis. 2020, 12, 5617–5627. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Luo, F.; Lei, K. Exosomes Containing LINC00636 Inhibit MAPK1 through the miR-450a-2-3p Overexpression in Human Pericardial Fluid and Improve Cardiac Fibrosis in Patients with Atrial Fibrillation. Mediat. Inflamm. 2021, 2021, 9960241. [Google Scholar] [CrossRef] [PubMed]
- Khudiakov, A.A.; Panshin, D.D.; Fomicheva, Y.V.; Knyazeva, A.A.; Simonova, K.A.; Lebedev, D.S.; Mikhaylov, E.N.; Kostareva, A.A. Different Expressions of Pericardial Fluid MicroRNAs in Patients With Arrhythmogenic Right Ventricular Cardiomyopathy and Ischemic Heart Disease Undergoing Ventricular Tachycardia Ablation. Front. Cardiovasc. Med. 2021, 8, 647812. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Usami, S.; Kuwabara, Y.; Horie, T.; Baba, O.; Hakuno, D.; Nakashima, Y.; Nishiga, M.; Izuhara, M.; Nakao, T.; et al. Expression Patterns of miRNA-423-5p in the Serum and Pericardial Fluid in Patients Undergoing Cardiac Surgery. PLoS ONE 2015, 10, e0142904. [Google Scholar] [CrossRef] [PubMed]
- Schoettler, F.I.; Hassanabad, A.F.; Jadli, A.S.; Patel, V.B.; Fedak, P.W.M. Exploring the role of pericardial miRNAs and exosomes in modulating cardiac fibrosis. Cardiovasc. Pathol. Off. J. Soc. Cardiovasc. Pathol. 2024, 107671. [Google Scholar] [CrossRef]
- Fiedler, J.; Thum, T. MicroRNAs in Myocardial Infarction. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Zhang, C. MicroRNA-21 in cardiovascular disease. J. Cardiovasc. Transl. Res. 2010, 3, 251–255. [Google Scholar] [CrossRef]
- Huang, S.; Chen, M.; Li, L.; He, M.; Hu, D.; Zhang, X.; Li, J.; Tanguay, R.M.; Feng, J.; Cheng, L.; et al. Circulating MicroRNAs and the occurrence of acute myocardial infarction in Chinese populations. Circ. Cardiovasc. Genet. 2014, 7, 189–198. [Google Scholar] [CrossRef]
- Vegter, E.L.; Ovchinnikova, E.S.; van Veldhuisen, D.J.; Jaarsma, T.; Berezikov, E.; van der Meer, P.; Voors, A.A. Low circulating microRNA levels in heart failure patients are associated with atherosclerotic disease and cardiovascular-related rehospitalizations. Clin. Res. Cardiol. 2017, 106, 598–609. [Google Scholar] [CrossRef] [PubMed]
- Vegter, E.L.; van der Meer, P.; de Windt, L.J.; Pinto, Y.M.; Voors, A.A. MicroRNAs in heart failure: From biomarker to target for therapy. Eur. J. Heart Fail. 2016, 18, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Ramanujam, D.; Sassi, Y.; Laggerbauer, B.; Engelhardt, S. Viral Vector-Based Targeting of miR-21 in Cardiac Nonmyocyte Cells Reduces Pathologic Remodeling of the Heart. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 1939–1948. [Google Scholar] [CrossRef] [PubMed]
