
Insulin–Heart Axis: Bridging Physiology to Insulin Resistance
 , ,
, ,  , , , , and
, , , , and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Insulin Signaling and Metabolic Regulation in Cardiac Function
3. Insulin Signaling and the Regulation of Physiological Cardiac Hypertrophy
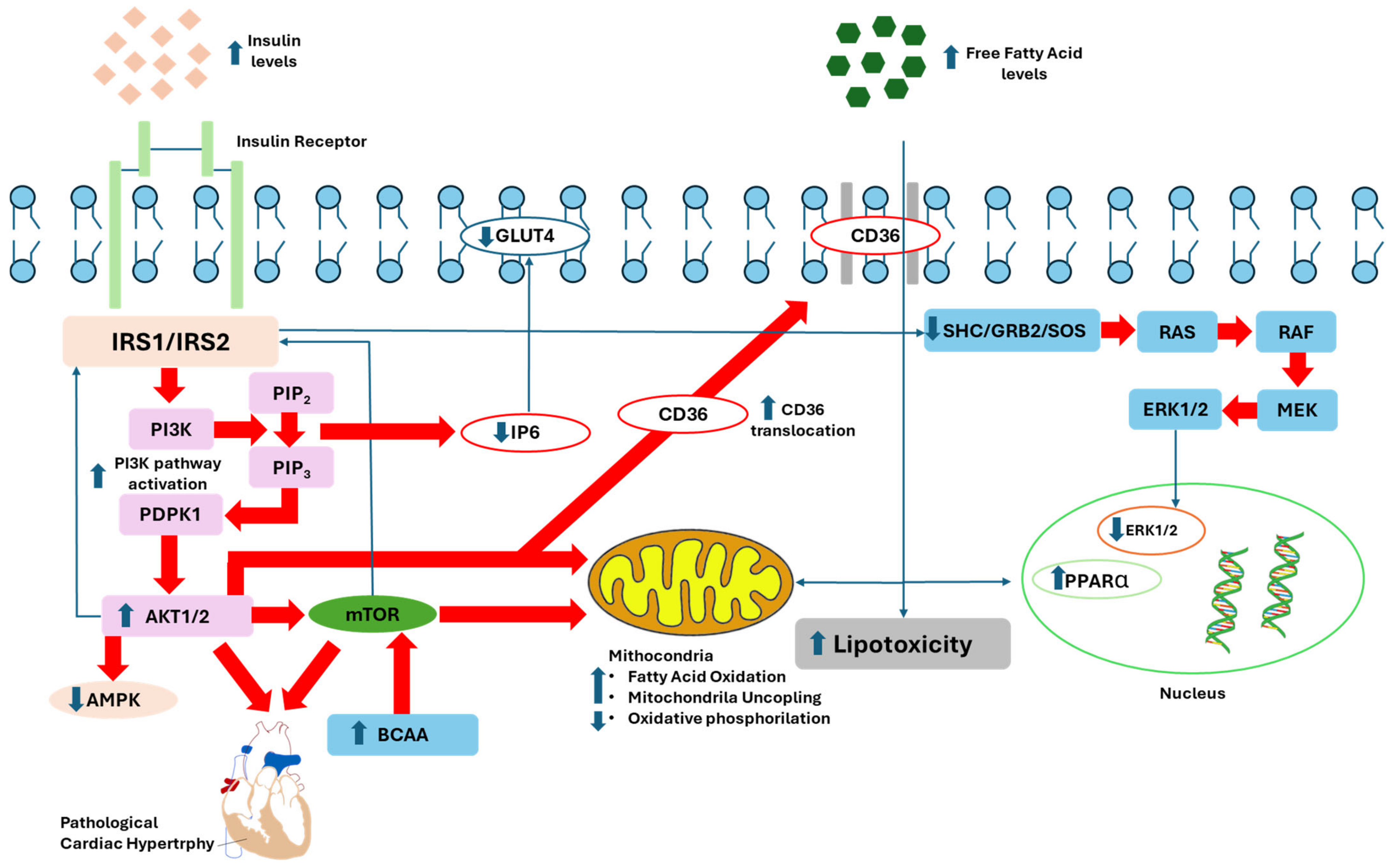
4. Pathophysiology of Insulin Resistance
4.1. Pathophysiological Mechanisms of Proximal Insulin Signaling Impairment
4.2. Pathophysiological Mechanisms of Distal Insulin Signaling Impairment
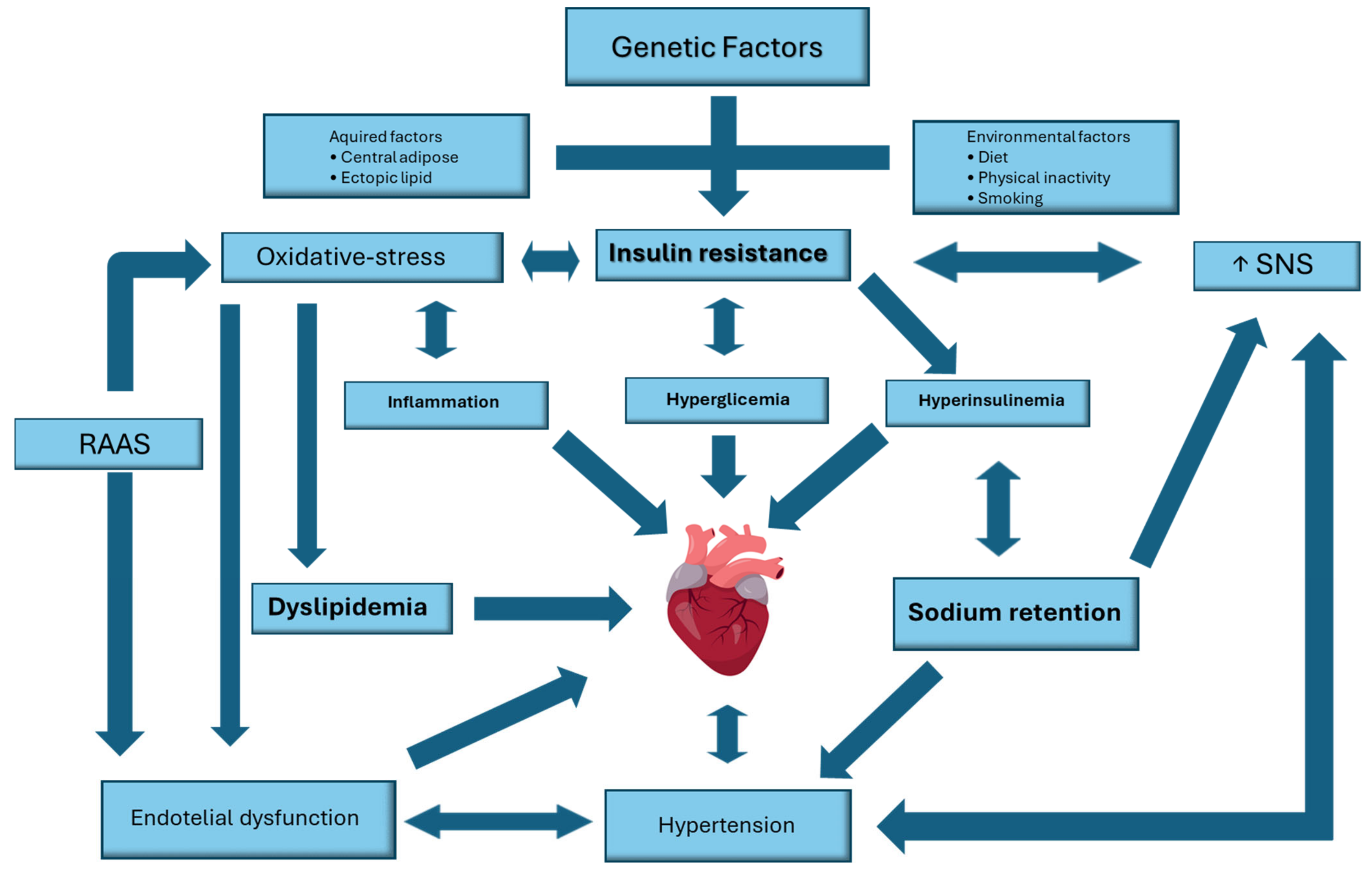
5. Cardiovascular Manifestations of Insulin Resistance
5.1. Cardiac Metabolic Disturbances
5.2. Autonomic Dysfunction
5.3. Subcellular Signalling Abnormalities
5.4. Renin–Angiotensin–Aldosterone System
5.5. Inflammation and Maladaptive Immune Response
6. Cardiovascular Diseases
6.1. Epidemiology Variations in the Context of Diabetes and Cardiovascular Disease Risks
6.1.1. Variations by Age
6.1.2. Variations by Sex
6.1.3. Ethnic Variations
6.2. Impact of Insulin Resistance on Cardiac Remodeling and Hypertrophy
6.3. The Role of Insulin Resistance in Altered Lipid Metabolism and Cardiovascular Risk
6.4. The Impact of Insulin Resistance on Vascular Health and Abdominal Aortic Aneurysm Risk
7. Cardiovascular Advantages of Insulin Therapy
7.1. Experimental Models
7.2. Clinical Setting
8. Future Directions
9. Research Methodology
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Abel, E.D. Insulin signaling in the heart. Am. J. Physiol. Endocrinol. Metab. 2021, 321, E130–E145. [Google Scholar] [CrossRef] [PubMed]
- Belke, D.D.; Betuing, S.; Tuttle, M.J.; Graveleau, C.; Young, M.E.; Pham, M.; Zhang, D.; Cooksey, R.C.; McClain, D.A.; Litwin, S.E.; et al. Insulin signaling coordinately regulates cardiac size, metabolism, and contractile protein isoform expression. J. Clin. Investig. 2002, 109, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Opie, L.H.; Lopaschuk, G.D. Fuels: Aerobic and anaerobic metabolism. In Heart Physiology, from Cell to Circulation; Opie, L.H., Ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2004; pp. 306–354. [Google Scholar]
- Kolwicz, S.C., Jr.; Purohit, S.; Tian, R. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ. Res. 2013, 113, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Randle, P.J.; Garland, P.B.; Hales, C.N.; Newsholme, E.A. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963, 1, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Opie, L.H. Cardiac metabolism–emergence, decline, and resurgence. Part II. Cardiovasc. Res. 1992, 26, 817–830. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, L.; Horman, S.; Beauloye, C.; Vanoverschelde, J.L. Insulin signalling in the heart. Cardiovasc. Res. 2008, 79, 238–248. [Google Scholar] [CrossRef]
- Chen, Y.X.; Zhao, A.R.; Wei, T.W.; Wang, H.; Wang, L.S. Progress of Mitochondrial Function Regulation in Cardiac Regeneration. J. Cardiovasc. Transl. Res. 2024. [Google Scholar] [CrossRef]
- Wang, L.Y.; Chen, C. Energy metabolism homeostasis in cardiovascular diseases. J. Geriatr. Cardiol. 2021, 18, 1044–1057. [Google Scholar] [CrossRef] [PubMed]
- Ritterhoff, J.; Tian, R. Metabolic mechanisms in physiological and pathological cardiac hypertrophy: New paradigms and challenges. Nat. Rev. Cardiol. 2023, 20, 812–829. [Google Scholar] [CrossRef]
- Wilhelmi de Toledo, F.; Grundler, F.; Sirtori, C.R.; Ruscica, M. Unravelling the health effects of fasting: A long road from obesity treatment to healthy life span increase and improved cognition. Ann. Med. 2020, 52, 147–161. [Google Scholar] [CrossRef]
- Ikeda, H.; Shiojima, I.; Ozasa, Y.; Yoshida, M.; Holzenberger, M.; Kahn, C.R.; Walsh, K.; Igarashi, T.; Abel, E.D.; Komuro, I. Interaction of myocardial insulin receptor and IGF receptor signaling in exercise-induced cardiac hypertrophy. J. Mol. Cell. Cardiol. 2009, 47, 664–675. [Google Scholar] [CrossRef] [PubMed]
- Hermida, M.A.; Dinesh Kumar, J.; Leslie, N.R. GSK3 and its interactions with the PI3K/AKT/mTOR signalling network. Adv. Biol. Regul. 2017, 65, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Vidal, S.; Bouzaher, Y.H.; El Motiam, A.; Seoane, R.; Rivas, C. Overview of the regulation of the class IA PI3K/AKT pathway by SUMO. Semin. Cell Dev. Biol. 2022, 132, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Shiojima, I.; Yefremashvili, M.; Luo, Z.; Kureishi, Y.; Takahashi, A.; Tao, J.; Rosenzweig, A.; Kahn, C.R.; Abel, E.D.; Walsh, K. Akt signaling mediates postnatal heart growth in response to insulin and nutritional status. J. Biol. Chem. 2002, 277, 37670–37677. [Google Scholar] [CrossRef] [PubMed]
- Skurk, C.; Izumiya, Y.; Maatz, H.; Razeghi, P.; Shiojima, I.; Sandri, M.; Sato, K.; Zeng, L.; Schiekofer, S.; Pimentel, D.; et al. The FOXO3a transcription factor regulates cardiac myocyte size downstream of AKT signaling. J. Biol. Chem. 2005, 280, 20814–20823. [Google Scholar] [CrossRef] [PubMed]
- Safaroghli-Azar, A.; Sanaei, M.J.; Pourbagheri-Sigaroodi, A.; Bashash, D. Phosphoinositide 3-kinase (PI3K) classes: From cell signaling to endocytic recycling and autophagy. Eur. J. Pharmacol. 2023, 953, 175827. [Google Scholar] [CrossRef] [PubMed]
- Riehle, C.; Abel, E.D. Insulin signaling and heart failure. Circ. Res. 2016, 118, 1151–1169. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Folmes, C.D.; Stanley, W.C. Cardiac energy metabolism in obesity. Circ. Res. 2007, 101, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Stanley, W.C.; Recchia, F.A.; Lopaschuk, G.D. Myocardial substrate metabolism in the normal and failing heart. Physiol. Rev. 2005, 85, 1093–1129. [Google Scholar] [CrossRef]
- Kodde, I.F.; van der Stok, J.; Smolenski, R.T.; de Jong, J.W. Metabolic and genetic regulation of cardiac energy substrate preference. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2007, 146, 26–39. [Google Scholar] [CrossRef]
- Ventura-Clapier, R.; Garnier, A.; Veksler, V. Energy metabolism in heart failure. J. Physiol. 2004, 555, 1–13. [Google Scholar] [CrossRef]
- Siddle, K.; Urso, B.; Niesler, C.A.; Cope, D.L.; Molina, L.; Surinya, K.H.; Soos, M.A. Specificity in ligand binding and intracellular signalling by insulin and insulin-like growth factor receptors. Biochem. Soc. Trans. 2001, 29, 513–525. [Google Scholar] [CrossRef]
- Nakae, J.; Kido, Y.; Accili, D. Distinct and overlapping functions of insulin and IGF-I receptors. Endocr. Rev. 2001, 22, 818–835. [Google Scholar] [CrossRef]
- Avruch, J. Insulin signal transduction through protein kinase cascades. Mol. Cell. Biochem. 1998, 182, 31–48. [Google Scholar] [CrossRef] [PubMed]
- Saltiel, A.R.; Kahn, C.R. Insulin signalling and the regulation of glucose and lipid metabolism. Nature 2001, 414, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Muniyappa, R.; Montagnani, M.; Koh, K.K.; Quon, M.J. Cardiovascular actions of insulin. Endocr. Rev. 2007, 28, 463–491. [Google Scholar] [CrossRef]
- Thirone, A.C.; Huang, C.; Klip, A. Tissue-specific roles of IRS proteins in insulin signaling and glucose transport. Trends Endocrinol. Metab. 2006, 17, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Heineke, J.; Molkentin, J.D. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat. Rev. Mol. Cell Biol. 2006, 7, 589–600. [Google Scholar] [CrossRef]