- de Jong, S.; van Veen, T.A.; de Bakker, J.M.; Vos, M.A.; van Rijen, H.V. Biomarkers of myocardial fibrosis. J. Cardiovasc. Pharmacol. 2011, 57, 522–535. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Chang, H.; Zhang, H.; Zhang, L. MiR-30e Attenuates Isoproterenol-induced Cardiac Fibrosis Through Suppressing Snai1/TGF-β Signaling. J. Cardiovasc. Pharmacol. 2017, 70, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, V.; Rai, R.; Place, A.T.; Murphy, S.B.; Verma, S.K.; Ghosh, A.K.; Vaughan, D.E. MiR-125b Is Critical for Fibroblast-to-Myofibroblast Transition and Cardiac Fibrosis. Circulation 2016, 133, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Chen, H.; Ge, D.; Xu, Y.; Xu, H.; Yang, Y.; Gu, M.; Zhou, Y.; Zhu, J.; Ge, T.; et al. Mir-21 Promotes Cardiac Fibrosis After Myocardial Infarction Via Targeting Smad7. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2017, 42, 2207–2219. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Wang, C.M.; Qin, J.Y.; Zhang, Y.J.; Xia, D.S.; Chen, X.; Guo, S.Z.; Zhao, X.D.; Guo, Q.Y.; Lu, C.Z. Effect of miR-203 expression on myocardial fibrosis. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 837–842. [Google Scholar] [PubMed]
- Bayoumi, A.S.; Teoh, J.P.; Aonuma, T.; Yuan, Z.; Ruan, X.; Tang, Y.; Su, H.; Weintraub, N.L.; Kim, I.M. MicroRNA-532 protects the heart in acute myocardial infarction, and represses prss23, a positive regulator of endothelial-to-mesenchymal transition. Cardiovasc. Res. 2017, 113, 1603–1614. [Google Scholar] [CrossRef]
- Hong, Y.; Cao, H.; Wang, Q.; Ye, J.; Sui, L.; Feng, J.; Cai, X.; Song, H.; Zhang, X.; Chen, X. MiR-22 may Suppress Fibrogenesis by Targeting TGFβR I in Cardiac Fibroblasts. Cell Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2016, 40, 1345–1353. [Google Scholar] [CrossRef]
- Wang, X.; Wang, L.; Sun, Y.; Chen, B.; Xiong, L.; Chen, J.; Huang, M.; Wu, J.; Tan, X.; Zheng, Y.; et al. MiR-22-3p inhibits fibrotic cataract through inactivation of HDAC6 and increase of α-tubulin acetylation. Cell Prolif. 2020, 53, e12911. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wen, H.; Huang, Y. MicroRNA-146a attenuates isoproterenol-induced cardiac fibrosis by inhibiting FGF2. Exp. Ther. Med. 2022, 24, 506. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Cao, X.; Ge, L.; Gu, Y.; Lv, X.; Getachew, T.; Mwacharo, J.M.; Haile, A.; Sun, W. MiR-22-3p Inhibits Proliferation and Promotes Differentiation of Skeletal Muscle Cells by Targeting IGFBP3 in Hu Sheep. Animals 2022, 12, 114. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K.; Foinquinos, A.; Thum, S.; Remke, J.; Zimmer, K.; Bauters, C.; Groote, P.d.; Boon, R.A.; Windt, L.J.d.; Preissl, S.; et al. Preclinical Development of a MicroRNA-Based Therapy for Elderly Patients With Myocardial Infarction. J. Am. Coll. Cardiol. 2016, 68, 1557–1571. [Google Scholar] [CrossRef] [PubMed]
- Fatehi Hassanabad, A.; Zarzycki, A.; Deniset, J.F.; Fedak, P.W. An overview of human pericardial space and pericardial fluid. Cardiovasc. Pathol. 2021, 53, 107346. [Google Scholar] [CrossRef] [PubMed]
- Suryadevara, K.; George, M.; Jena, A.; Dhandapani, V.E.; Damodharan, N.; J, J. Evaluation of soluble ST2 as a novel cardiovascular biomarker in patients with acute myocardial infarction. Int. J. Res. Med. Sci. 2016, 4, 5297–5301. [Google Scholar] [CrossRef]
- Kakkar, R.; Lee, R.T. The IL-33/ST2 pathway: Therapeutic target and novel biomarker. Nat. Rev. Drug Discov. 2008, 7, 827–840. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Li, H.; Sun, Y.; Qiu, J.; Xu, H.; Liu, J.; Zhou, M.; Chen, A.; Ye, X.; Wang, Z.; et al. Pericardial fluid levels of growth differentiation factor 15 in patients with or without coronary artery disease: A prospective study. Ann. Transl. Med. 2020, 8, 113. [Google Scholar] [CrossRef]
- Kempf, T.; Björklund, E.; Olofsson, S.; Lindahl, B.; Allhoff, T.; Peter, T.; Tongers, J.; Wollert, K.C.; Wallentin, L. Growth-differentiation factor-15 improves risk stratification in ST-segment elevation myocardial infarction. Eur. Heart J. 2007, 28, 2858–2865. [Google Scholar] [CrossRef]
- Ege, T.; Canbaz, S.; Yuksel, V.; Duran, E. Effect of pericardial fluid pro-inflammatory cytokines on hemodynamic parameters. Cytokine 2003, 23, 47–51. [Google Scholar] [CrossRef]
- Oyama, J.-I.; Shimokawa, H.; Morita, S.; Yasui, H.; Takeshita, A. Elevated interleukin-1β in pericardial fluid of patients with ischemic heart disease. Coron. Artery Dis. 2001, 12, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, Z.; Cziraki, A.; Szabados, S.; Horvath, I.; Koller, A. Pericardial fluid of cardiac patients elicits arterial constriction: Role of endothelin-1. Can. J. Physiol. Pharmacol. 2015, 93, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Böhm, F.; Pernow, J. The importance of endothelin-1 for vascular dysfunction in cardiovascular disease. Cardiovasc. Res. 2007, 76, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Chinyere, I.R.; Bradley, P.; Uhlorn, J.; Eason, J.; Mohran, S.; Repetti, G.G.; Daugherty, S.; Koevary, J.W.; Goldman, S.; Lancaster, J.J. Epicardially Placed Bioengineered Cardiomyocyte Xenograft in Immune-Competent Rat Model of Heart Failure. Stem Cells Int. 2021, 2021, 9935679. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, X.; Lin, Q.; Xu, Q. Up-regulation of microRNA-203 inhibits myocardial fibrosis and oxidative stress in mice with diabetic cardiomyopathy through the inhibition of PI3K/Akt signaling pathway via PIK3CA. Gene 2019, 715, 143995. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.L.; Xu, H.; Liu, Z.B.; Wu, Q.C.; Zhu, R.R.; Liu, J.C. miR-21 promotes cardiac fibroblast-to-myofibroblast transformation and myocardial fibrosis by targeting Jagged1. J. Cell. Mol. Med. 2018, 22, 3816–3824. [Google Scholar] [CrossRef] [PubMed]
- Thum, T.; Gross, C.; Fiedler, J.; Fischer, T.; Kissler, S.; Bussen, M.; Galuppo, P.; Just, S.; Rottbauer, W.; Frantz, S.; et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 2008, 456, 980–984. [Google Scholar] [CrossRef] [PubMed]
- Abbas, N.; Haas, J.A.; Xiao, K.; Fuchs, M.; Just, A.; Pich, A.; Perbellini, F.; Werlein, C.; Ius, F.; Ruhparwar, A.; et al. Inhibition of miR-21: Cardioprotective effects in human failing myocardium ex vivo. Eur. Heart J. 2024, 45, ehae102. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Hu, Y.; Li, H.; Guo, X.; Zhong, J.; He, S. miR-22-3p as a potential biomarker for coronary artery disease based on integrated bioinformatics analysis. Front. Genet. 2022, 13, 936937. [Google Scholar] [CrossRef]