- McMullen, J.R.; Jennings, G.L. Differences between pathological and physiological cardiac hypertrophy: Novel therapeutic strategies to treat heart failure. Clin. Exp. Pharmacol. Physiol. 2007, 34, 255–262. [Google Scholar] [CrossRef]
- Proud, C.G. Ras, PI3-kinase and mTOR signaling in cardiac hypertrophy. Cardiovasc. Res. 2004, 63, 403–413. [Google Scholar] [CrossRef]
- Wang, Y. Mitogen-activated protein kinases in heart development and diseases. Circulation 2007, 116, 1413–1423. [Google Scholar] [CrossRef] [PubMed]
- DeBosch, B.; Sambandam, N.; Weinheimer, C.; Courtois, M.; Muslin, A.J. Akt2 regulates cardiac metabolism and cardiomyocyte survival. J. Biol. Chem. 2006, 281, 32841–32851. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Yan, X.; Dai, X.; Wang, Y.; Lin, Q.; Xiao, J.; Zhou, S.; Zhang, J.; Wang, K.; Zeng, J.; et al. Metallothionein Preserves Akt2 Activity and Cardiac Function via Inhibiting TRB3 in Diabetic Hearts. Diabetes 2018, 67, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Abel, E.D.; Kaulbach, H.C.; Tian, R.; Hopkins, J.C.; Duffy, J.; Doetschman, T.; Minnemann, T.; Boers, M.E.; Hadro, E.; Oberste-Berghaus, C.; et al. Cardiac hypertrophy with preserved contractile function after selective deletion of GLUT4 from the heart. J. Clin. Investig. 1999, 104, 1703–1714. [Google Scholar] [CrossRef] [PubMed]
- Tian, R.; Abel, E.D. Responses of GLUT4-deficient hearts to ischemia underscore the importance of glycolysis. Circulation 2001, 103, 2961–2966. [Google Scholar] [CrossRef] [PubMed]
- Wende, A.R.; Kim, J.; Holland, W.L.; Wayment, B.E.; O’Neill, B.T.; Tuinei, J.; Brahma, M.K.; Pepin, M.E.; McCrory, M.A.; Luptak, I.; et al. Glucose transporter 4-deficient hearts develop maladaptive hypertrophy in response to physiological or pathological stresses. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H1098–H1108. [Google Scholar] [CrossRef] [PubMed]
- Fischer-Rasokat, U.; Beyersdorf, F.; Doenst, T. Insulin addition after ischemia improves recovery of function equal to ischemic preconditioning in rat heart. Basic Res. Cardiol. 2003, 98, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Fischer-Rasokat, U.; Doenst, T. Insulin-induced improvement of postischemic recovery is abolished by inhibition of protein kinase C in rat heart. J. Thorac. Cardiovasc. Surg. 2003, 126, 1806–1812. [Google Scholar] [CrossRef] [PubMed]
- Zaha, V.; Francischetti, I.; Doesn’t, T. Insulin improves postischemic recovery of function through PI3K in isolated working rat heart. Mol. Cell. Biochem. 2003, 247, 229–232. [Google Scholar] [CrossRef]
- Fullmer, T.M.; Pei, S.; Zhu, Y.; Sloan, C.; Manzanares, R.; Henrie, B.; Pires, K.M.; Cox, J.E.; Abel, E.D.; Boudina, S. Insulin suppresses ischemic preconditioning-mediated cardioprotection through Akt-dependent mechanisms. J. Mol. Cell. Cardiol. 2013, 64, 20–29. [Google Scholar] [CrossRef]
- Watson, R.T.; Pessin, J.E. Bridging the GAP between insulin signaling and GLUT4 translocation. Trends Biochem. Sci. 2006, 31, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Watson, R.T.; Pessin, J.E. GLUT4 translocation: The last 200 nanometers. Cell Signal. 2007, 19, 2209–2217. [Google Scholar] [CrossRef]
- Huang, S.; Czech, M.P. The GLUT4 glucose transporter. Cell Metab. 2007, 5, 237–252. [Google Scholar] [CrossRef] [PubMed]
- He, A.; Liu, X.; Liu, L.; Chang, Y.; Fang, F. How many signals impinge on GLUT4 activation by insulin? Cell Signal. 2007, 19, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Berwick, D.C.; Dell, G.C.; Welsh, G.I.; Heesom, K.J.; Hers, I.; Fletcher, L.M.; Cooke, F.T.; Tavaré, J.M. Protein kinase B phosphorylation of PIKfyve regulates the trafficking of GLUT4 vesicles. J. Cell Sci. 2004, 117, 5985–5993. [Google Scholar] [CrossRef]
- Yamada, E.; Okada, S.; Saito, T.; Ohshima, K.; Sato, M.; Tsuchiya, T.; Uehara, Y.; Shimizu, H.; Mori, M. Akt2 phosphorylates Synip to regulate docking and fusion of GLUT4-containing vesicles. J. Cell Biol. 2005, 168, 921–928. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, V.; Mechin, M.C.; Louckx, M.P.; Rider, M.H.; Hue, L. Signaling pathway involved in the activation of heart 6-phosphofructo-2-kinase by insulin. J. Biol. Chem. 1996, 271, 22289–22292. [Google Scholar] [CrossRef]
- Hue, L.; Beauloye, C.; Marsin, A.S.; Bertrand, L.; Horman, S.; Rider, M.H. Insulin and ischemia stimulate glycolysis by acting on the same targets through different and opposing signaling pathways. J. Mol. Cell. Cardiol. 2002, 34, 1091–1097. [Google Scholar] [CrossRef]
- Rider, M.H.; Bertrand, L.; Vertommen, D.; Michels, P.A.; Rousseau, G.G.; Hue, L. 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase: Head-to-head with a bifunctional enzyme that controls glycolysis. Biochem. J. 2004, 381, 561–579. [Google Scholar] [CrossRef]
- Rider, M.H.; Hue, L. Activation of rat heart phosphofructokinase-2 by insulin in vivo. FEBS Lett. 1984, 176, 484–488. [Google Scholar] [CrossRef]
- Murashige, D.; Jang, C.; Neinast, M.; Edwards, J.J.; Cowan, A.; Hyman, M.C.; Rabinowitz, J.D.; Frankel, D.S.; Arany, Z. Comprehensive quantification of fuel use by the failing and nonfailing human heart. Science 2020, 370, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Barrett, E.J.; Schwartz, R.G.; Francis, C.K.; Zaret, B.L. Regulation by insulin of myocardial glucose and fatty acid metabolism in the conscious dog. J. Clin. Investig. 1984, 74, 1073–1079. [Google Scholar] [CrossRef] [PubMed]
- Peterson, L.R.; Herrero, P.; McGill, J.; Schechtman, K.B.; Kisrieva-Ware, Z.; Lesniak, D.; Gropler, R.J. Fatty acids and insulin modulate myocardial substrate metabolism in humans with type 1 diabetes. Diabetes 2008, 57, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Labbé, S.M.; Grenier-Larouche, T.; Noll, C.; Phoenix, S.; Guérin, B.; Turcotte, E.E.; Carpentier, A.C. Increased myocardial uptake of dietary fatty acids linked to cardiac dysfunction in glucose-intolerant humans. Diabetes 2012, 61, 2701–2710. [Google Scholar] [CrossRef]
- Labbé, S.M.; Grenier-Larouche, T.; Croteau, E.; Normand-Lauzière, F.; Frisch, F.; Ouellet, R.; Guérin, B.; Turcotte, E.E.; Carpentier, A.C. Organ-specific dietary fatty acid uptake in humans using positron emission tomography coupled to computed tomography. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E445–E453. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.R., 3rd; Cline, G.W.; Guthrie, P.H.; Goodwin, G.W.; Shulman, G.I.; Taegtmeyer, H. Regulation of exogenous and endogenous glucose metabolism by insulin and acetoacetate in the isolated working rat heart. A three tracer study of glycolysis, glycogen metabolism, and glucose oxidation. J. Clin. Investig. 1997, 100, 2892–2899. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.M.; Goodwin, G.W.; Guthrie, P.H.; Taegtmeyer, H. Effects of insulin on glucose uptake by rat hearts during and after coronary flow reduction. Am. J. Physiol. 1997, 273, H2170–H2177. [Google Scholar] [CrossRef]
- Mazumder, P.K.; O’Neill, B.T.; Roberts, M.W.; Buchanan, J.; Yun, U.J.; Cooksey, R.C.; Boudina, S.; Abel, E.D. Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes 2004, 53, 2366–2374. [Google Scholar] [CrossRef]
- Karwi, Q.G.; Wagg, C.S.; Altamimi, T.R.; Uddin, G.M.; Ho, K.L.; Darwesh, A.M.; Seubert, J.M.; Lopaschuk, G.D. Insulin directly stimulates mitochondrial glucose oxidation in the heart. Cardiovasc. Diabetol. 2020, 19, 207. [Google Scholar] [CrossRef]