- Maciejak, A.; Kiliszek, M.; Opolski, G.; Segiet, A.; Matlak, K.; Dobrzycki, S.; Tulacz, D.; Sygitowicz, G.; Burzynska, B.; Gora, M. miR-22-5p revealed as a potential biomarker involved in the acute phase of myocardial infarction via profiling of circulating microRNAs. Mol. Med. Rep. 2016, 14, 2867–2875. [Google Scholar] [CrossRef]
- Wang, Y.; Chang, W.; Zhang, Y.; Zhang, L.; Ding, H.; Qi, H.; Xue, S.; Yu, H.; Hu, L.; Liu, D.; et al. Circulating miR-22-5p and miR-122-5p are promising novel biomarkers for diagnosis of acute myocardial infarction. J. Cell. Physiol. 2019, 234, 4778–4786. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.S.; Ren, Y.; Wu, Y.; Ren, H.K.; Chen, H. MiR-30b-5p and miR-22-3p restrain the fibrogenesis of post-myocardial infarction in mice via targeting PTAFR. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 3993–4004. [Google Scholar] [CrossRef] [PubMed]
- Jazbutyte, V.; Fiedler, J.; Kneitz, S.; Galuppo, P.; Just, A.; Holzmann, A.; Bauersachs, J.; Thum, T. MicroRNA-22 increases senescence and activates cardiac fibroblasts in the aging heart. Age 2013, 35, 747–762. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.; Law, J.P.; Reyat, J.S.; Cumberland, M.J.; Hang, S.; Vo, N.T.N.; Raniga, K.; Weston, C.J.; O’Shea, C.; Townend, J.N.; et al. Chronic activation of human cardiac fibroblasts in vitro attenuates the reversibility of the myofibroblast phenotype. Sci. Rep. 2023, 13, 12137. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.P.; Wang, D.Z. miR-22 in cardiac remodeling and disease. Trends Cardiovasc. Med. 2014, 24, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Siklenka, K.; Arora, S.K.; Ribeiro, P.; Kimmins, S.; Xia, J. miRNet—Dissecting miRNA-target interactions and functional associations through network-based visual analysis. Nucleic Acids Res. 2016, 44, W135–W141. [Google Scholar] [CrossRef] [PubMed]
- Filgueira, C.S.; Igo, S.R.; Wang, D.K.; Hirsch, M.; Schulz, D.G.; Bruckner, B.A.; Grattoni, A. Technologies for intrapericardial delivery of therapeutics and cells. Adv. Drug Deliv. Rev. 2019, 151, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Vitsios, D.M.; Enright, A.J. Chimira: Analysis of small RNA sequencing data and microRNA modifications. Bioinformatics 2015, 31, 3365–3367. [Google Scholar] [CrossRef]
- Babicki, S.; Arndt, D.; Marcu, A.; Liang, Y.; Grant, J.R.; Maciejewski, A.; Wishart, D.S. Heatmapper: Web-enabled heat mapping for all. Nucleic Acids Res. 2016, 44, W147–W153. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CTRL (n = 9) | NSTEMI (n = 25) | STEMI (n = 8) | ANOVA p-Value | Dunn’s Multiple Comparisons Test p-Value | |||
|---|---|---|---|---|---|---|---|
| CTRL vs. NSTEMI | CTRL vs. STEMI | NSTEMI vs. STEMI | |||||
| Age (years) | 65.6 ± 8.8 | 68.8 ± 7.1 | 62.2 ± 9.2 | NS | NS | NS | NS |
| Female (%) | 1 (11) | 1 (4) | 1 (13) | NS | NS | NS | NS |
| LV EF (%) | 55.75 ± 5.97 | 45.04 ± 13.07 | 44.75 ± 10.74 | 0.005 | 0.06 | 0.14 | NS |
| IHD Risk Factors | |||||||
| BMI (Kg·m−2) | 28.23 ± 2.56 | 27.50 ± 4.10 | 26.89 ± 4.00 | NS | NS | NS | NS |
| Diabetes mellitus (%) | 4 (44) | 13 (52) | 5 (62) | NS | NS | NS | NS |
| Hypertension (%) | 5 (56) | 21 (84) | 6 (75) | NS | NS | NS | NS |
| Dyslipedemia (%) | 8 (89) | 18 (72) | 7 (88) | NS | NS | NS | NS |
| Smokers (%) | 1 (11) | 4 (16) | 2 (25) | NS | NS | NS | NS |
| Ex-smokers (%) | 5 (56) | 10 (40) | 4 (50) | NS | NS | NS | NS |
| Previous IHD (%) | 0 (0) | 4 (16) | 1 (13) | NS | NS | NS | NS |
| Control (n = 9) | NSTEMI (n = 25) | STEMI (n = 8) | ANOVA p-Value | Dunn’s Multiple Comparisons Test p-Value | |||
|---|---|---|---|---|---|---|---|
| CTRL vs. MI | CTRL vs. MI | CTRL vs. MI | |||||
| Aspirin (%) | 6 (66.66) | 16 (64.00) | 5 (62.50) | NS | NS | NS | NS |
| β-blockers (%) | 6 (66.66) | 14 (56.00) | 3 (37.50 | NS | NS | NS | NS |
| Statins (%) | 7 (77.78) | 16 (64.00) | 8 (100.0) | NS | NS | NS | NS |
| ACEI (%) | 3 (33.33) | 9 (36.00) | 2 (25.00) | NS | NS | NS | NS |
| Antidiabetics (%) | 3 (33.33) | 11 (44.00) | 6 (75.00) | NS | NS | NS | NS |
| Fibrates (%) | 0 (0.00) | 0 (0.00) | 1 (12.50) | NS | NS | NS | NS |
| Ca2+ Channel Blockers (%) | 5 (55.55) | 5 (20.00) | 0 (0.00) | NS | NS | NS | NS |
| Antiaggregant (%) | 1 (11.11) | 6 (24.00) | 2 (25.00) | NS | NS | NS | NS |
| Nitrates (%) | 3 (33.33) | 9 (36.00) | 1 (12.50) | NS | NS | NS | NS |
| ARB (%) | 1 (11.11) | 7 (28.00) | 0 (0.00) | NS | NS | NS | NS |
| Spironolactone (%) | 0 (0.00) | 3 (12.00) | 0 (0.00) | NS | NS | NS | NS |
| Bronchodilators (%) | 2 (22.22) | 3 (12.00) | 0 (0.00) | NS | NS | NS | NS |
| Anticoagulants (%) | 1 (11.11) | 3 (12.00) | 1 (12.50) | NS | NS | NS | NS |
| Corticosteroids (%) | 1 (11.11) | 2 (8.00) | 0 (0.00) | NS | NS | NS | NS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, E.D.; Pereira-Sousa, D.; Ribeiro-Costa, F.; Cerqueira, R.; Enguita, F.J.; Gomes, R.N.; Dias-Ferreira, J.; Pereira, C.; Castanheira, A.; Pinto-do-Ó, P.; et al. Pericardial Fluid Accumulates microRNAs That Regulate Heart Fibrosis after Myocardial Infarction. Int. J. Mol. Sci. 2024, 25, 8329. https://doi.org/10.3390/ijms25158329
Silva ED, Pereira-Sousa D, Ribeiro-Costa F, Cerqueira R, Enguita FJ, Gomes RN, Dias-Ferreira J, Pereira C, Castanheira A, Pinto-do-Ó P, et al. Pericardial Fluid Accumulates microRNAs That Regulate Heart Fibrosis after Myocardial Infarction. International Journal of Molecular Sciences. 2024; 25(15):8329. https://doi.org/10.3390/ijms25158329
Chicago/Turabian StyleSilva, Elsa D., Daniel Pereira-Sousa, Francisco Ribeiro-Costa, Rui Cerqueira, Francisco J. Enguita, Rita N. Gomes, João Dias-Ferreira, Cassilda Pereira, Ana Castanheira, Perpétua Pinto-do-Ó, and et al. 2024. "Pericardial Fluid Accumulates microRNAs That Regulate Heart Fibrosis after Myocardial Infarction" International Journal of Molecular Sciences 25, no. 15: 8329. https://doi.org/10.3390/ijms25158329