- Kovacic, S.; Soltys, C.L.; Barr, A.J.; Shiojima, I.; Walsh, K.; Dyck, J.R. Akt activity negatively regulates phosphorylation of AMP-activated protein kinase in the heart. J. Biol. Chem. 2003, 278, 39422–39427. [Google Scholar] [CrossRef]
- Gamble, J.; Lopaschuk, G.D. Insulin inhibition of 5′ adenosine monophosphate-activated protein kinase in the heart results in activation of acetyl coenzyme A carboxylase and inhibition of fatty acid oxidation. Metabolism 1997, 46, 1270–1274. [Google Scholar] [CrossRef] [PubMed]
- Luiken, J.J.; Ouwens, D.M.; Habets, D.D.; van der Zon, G.C.; Coumans, W.A.; Schwenk, R.W.; Bonen, A.; Glatz, J.F. Permissive action of protein kinase C-zeta in insulin-induced CD36- and GLUT4 translocation in cardiac myocytes. J. Endocrinol. 2009, 201, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.S.; Luiken, J.J.; Snook, L.A.; Han, X.X.; Holloway, G.P.; Glatz, J.F.; Bonen, A. Fatty acid transport and transporters in muscle are critically regulated by Akt2. FEBS Lett. 2015, 589, 2769–2775. [Google Scholar] [CrossRef] [PubMed]
- Banke, N.H.; Wende, A.R.; Leone, T.C.; O’Donnell, J.M.; Abel, E.D.; Kelly, D.P.; Lewandowski, E.D. Preferential oxidation of triacylglyceride-derived fatty acids in heart is augmented by the nuclear receptor PPARalpha. Circ. Res. 2010, 107, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Hedhli, N.; Pelat, M.; Depre, C. Protein turnover in cardiac cell growth and survival. Cardiovasc. Res. 2005, 68, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Proud, C.G. Signalling to translation: How signal transduction pathways control the protein synthetic machinery. Biochem. J. 2007, 403, 217–234. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef]
- Potter, C.J.; Pedraza, L.G.; Xu, T. Akt regulates growth by directly phosphorylating Tsc2. Nat. Cell Biol. 2002, 4, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Cantley, L.C. Rheb fills a GAP between TSC and TOR. Trends Biochem. Sci. 2003, 28, 573–576. [Google Scholar] [CrossRef]
- Rolfe, M.; McLeod, L.E.; Pratt, P.F.; Proud, C.G. Activation of protein synthesis in cardiomyocytes by the hypertrophic agent phenylephrine requires the activation of ERK and involves phosphorylation of tuberous sclerosis complex 2 (TSC2). Biochem. J. 2005, 388, 973–984. [Google Scholar] [CrossRef]
- Pham, F.H.; Sugden, P.H.; Clerk, A. Regulation of protein kinase B and 4E-BP1 by oxidative stress in cardiac myocytes. Circ. Res. 2000, 86, 1252–1258. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Guthrie, P.H.; Chan, S.S.; Haq, S.; Taegtmeyer, H. Glucose phosphorylation is required for insulin-dependent mTOR signalling in the heart. Cardiovasc. Res. 2007, 76, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, X.; Proud, C.G. Activation of mRNA translation in rat cardiac myocytes by insulin involves multiple rapamycin-sensitive steps. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H1056–H1068. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Wende, A.R.; Sena, S.; Theobald, H.A.; Soto, J.; Sloan, C.; Wayment, B.E.; Litwin, S.E.; Holzenberger, M.; LeRoith, D.; et al. Insulin-like growth factor I receptor signaling is required for exercise-induced cardiac hypertrophy. Mol. Endocrinol. 2008, 22, 2531–2543. [Google Scholar] [CrossRef] [PubMed]
- Riehle, C.; Wende, A.R.; Zhu, Y.; Oliveira, K.J.; Pereira, R.O.; Jaishy, B.P.; Bevins, J.; Valdez, S.; Noh, J.; Kim, B.J.; et al. Insulin receptor substrates are essential for the bioenergetic and hypertrophic response of the heart to exercise training. Mol. Cell. Biol. 2014, 34, 3450–3460. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chi, X.; Wang, Y.; Setrerrahmane, S.; Xie, W.; Xu, H. Trends in insulin resistance: Insights into mechanisms and therapeutic strategy. Signal Transduct. Target. Ther. 2022, 7, 216. [Google Scholar] [CrossRef]
- Gojda, J.; Patková, J.; Jaček, M.; Potočková, J.; Trnka, J.; Kraml, P.; Anděl, M. Higher insulin sensitivity in vegans is not associated with higher mitochondrial density. Eur. J. Clin. Nutr. 2013, 67, 1310–1315. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, P.; Weiskirchen, R. Cellular and Molecular Mechanisms of Insulin Resistance. Curr. Tissue Microenviron. Rep. 2024. [Google Scholar] [CrossRef]
- Hsu, P.P.; Kang, S.A.; Rameseder, J.; Zhang, Y.; Ottina, K.A.; Lim, D.; Peterson, T.R.; Choi, Y.; Gray, N.S.; Yaffe, M.B.; et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science 2011, 332, 1317–1322. [Google Scholar] [CrossRef]
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef]
- Caro, J.F.; Sinha, M.K.; Raju, S.M.; Ittoop, O.; Pories, W.J.; Flickinger, E.G.; Meelheim, D.; Dohm, G.L. Insulin receptor kinase in human skeletal muscle from obese subjects with and without noninsulin dependent diabetes. J. Clin. Investig. 1987, 79, 1330–1337. [Google Scholar] [CrossRef] [PubMed]
- Fröjdö, S.; Vidal, H.; Pirola, L. Alterations of insulin signaling in type 2 diabetes: A review of the current evidence from humans. Biochim. Biophys. Acta 2009, 1792, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Fisher, S.J.; Kahn, C.R. Insulin signaling is required for insulin’s direct and indirect action on hepatic glucose production. J. Clin. Investig. 2003, 111, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Michael, M.D.; Kulkarni, R.N.; Postic, C.; Previs, S.F.; Shulman, G.I.; Magnuson, M.A.; Kahn, C.R. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol. Cell. 2000, 6, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, V.; Uchida, T.; Yenush, L.; Davis, R.; White, M.F. The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307). J. Biol. Chem. 2000, 275, 9047–9054. [Google Scholar] [CrossRef] [PubMed]
- Carvalho-Filho, M.A.; Ueno, M.; Hirabara, S.M.; Seabra, A.B.; Carvalheira, J.B.; de Oliveira, M.G.; Velloso, L.A.; Curi, R.; Saad, M.J. S-nitrosation of the insulin receptor, insulin receptor substrate 1, and protein kinase B/Akt: A novel mechanism of insulin resistance. Diabetes 2005, 54, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.; Bai, X.C. The Activation Mechanism of the Insulin Receptor: A Structural Perspective. Annu. Rev. Biochem. 2023, 92, 247–272. [Google Scholar] [CrossRef] [PubMed]
- Yunn, N.O.; Kim, J.; Ryu, S.H.; Cho, Y. A stepwise activation model for the insulin receptor. Exp. Mol. Med. 2023, 55, 2147–2161. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical nodes in signalling pathways: Insights into insulin action. Nat. Rev. Mol. Cell. Biol. 2006, 7, 85–96. [Google Scholar] [CrossRef]
- Brachmann, S.M.; Ueki, K.; Engelman, J.A.; Kahn, R.C.; Cantley, L.C. Phosphoinositide 3-kinase catalytic subunit deletion and regulatory subunit deletion have opposite effects on insulin sensitivity in mice. Mol. Cell. Biol. 2005, 25, 1596–1607. [Google Scholar] [CrossRef]
- Cheatham, B.; Vlahos, C.J.; Cheatham, L.; Wang, L.; Blenis, J.; Kahn, C.R. Phosphatidylinositol 3-kinase activation is required for insulin stimulation of pp70 S6 kinase, DNA synthesis, and glucose transporter translocation. Mol. Cell. Biol. 1994, 14, 4902–4911. [Google Scholar] [CrossRef] [PubMed]
- Czech, M.P.; Corvera, S. Signaling mechanisms that regulate glucose transport. J. Biol. Chem. 1999, 274, 1865–1868. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Sobkiw, C.L.; Hirshman, M.F.; Logsdon, M.N.; Li, T.Q.; Goodyear, L.J.; Cantley, L.C. Loss of class IA PI3K signaling in muscle leads to impaired muscle growth, insulin response, and hyperlipidemia. Cell. Metab. 2006, 3, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.N.; Chen, H.; Li, Y.; Zhou, L.; McGibbon, M.A.; Taylor, S.I.; Quon, M.J. Physiological role of Akt in insulin-stimulated translocation of GLUT4 in transfected rat adipose cells. Mol. Endocrinol. 1997, 11, 1881–1890. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.Y.; Holland, W.L.; Kusminski, C.M.; Sun, K.; Sharma, A.X.; Pearson, M.J.; Sifuentes, A.J.; McDonald, J.G.; Gordillo, R.; Scherer, P.E. Targeted Induction of Ceramide Degradation Leads to Improved Systemic Metabolism and Reduced Hepatic Steatosis. Cell Metab. 2015, 22, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Le Marchand-Brustel, Y.; Grémeaux, T.; Ballotti, R.; Van Obberghen, E. Insulin receptor tyrosine kinase is defective in skeletal muscle of insulin-resistant obese mice. Nature 1985, 315, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Vetrano, E.; Rinaldi, L.; Mormone, A.; Giorgione, C.; Galiero, R.; Caturano, A.; Nevola, R.; Marfella, R.; Sasso, F.C. Non-alcoholic Fatty Liver Disease (NAFLD), Type 2 Diabetes, and Non-viral Hepatocarcinoma: Pathophysiological Mechanisms and New Therapeutic Strategies. Biomedicines 2023, 11, 468. [Google Scholar] [CrossRef]
- Galiero, R.; Caturano, A.; Vetrano, E.; Cesaro, A.; Rinaldi, L.; Salvatore, T.; Marfella, R.; Sardu, C.; Moscarella, E.; Gragnano, F.; et al. Pathophysiological mechanisms and clinical evidence of relationship between Nonalcoholic fatty liver disease (NAFLD) and cardiovascular disease. Rev. Cardiovasc. Med. 2021, 22, 755–768. [Google Scholar] [CrossRef]
- Brozinick, J.T., Jr.; Roberts, B.R.; Dohm, G.L. Defective signaling through Akt-2 and -3 but not Akt-1 in insulin-resistant human skeletal muscle: Potential role in insulin resistance. Diabetes 2003, 52, 935–941. [Google Scholar] [CrossRef]
- Kruszynska, Y.T.; Worrall, D.S.; Ofrecio, J.; Frias, J.P.; Macaraeg, G.; Olefsky, J.M. Fatty acid-induced insulin resistance: Decreased muscle PI3K activation but unchanged Akt phosphorylation. J. Clin. Endocrinol. Metab. 2002, 87, 226–234. [Google Scholar] [CrossRef]
- Mackenzie, R.W.; Elliott, B.T. Akt/PKB activation and insulin signaling: A novel insulin signaling pathway in the treatment of type 2 diabetes. Diabetes Metab. Syndr. Obes. 2014, 7, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.; Kim, Y.B. Molecular mechanism of insulin resistance in obesity and type 2 diabetes. Korean J. Intern. Med. 2010, 25, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Kahn, B.B. Lilly lecture 1995. Glucose transport: Pivotal step in insulin action. Diabetes 1996, 45, 1644–1654. [Google Scholar] [CrossRef] [PubMed]
- Dimitriadis, G.; Mitrou, P.; Lambadiari, V.; Maratou, E.; Raptis, S.A. Insulin effects in muscle and adipose tissue. Diabetes Res. Clin. Pract. 2011, 93, S52–S59. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Houseknecht, K.L.; Stenbit, A.E.; Katz, E.B.; Charron, M.J. Reduced glucose uptake precedes insulin signaling defects in adipocytes from heterozygous GLUT4 knockout mice. FASEB J. 2000, 14, 1117–1125. [Google Scholar] [CrossRef] [PubMed]
- Klip, A.; Ramlal, T.; Bilan, P.J.; Cartee, G.D.; Gulve, E.A.; Holloszy, J.O. Recruitment of GLUT-4 glucose transporters by insulin in diabetic rat skeletal muscle. Biochem. Biophys. Res. Commun. 1990, 172, 728–736. [Google Scholar] [CrossRef]
- Etgen, G.J., Jr.; Jensen, J.; Wilson, C.M.; Hunt, D.G.; Cushman, S.W.; Ivy, J.L. Exercise training reverses insulin resistance in muscle by enhanced recruitment of GLUT-4 to the cell surface. Am. J. Physiol. 1997, 272, E864–E869. [Google Scholar] [CrossRef] [PubMed]
- Ryder, J.W.; Yang, J.; Galuska, D.; Rincón, J.; Björnholm, M.; Krook, A.; Lund, S.; Pedersen, O.; Wallberg-Henriksson, H.; Zierath, J.R.; et al. Use of a novel impermeable biotinylated photolabeling reagent to assess insulin- and hypoxia-stimulated cell surface GLUT4 content in skeletal muscle from type 2 diabetic patients. Diabetes 2000, 49, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Garvey, W.T.; Maianu, L.; Zhu, J.H.; Hancock, J.A.; Golichowski, A.M. Multiple defects in the adipocyte glucose transport system cause cellular insulin resistance in gestational diabetes. Heterogeneity in the number and a novel abnormality in subcellular localization of GLUT4 glucose transporters. Diabetes 1993, 42, 1773–1785. [Google Scholar] [CrossRef]
- Salvatore, T.; Galiero, R.; Caturano, A.; Vetrano, E.; Rinaldi, L.; Coviello, F.; Di Martino, A.; Albanese, G.; Colantuoni, S.; Medicamento, G.; et al. Dysregulated Epicardial Adipose Tissue as a Risk Factor and Potential Therapeutic Target of Heart Failure with Preserved Ejection Fraction in Diabetes. Biomolecules 2022, 12, 176. [Google Scholar] [CrossRef]
- D’Andrea, A.; Ilardi, F.; D’Ascenzi, F.; Bandera, F.; Benfari, G.; Esposito, R.; Malagoli, A.; Mandoli, G.E.; Santoro, C.; Russo, V.; et al. Impaired myocardial work efficiency in heart failure with preserved ejection fraction. Eur. Heart J. Cardiovasc. Imaging 2021, 22, 1312–1320. [Google Scholar] [CrossRef] [PubMed]
- Chadt, A.; Immisch, A.; de Wendt, C.; Springer, C.; Zhou, Z.; Stermann, T.; Holman, G.D.; Loffing-Cueni, D.; Loffing, J.; Joost, H.G.; et al. Deletion of both Rab-GTPase–activating proteins TBC1D1 and TBC1D4 in mice eliminates insulin- and AICAR-stimulated glucose transport. Diabetes 2015, 64, 746–759. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wasserman, D.H.; MacKintosh, C.; Sakamoto, K. Mice with AS160/TBC1D4-Thr649Ala knockin mutation are glucose intolerant with reduced insulin sensitivity and altered GLUT4 trafficking. Cell Metab. 2011, 13, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Chatree, S.; Thongmaen, N.; Tantivejkul, K.; Sitticharoon, C.; Vucenik, I. Role of Inositols and Inositol Phosphates in Energy Metabolism. Molecules 2020, 25, 5079. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, H.; Liu, J. Akt activation: A potential strategy to ameliorate insulin resistance. Diabetes Res. Clin. Pract. 2019, 156, 107092. [Google Scholar] [CrossRef] [PubMed]
- Lemmer, I.L.; Willemsen, N.; Hilal, N.; Bartelt, A. A guide to understanding endoplasmic reticulum stress in metabolic disorders. Mol. Metab. 2021, 47, 101169. [Google Scholar] [CrossRef] [PubMed]
- Alicka, M.; Marycz, K. The Effect of Chronic Inflammation and Oxidative and Endoplasmic Reticulum Stress. in the Course of Metabolic Syndrome and Its Therapy. Stem Cells Int. 2018, 2018, 4274361. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, L.; Tabas, I. Role of endoplasmic reticulum stress in metabolic disease and other disorders. Annu. Rev. Med. 2012, 63, 317–328. [Google Scholar] [CrossRef]
- Kim, J.N.; Han, S.N.; Kim, H.K. Phytic acid and myo-inositol support adipocyte differentiation and improve insulin sensitivity in 3T3-L1 cells. Nutr. Res. 2014, 34, 723–731. [Google Scholar] [CrossRef]
- Alvarez, L.; Avila, M.A.; Mato, J.M.; Castaño, J.G.; Varela-Nieto, I. Insulin-like effects of inositol phosphate-glycan on messenger RNA expression in rat hepatocytes. Mol. Endocrinol. 1991, 5, 1062–1068. [Google Scholar] [CrossRef]
- Shamsuddin, A.K.; Bose, S. IP6 (Inositol Hexaphosphate) as a Signaling Molecule. Curr. Cancer Rev. 2012, 7, 289–304. [Google Scholar] [CrossRef]
- Zeke, A.; Misheva, M.; Reményi, A.; Bogoyevitch, M.A. JNK Signaling: Regulation and Functions Based on Complex Protein-Protein Partnerships. Microbiol. Mol. Biol. Rev. 2016, 80, 793–835. [Google Scholar] [CrossRef] [PubMed]
- Papaconstantinou, J. The Role of Signaling Pathways of Inflammation and Oxidative Stress in Development of Senescence and Aging Phenotypes in Cardiovascular Disease. Cells 2019, 8, 1383. [Google Scholar] [CrossRef] [PubMed]
- Min, R.W.M.; Aung, F.W.M.; Liu, B.; Arya, A.; Win, S. Mechanism and Therapeutic Targets of c-Jun-N-Terminal Kinases Activation in Nonalcoholic Fatty Liver Disease. Biomedicines 2022, 10, 2035. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; DeMarco, V.G.; Sowers, J.R. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat. Rev. Endocrinol. 2016, 12, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Huo, J.L.; Feng, Q.; Pan, S.; Fu, W.J.; Liu, Z.; Liu, Z. Diabetic cardiomyopathy: Early diagnostic biomarkers, pathogenetic mechanisms, and therapeutic interventions. Cell Death Discov. 2023, 9, 256. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Shen, W.; Zhao, B.; Wang, Y.; Wertz, K.; Weber, P.; Zhang, P. Targeting mitochondrial biogenesis for preventing and treating insulin resistance in diabetes and obesity: Hope from natural mitochondrial nutrients. Adv. Drug Deliv. Rev. 2009, 61, 1343–1352. [Google Scholar] [CrossRef] [PubMed]
- Caturano, A.; D’Angelo, M.; Mormone, A.; Russo, V.; Mollica, M.P.; Salvatore, T.; Galiero, R.; Rinaldi, L.; Vetrano, E.; Marfella, R.; et al. Oxidative Stress in Type 2 Diabetes: Impacts from Pathogenesis to Lifestyle Modifications. Curr. Issues Mol. Biol. 2023, 45, 6651–6666. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Liu, X.; Panagia, V.; Takeda, N. Subcellular remodeling and heart dysfunction in chronic diabetes. Cardiovasc. Res. 1998, 40, 239–247. [Google Scholar] [CrossRef]
- Di Francia, R.; Rinaldi, L.; Cillo, M.; Varriale, E.; Facchini, G.; D’Aniello, C.; Marotta, G.; Berretta, M. Antioxidant diet and genotyping as tools for the prevention of liver disease. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 5155–5163. [Google Scholar]
- Jain, S.S.; Paglialunga, S.; Vigna, C.; Ludzki, A.; Herbst, E.A.; Lally, J.S.; Schrauwen, P.; Hoeks, J.; Tupling, A.R.; Bonen, A.; et al. High-fat diet-induced mitochondrial biogenesis is regulated by mitochondrial-derived reactive oxygen species activation of CaMKII. Diabetes 2014, 63, 1907–1913. [Google Scholar] [CrossRef] [PubMed]
- Adameova, A.; Dhalla, N.S. Role of microangiopathy in diabetic cardiomyopathy. Heart Fail. Rev. 2014, 19, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, T.; Galiero, R.; Caturano, A.; Vetrano, E.; Loffredo, G.; Rinaldi, L.; Catalini, C.; Gjeloshi, K.; Albanese, G.; Di Martino, A.; et al. Coronary Microvascular Dysfunction in Diabetes Mellitus: Pathogenetic Mechanisms and Potential Therapeutic Options. Biomedicines 2022, 10, 2274. [Google Scholar] [CrossRef] [PubMed]
- Battiprolu, P.K.; Lopez-Crisosto, C.; Wang, Z.V.; Nemchenko, A.; Lavandero, S.; Hill, J.A. Diabetic cardiomyopathy and metabolic remodeling of the heart. Life Sci. 2013, 92, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Mytas, D.Z.; Stougiannos, P.N.; Zairis, M.N.; Foussas, S.G.; Pyrgakis, V.N.; Kyriazis, I.A. Diabetic myocardial disease: Pathophysiology, early diagnosis and therapeutic options. J. Diabetes Complicat. 2009, 23, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Harmancey, R.; Lam, T.N.; Lubrano, G.M.; Guthrie, P.H.; Vela, D.; Taegtmeyer, H. Insulin resistance improves metabolic and contractile efficiency in stressed rat heart. FASEB J. 2012, 26, 3118–3126. [Google Scholar] [CrossRef] [PubMed]
- Mandavia, C.H.; Pulakat, L.; DeMarco, V.; Sowers, J.R. Over-nutrition and metabolic cardiomyopathy. Metabolism 2012, 61, 1205–1210. [Google Scholar] [CrossRef] [PubMed]
- Sasso, F.C.; Salvatore, T.; Tranchino, G.; Cozzolino, D.; Caruso, A.A.; Persico, M.; Gentile, S.; Torella, D.; Torella, R. Cochlear dysfunction in type 2 diabetes: A complication independent of neuropathy and acute hyperglycemia. Metabolism 1999, 48, 1346–1350. [Google Scholar] [CrossRef] [PubMed]
- Galiero, R.; Caturano, A.; Vetrano, E.; Beccia, D.; Brin, C.; Alfano, M.; Di Salvo, J.; Epifani, R.; Piacevole, A.; Tagliaferri, G.; et al. Peripheral Neuropathy in Diabetes Mellitus: Pathogenetic Mechanisms and Diagnostic Options. Int. J. Mol. Sci. 2023, 24, 3554. [Google Scholar] [CrossRef]
- Spallone, V.; Ziegler, D.; Freeman, R.; Bernardi, L.; Frontoni, S.; Pop-Busui, R.; Stevens, M.; Kempler, P.; Hilsted, J.; Tesfaye, S.; et al. Toronto Consensus Panel on Diabetic Neuropathy. Cardiovascular autonomic neuropathy in diabetes: Clinical impact, assessment, diagnosis, and management. Diabetes Metab. Res. Rev. 2011, 27, 639–653. [Google Scholar] [CrossRef]
- La Rovere, M.T.; Pinna, G.D.; Raczak, G. Baroreflex sensitivity: Measurement and clinical implications. Ann. Noninvasive Electrocardiol. 2008, 13, 191–207. [Google Scholar] [CrossRef] [PubMed]
- Paulson, D.J.; Light, K.E. Elevation of serum and ventricular norepinephrine content in the diabetic rat. Res. Commun. Chem. Pathol. Pharmacol. 1981, 33, 559–562. [Google Scholar] [PubMed]
- Givertz, M.M.; Sawyer, D.B.; Colucci, W.S. Antioxidants and myocardial contractility: Illuminating the “Dark Side” of beta-adrenergic receptor activation? Circulation 2001, 103, 782–783. [Google Scholar] [CrossRef] [PubMed]
- Kellogg, A.P.; Converso, K.; Wiggin, T.; Stevens, M.; Pop-Busui, R. Effects of cyclooxygenase-2 gene inactivation on cardiac autonomic and left ventricular function in experimental diabetes. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H453–H461. [Google Scholar] [CrossRef] [PubMed]
- Communal, C.; Singh, K.; Pimentel, D.R.; Colucci, W.S. Norepinephrine stimulates apoptosis in adult rat ventricular myocytes by activation of the beta-adrenergic pathway. Circulation 1998, 98, 1329–1334. [Google Scholar] [CrossRef] [PubMed]
- Schnell, O.; Kirsch, C.M.; Stemplinger, J.; Haslbeck, M.; Standl, E. Scintigraphic evidence for cardiac sympathetic dysinnervation in long-term IDDM patients with and without ECG-based autonomic neuropathy. Diabetologia 1995, 38, 1345–1352. [Google Scholar] [CrossRef] [PubMed]
- Sasso, F.C.; Rinaldi, L.; Lascar, N.; Marrone, A.; Pafundi, P.C.; Adinolfi, L.E.; Marfella, R. Role of Tight Glycemic Control during Acute Coronary Syndrome on CV Outcome in Type 2 Diabetes. J. Diabetes Res. 2018, 2018, 3106056. [Google Scholar] [CrossRef] [PubMed]
- Caturano, A.; Galiero, R.; Pafundi, P.C.; Cesaro, A.; Vetrano, E.; Palmiero, G.; Rinaldi, L.; Salvatore, T.; Marfella, R.; Sardu, C.; et al. Does a strict glycemic control during acute coronary syndrome play a cardioprotective effect? Pathophysiology and clinical evidence. Diabetes Res. Clin. Pract. 2021, 178, 108959. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.Y.; Prins, J.B.; Marwick, T.H. Diabetic cardiomyopathy: Evidence, mechanisms, and therapeutic implications. Endocr. Rev. 2004, 25, 543–567. [Google Scholar] [CrossRef]
- Borovac, J.A.; D’Amario, D.; Bozic, J.; Glavas, D. Sympathetic nervous system activation and heart failure: Current state of evidence and the pathophysiology in the light of novel biomarkers. World J. Cardiol. 2020, 12, 373–408. [Google Scholar] [CrossRef]
- da Silva, A.A.; do Carmo, J.M.; Li, X.; Wang, Z.; Mouton, A.J.; Hall, J.E. Role of Hyperinsulinemia and Insulin Resistance in Hypertension: Metabolic Syndrome Revisited. Can. J. Cardiol. 2020, 36, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Larkin, D.; Yamada, E.; Raffel, D.M.; Stevens, M.J. Sympathetic dysfunction in type 1 diabetes: Association with impaired myocardial blood flow reserve and diastolic dysfunction. J. Am. Coll. Cardiol. 2004, 44, 2368–2374. [Google Scholar] [CrossRef]
- Dinh, W.; Füth, R.; Lankisch, M.; Bansemir, L.; Nickl, W.; Scheffold, T.; Bufe, A.; Krahn, T.; Ziegler, D. Cardiovascular autonomic neuropathy contributes to left ventricular diastolic dysfunction in subjects with Type 2 diabetes and impaired glucose tolerance undergoing coronary angiography. Diabet. Med. 2011, 28, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Pop-Busui, R.; Cleary, P.A.; Braffett, B.H.; Martin, C.L.; Herman, W.H.; Low, P.A.; Lima, J.A.C.; Bluemke, D.A.; DCCT/EDIC Research Group. Association between cardiovascular autonomic neuropathy and left ventricular dysfunction: DCCT/EDIC study (Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications). J. Am. Coll. Cardiol. 2013, 61, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Rowe, G.C.; Jiang, A.; Arany, Z. PGC-1 coactivators in cardiac development and disease. Circ. Res. 2010, 107, 825–838. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Zhao, Y.; He, M.; Li, H.; Fan, J.; Nie, X.; Yan, M.; Chen, C.; Wang, D.W. MiR30c/PGC-1β protects against diabetic cardiomyopathy via PPARα. Cardiovasc. Diabetol. 2019, 18, 7. [Google Scholar] [CrossRef] [PubMed]
- Mandavia, C.H.; Aroor, A.R.; Demarco, V.G.; Sowers, J.R. Molecular and metabolic mechanisms of cardiac dysfunction in diabetes. Life Sci. 2013, 92, 601–608. [Google Scholar] [CrossRef]
- Muoio, D.M.; Way, J.M.; Tanner, C.J.; Winegar, D.A.; Kliewer, S.A.; Houmard, J.A.; Kraus, W.E.; Dohm, G.L. Peroxisome proliferator-activated receptor-alpha regulates fatty acid utilization in primary human skeletal muscle cells. Diabetes 2002, 51, 901–909. [Google Scholar] [CrossRef] [PubMed]
- Lam, V.H.; Zhang, L.; Huqi, A.; Fukushima, A.; Tanner, B.A.; Onay-Besikci, A.; Keung, W.; Kantor, P.F.; Jaswal, J.S.; Rebeyka, I.M.; et al. Activating PPARα prevents post-ischemic contractile dysfunction in hypertrophied neonatal hearts. Circ. Res. 2015, 117, 41–51. [Google Scholar] [CrossRef]
- Buchanan, J.; Mazumder, P.K.; Hu, P.; Chakrabarti, G.; Roberts, M.W.; Yun, U.J.; Cooksey, R.C.; Litwin, S.E.; Abel, E.D. Reduced cardiac efciency and altered substrate metabolism precedes the onset of hyperglycemia andcontractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology 2005, 146, 5341–5349. [Google Scholar] [CrossRef]
- Salvatore, T.; Pafundi, P.C.; Galiero, R.; Albanese, G.; Di Martino, A.; Caturano, A.; Vetrano, E.; Rinaldi, L.; Sasso, F.C. The Diabetic Cardiomyopathy: The Contributing Pathophysiological Mechanisms. Front. Med. 2021, 8, 695792. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Whaley-Connell, A.; Sowers, J.R. Diabetic cardiomyopathy: A hyperglycaemia- and insulin-resistance-induced heart disease. Diabetologia 2018, 61, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Wollert, K.C.; Drexler, H. The renin-angiotensin system and experimental heart failure. Cardiovasc. Res. 1999, 43, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Bai, T.; Xu, Z.; Liu, Q.; Zheng, Y.; Cai, L. Pathophysiological Fundamentals of Diabetic Cardiomyopathy. Compr. Physiol. 2017, 7, 693–711. [Google Scholar] [CrossRef] [PubMed]
- Huynh, K.; Kiriazis, H.; Du, X.J.; Love, J.E.; Gray, S.P.; Jandeleit-Dahm, K.A.; McMullen, J.R.; Ritchie, R.H. Targeting the upregulation of reactive oxygen species subsequent to hyperglycemia prevents type 1 diabetic cardiomyopathy in mice. Free Radic. Biol. Med. 2013, 60, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, M.; Nakamura, T.; Kataoka, K.; Nako, H.; Tokutomi, Y.; Dong, Y.F.; Ogawa, H.; Kim-Mitsuyama, S. Potentiation by candesartan of protective effects of pioglitazone against type 2 diabetic cardiovascular and renal complications in obese mice. J. Hypertens. 2010, 28, 340–352. [Google Scholar] [CrossRef]
- Kim, J.A.; Jang, H.J.; Martinez-Lemus, L.A.; Sowers, J.R. Activation of mTOR/p70S6 kinase by ANG II inhibits insulin-stimulated endothelial nitric oxide synthase and vasodilation. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E201–E208. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Habibi, J.; DeMarco, V.G.; Martinez-Lemus, L.A.; Ma, L.; Whaley-Connell, A.T.; Aroor, A.R.; Domeier, T.L.; Zhu, Y.; Meininger, G.A.; et al. Endothelial Mineralocorticoid Receptor Deletion Prevents Diet-Induced Cardiac Diastolic Dysfunction in Females. Hypertension 2015, 66, 1159–1167. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Aroor, A.R.; Martinez-Lemus, L.A.; Sowers, J.R. Overnutrition, mTOR signaling, and cardiovascular diseases. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 307, R1198–R1206. [Google Scholar] [CrossRef]
- Tirosh, A.; Garg, R.; Adler, G.K. Mineralocorticoid receptor antagonists and the metabolic syndrome. Curr. Hypertens. Rep. 2010, 12, 252–257. [Google Scholar] [CrossRef]
- Falcao-Pires, I.; Leite-Moreira, A.F. Diabetic cardiomyopathy: Understanding the molecular and cellular basis to progress in diagnosis and treatment. Heart Fail. Rev. 2012, 17, 325–344. [Google Scholar] [CrossRef] [PubMed]
- Kellogg, A.P.; Wiggin, T.D.; Larkin, D.D.; Hayes, J.M.; Stevens, M.J.; Pop-Busui, R. Protective effects of cyclooxygenase-2 gene inactivation against peripheral nerve dysfunction and intraepidermal nerve fiber loss in experimental diabetes. Diabetes 2007, 56, 2997–3005. [Google Scholar] [CrossRef] [PubMed]
- Asrih, M.; Mach, F.; Nencioni, A.; Dallegri, F.; Quercioli, A.; Montecucco, F. Role of mitogen-activated protein kinase pathways in multifactorial adverse cardiac remodeling associated with metabolic syndrome. Mediators Inflamm. 2013, 2013, 367245. [Google Scholar] [CrossRef] [PubMed]
- Weirather, J.; Hofmann, U.D.; Beyersdorf, N.; Ramos, G.C.; Vogel, B.; Frey, A.; Ertl, G.; Kerkau, T.; Frantz, S. Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ. Res. 2014, 115, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Sell, H.; Habich, C.; Eckel, J. Adaptive immunity in obesity and insulin resistance. Nat. Rev. Endocrinol. 2012, 8, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Ait-Oufella, H.; Salomon, B.L.; Potteaux, S.; Robertson, A.K.; Gourdy, P.; Zoll, J.; Merval, R.; Esposito, B.; Cohen, J.L.; Fisson, S.; et al. Natural regulatory T cells control the development of atherosclerosis in mice. Nat. Med. 2006, 12, 178–180. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Vazquez, R.; Zabadi, S.; Watson, R.R.; Larson, D.F. T-lymphocytes mediate left ventricular fibrillar collagen cross-linking and diastolic dysfunction in mice. Matrix Biol. 2010, 29, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, O.; Picatoste, B.; Ares-Carrasco, S.; Ramírez, E.; Egido, J.; Tuñón, J. Potential role of nuclear factor κB in diabetic cardiomyopathy. Mediators Infamm. 2011, 2011, 65209. [Google Scholar]
- Jones, W.K.; Brown, M.; Ren, X.; He, S.; McGuinness, M. NF-kappaB as an integrator of diverse signaling pathways: The heart of myocardial signaling? Cardiovasc. Toxicol. 2003, 3, 229–254. [Google Scholar] [CrossRef]
- Chia, C.W.; Egan, J.M.; Ferrucci, L. Age-Related Changes in Glucose Metabolism, Hyperglycemia, and Cardiovascular Risk. Circ. Res. 2018, 123, 886–904. [Google Scholar] [CrossRef]
- Abdul-Ghani, M.A.; Jayyousi, A.; DeFronzo, R.A.; Asaad, N.; Al-Suwaidi, J. Insulin Resistance the Link between T2DM and CVD: Basic Mechanisms and Clinical Implications. Curr. Vasc. Pharmacol. 2019, 17, 153–163. [Google Scholar] [CrossRef]
- Ghodeshwar, G.K.; Dube, A.; Khobragade, D. Impact of Lifestyle Modifications on Cardiovascular Health: A Narrative Review. Cureus 2023, 15, e42616. [Google Scholar] [CrossRef]
- Qiu, J.; Bosch, M.A.; Meza, C.; Navarro, U.V.; Nestor, C.C.; Wagner, E.J.; Rønnekleiv, O.K.; Kelly, M.J. Estradiol Protects Proopiomelanocortin Neurons Against Insulin Resistance. Endocrinology 2018, 159, 647–664. [Google Scholar] [CrossRef]
- Zidon, T.M.; Padilla, J.; Fritsche, K.L.; Welly, R.J.; McCabe, L.T.; Stricklin, O.E.; Frank, A.; Park, Y.; Clegg, D.J.; Lubahn, D.B.; et al. Effects of ERβ and ERα on OVX-induced changes in adiposity and insulin resistance. J. Endocrinol. 2020, 245, 165–178. [Google Scholar] [CrossRef]
- Ikeda, K.; Horie-Inoue, K.; Inoue, S. Functions of estrogen and estrogen receptor signaling on skeletal muscle. J. Steroid Biochem. Mol. Biol. 2019, 191, 105375. [Google Scholar] [CrossRef] [PubMed]
- Gerdts, E.; Regitz-Zagrosek, V. Sex differences in cardiometabolic disorders. Nat. Med. 2019, 25, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Faulkner, J.L.; Belin de Chantemèle, E.J. Sex hormones, aging and cardiometabolic syndrome. Biol. Sex. Differ. 2019, 10, 30. [Google Scholar] [CrossRef]
- Geer, E.B.; Shen, W. Gender differences in insulin resistance, body composition, and energy balance. Gend. Med. 2009, 6, 60–75. [Google Scholar] [CrossRef] [PubMed]
- Christen, T.; Trompet, S.; Noordam, R.; van Klinken, J.B.; van Dijk, K.W.; Lamb, H.J.; Cobbaert, C.M.; den Heijer, M.; Jazet, I.M.; Jukema, J.W.; et al. Sex differences in body fat distribution are related to sex differences in serum leptin and adiponectin. Peptides 2018, 107, 25–31. [Google Scholar] [CrossRef]
- Ehtisham, S.; Crabtree, N.; Clark, P.; Shaw, N.; Barrett, T. Ethnic differences in insulin resistance and body composition in United Kingdom adolescents. J. Clin. Endocrinol. Metab. 2005, 90, 3963–3969. [Google Scholar] [CrossRef]
- Lear, S.A.; Kohli, S.; Bondy, G.P.; Tchernof, A.; Sniderman, A.D. Ethnic variation in fat and lean body mass and the association with insulin resistance. J. Clin. Endocrinol. Metab. 2009, 94, 4696–4702. [Google Scholar] [CrossRef] [PubMed]
- Hasson, B.R.; Apovian, C.; Istfan, N. Racial/Ethnic Differences in Insulin Resistance and Beta Cell Function: Relationship to Racial Disparities in Type 2 Diabetes among African Americans versus Caucasians. Curr. Obes. Rep. 2015, 4, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Gower, B.A.; Nagy, T.R.; Goran, M.I. Visceral fat, insulin sensitivity, and lipids in prepubertal children. Diabetes 1999, 48, 1515–1521. [Google Scholar] [CrossRef] [PubMed]
- Osei, K.; Cottrell, D.A.; Harris, B. Differences in basal and poststimulation glucose homeostasis in nondiabetic first degree relatives of black and white patients with type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 1992, 75, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Kodama, K.; Tojjar, D.; Yamada, S.; Toda, K.; Patel, C.J.; Butte, A.J. Ethnic differences in the relationship between insulin sensitivity and insulin response: A systematic review and meta-analysis. Diabetes Care 2013, 36, 1789–1796. [Google Scholar] [CrossRef]
- Proietti, R.; Russo, V.; Wu, M.A.; Maggioni, A.P.; Marfella, R. Diabete mellito e fibrillazione atriale: Evidenze di un’associazione fisiopatologica e clinico-epidemiologica che prescinde dal rischio tromboembolico [Diabetes mellitus and atrial fibrillation: Evidence of a pathophysiological, clinical and epidemiological association beyond the thromboembolic risk]. G. Ital. Cardiol. 2017, 18, 199–207. [Google Scholar] [CrossRef]
- Caturano, A.; Vetrano, E.; Galiero, R.; Salvatore, T.; Docimo, G.; Epifani, R.; Alfano, M.; Sardu, C.; Marfella, R.; Rinaldi, L.; et al. Cardiac Hypertrophy: From Pathophysiological Mechanisms to Heart Failure Development. Rev. Cardiovasc. Med. 2022, 23, 165. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Yan, Z.; Li, T.; Li, A.; Fan, X.; Qi, Z.; Zhang, J. Primordial Drivers of Diabetes Heart Disease: Comprehensive Insights into Insulin Resistance. Diabetes Metab. J. 2024, 48, 19–36. [Google Scholar] [CrossRef] [PubMed]
- Tong, L.; Li, W.; Zhang, Y.; Zhou, F.; Zhao, Y.; Zhao, L.; Liu, J.; Song, Z.; Yu, M.; Zhou, C.; et al. Tacrolimus inhibits insulin release and promotes apoptosis of Min6 cells through the inhibition of the PI3K/Akt/mTOR pathway. Mol. Med. Rep. 2021, 24, 658. [Google Scholar] [CrossRef]
- Forzisi, E.; Yu, W.; Rajwade, P.; Sesti, F. Antagonistic roles of RasMAPK and Akt signaling in integrin-K+ channel complexmediated cellular apoptosis. FASEB J. 2022, 36, e22292. [Google Scholar] [CrossRef]
- Adel, F.W.; Zheng, Y.; Wan, S.H.; Greason, C.; Pan, S.; Ameenuddin, S.; Chen, H.H. Insulin therapy is associated with increased myocardial interstitial fibrosis and cardiomyocyte apoptosis in a rodent model of experimental diabetes. Front. Physiol. 2022, 13, 890907. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Li, B.; Lin, S.; Chen, L.; Li, H. Role and mechanism of cardiac insulin resistance in occurrence of heart failure caused by myocardial hypertrophy. Aging 2019, 11, 6584–6590. [Google Scholar] [CrossRef] [PubMed]
- Knutti, D.; Kressler, D.; Kralli, A. Regulation of the transcriptional coactivator PGC-1 via MAPK-sensitive interaction with a repressor. Proc. Natl. Acad. Sci. USA 2001, 98, 9713–9718. [Google Scholar] [CrossRef] [PubMed]
- Finck, B.N.; Kelly, D.P. PGC-1 coactivators: Inducible regulators of energy metabolism in health and disease. J. Clin. Investig. 2006, 116, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Kianu Phanzu, B.; Nkodila Natuhoyila, A.; Kintoki Vita, E.; M’Buyamba Kabangu, J.R.; Longo-Mbenza, B. Association between insulin resistance and left ventricular hypertrophy in asymptomatic, Black, sub-Saharan African, hypertensive patients: A case-control study. BMC Cardiovasc. Disord. 2021, 21, 1. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, K.; Venkatesh, B.A.; Wu, C.O.; Mewton, N.; Gjesdal, O.; Kishi, S.; McClelland, R.L.; Bluemke, D.A.; Lima, J.A.C. Diabetes mellitus and insulin resistance associate with left ventricular shape and torsion by cardiovascular magnetic resonance imaging in asymptomatic individuals from the multi-ethnic study of atherosclerosis. J. Cardiovasc. Magn. Reson. 2018, 20, 53. [Google Scholar] [CrossRef] [PubMed]
- Russo, V.; Malvezzi Caracciolo D’Aquino, M.; Caturano, A.; Scognamiglio, G.; Pezzullo, E.; Fabiani, D.; Del Giudice, C.; Carbone, A.; Bottino, R.; Caso, V.; et al. Improvement of Global Longitudinal Strain and Myocardial Work in Type 2 Diabetes Patients on Sodium-Glucose Cotransporter 2 Inhibitors Therapy. J. Cardiovasc. Pharmacol. 2023, 82, 196–200. [Google Scholar] [CrossRef]
- Salzano, A.; D’Assante, R.; Iacoviello, M.; Triggiani, V.; Rengo, G.; Cacciatore, F.; Maiello, C.; Limongelli, G.; Masarone, D.; Sciacqua, A.; et al. Progressive right ventricular dysfunction and exercise impairment in patients with heart failure and diabetes mellitus: Insights from the T.O.S.CA. Registry. Cardiovasc. Diabetol. 2022, 21, 108. [Google Scholar] [CrossRef] [PubMed]
- Gudenkauf, B.; Shaya, G.; Mukherjee, M.; Michos, E.D.; Madrazo, J.; Mathews, L.; Shah, S.J.; Sharma, K.; Hays, A.G. Insulin resistance is associated with subclinical myocardial dysfunction and reduced functional capacity in heart failure with preserved ejection fraction. J. Cardiol. 2024, 83, 100–104. [Google Scholar] [CrossRef]
- Sasso, F.C.; Carbonara, O.; Cozzolino, D.; Rambaldi, P.; Mansi, L.; Torella, D.; Gentile, S.; Turco, S.; Torella, R.; Salvatore, T. Effects of insulin-glucose infusion on left ventricular function at rest and during dynamic exercise in healthy subjects and noninsulin dependent diabetic patients: A radionuclide ventriculographic study. J. Am. Coll. Cardiol. 2000, 36, 219–226. [Google Scholar] [CrossRef]
- Sasso, F.C.; Carbonara, O.; Nasti, R.; Marfella, R.; Esposito, K.; Rambaldi, P.; Mansi, L.; Salvatore, T.; Torella, R.; Cozzolino, D. Effects of insulin on left ventricular function during dynamic exercise in overweight and obese subjects. Eur. Heart J. 2005, 26, 1205–1212. [Google Scholar] [CrossRef] [PubMed]
- Palmiero, G.; Cesaro, A.; Galiero, R.; Loffredo, G.; Caturano, A.; Vetrano, E.; Rinaldi, L.; Salvatore, T.; Ruggiero, R.; Di Palo, M.R.; et al. Impact of gliflozins on cardiac remodeling in patients with type 2 diabetes mellitus & reduced ejection fraction heart failure: A pilot prospective study. GLISCAR study. Diabetes Res. Clin. Pract. 2023, 200, 110686. [Google Scholar] [CrossRef] [PubMed]
- Skals, R.; Krogager, M.L.; Appel, E.V.R.; Schnurr, T.M.; Have, C.T.; Gislason, G.; Poulsen, H.E.; Køber, L.; Engstrøm, T.; Stender, S.; et al. Insulin resistance genetic risk score and burden of coronary artery disease in patients referred for coronary angiography. PLoS ONE 2021, 16, e0252855. [Google Scholar] [CrossRef] [PubMed]
- Bjornstad, P.; Eckel, R.H. Pathogenesis of Lipid Disorders in Insulin Resistance: A Brief Review. Curr. Diab. Rep. 2018, 18, 127. [Google Scholar] [CrossRef] [PubMed]
- Taghibiglou, C.; Carpentier, A.; Van Iderstine, S.C.; Chen, B.; Rudy, D.; Aiton, A.; Lewis, G.F.; Adeli, K. Mechanisms of hepatic very low density lipoprotein overproduction in insulin resistance. Evidence for enhanced lipoprotein assembly, reduced intracellular ApoB degradation, and increased microsomal triglyceride transfer protein in a fructose-fed hamster model. J. Biol. Chem. 2000, 275, 8416–8425. [Google Scholar] [CrossRef] [PubMed]
- Gaggini, M.; Gorini, F.; Vassalle, C. Lipids in Atherosclerosis: Pathophysiology and the Role of Calculated Lipid Indices in Assessing Cardiovascular Risk in Patients with Hyperlipidemia. Int. J. Mol. Sci. 2022, 24, 75. [Google Scholar] [CrossRef] [PubMed]
- Ormazabal, V.; Nair, S.; Elfeky, O.; Aguayo, C.; Salomon, C.; Zuñiga, F.A. Association between insulin resistance and the development of cardiovascular disease. Cardiovasc. Diabetol. 2018, 17, 122. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, I.J. Clinical review 124: Diabetic dyslipidemia: Causes and consequences. J. Clin. Endocrinol. Metab. 2001, 86, 965–971. [Google Scholar] [CrossRef] [PubMed]
- Ikezaki, H.; Lim, E.; Cupples, L.A.; Liu, C.T.; Asztalos, B.F.; Schaefer, E.J. Small Dense Low-Density Lipoprotein Cholesterol Is the Most Atherogenic Lipoprotein Parameter in the Prospective Framingham Offspring Study. J. Am. Heart Assoc. 2021, 10, e019140. [Google Scholar] [CrossRef]
- Ginsberg, H.N. Insulin resistance and cardiovascular disease. J. Clin. Investig. 2000, 106, 453–458. [Google Scholar] [CrossRef]
- Mormone, A.; Tortorella, G.; Esposito, F.; Caturano, A.; Marrone, A.; Cozzolino, D.; Galiero, R.; Marfella, R.; Sasso, F.C.; Rinaldi, L. Advances in Pharmacological Approaches for Managing Hypercholesterolemia: A Comprehensive Overview of Novel Treatments. Biomedicines 2024, 12, 432. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.S.; Schulman, I.H.; Zeng, Q. Link between the renin-angiotensin system and insulin resistance: Implications for cardiovascular disease. Vasc. Med. 2012, 17, 330–341. [Google Scholar] [CrossRef]
- Muniyappa, R.; Sowers, J.R. Role of insulin resistance in endothelial dysfunction. Rev. Endocr. Metab. Disord. 2013, 14, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Lacolley, P.; Regnault, V.; Laurent, S. Mechanisms of Arterial Stiffening: From Mechanotransduction to Epigenetics. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.; Fang, J.; Liu, Z.; Shi, Y.; Agostini, M.; Bernassola, F.; Bove, P.; Candi, E.; Rovella, V.; Sica, G.; et al. Macrophage polarization and metabolism in atherosclerosis. Cell Death Dis. 2023, 14, 691. [Google Scholar] [CrossRef]
- Li, Y.; Liu, Y.; Liu, S.; Gao, M.; Wang, W.; Chen, K.; Huang, L.; Liu, Y. Diabetic vascular diseases: Molecular mechanisms and therapeutic strategies. Signal Transduct. Target. Ther. 2023, 8, 152. [Google Scholar] [CrossRef]
- Aune, D.; Schlesinger, S.; Norat, T.; Riboli, E. Diabetes mellitus and the risk of abdominal aortic aneurysm: A systematic review and meta-analysis of prospective studies. J. Diabetes Complicat. 2018, 32, 1169–1174. [Google Scholar] [CrossRef]
- Fishman, S.L.; Sonmez, H.; Basman, C.; Singh, V.; Poretsky, L. The role of advanced glycation end-products in the development of coronary artery disease in patients with and without diabetes mellitus: A review. Mol. Med. 2018, 24, 59. [Google Scholar] [CrossRef]
- Muniyappa, R.; Chen, H.; Montagnani, M.; Sherman, A.; Quon, M.J. Endothelial dysfunction due to selective insulin resistance in vascular endothelium: Insights from mechanistic modeling. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E629–E646. [Google Scholar] [CrossRef]
- Dongerkery, S.P.; Schroeder, P.R.; Shomali, M.E. Insulin and Its Cardiovascular Effects: What Is the Current Evidence? Curr. Diabetes Rep. 2017, 17, 120. [Google Scholar] [CrossRef]
- Steinberg, H.O.; Brechtel, G.; Johnson, A.; Fineberg, N.; Baron, A.D. Insulin-mediated skeletal muscle vasodilation is nitric oxide dependent. A novel action of insulin to increase nitric oxide release. J. Clin. Investig. 1994, 94, 1172–1179. [Google Scholar] [CrossRef] [PubMed]
- Scherrer, U.; Randin, D.; Vollenweider, P.; Vollenweider, L.; Nicod, P. Nitric oxide release accounts for insulin’s vascular effects in humans. J. Clin. Investig. 1994, 94, 2511–2515. [Google Scholar] [CrossRef] [PubMed]
- Vincent, M.A.; Clerk, L.H.; Rattigan, S.; Clark, M.G.; Barrett, E.J. Active role for the vasculature in the delivery of insulin to skeletal muscle. Clin. Exp. Pharmacol. Physiol. 2005, 32, 302–307. [Google Scholar] [CrossRef]
- Aljada, A.; Dandona, P. Effect of insulin on human aortic endothelial nitric oxide synthase. Metabolism 2000, 49, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Mendelsohn, M.E. Synergistic activation of endothelial nitric-oxide synthase (eNOS) by HSP90 and Akt: Calcium-independent eNOS activation involves formation of an HSP90-Akt-CaM-bound eNOS complex. J. Biol. Chem. 2003, 278, 30821–30827. [Google Scholar] [CrossRef] [PubMed]
- Shamir, R.; Shehadeh, N.; Rosenblat, M.; Eshach-Adiv, O.; Coleman, R.; Kaplan, M.; Hamoud, S.; Lischinsky, S.; Hayek, T. Oral insulin supplementation attenuates atherosclerosis progression in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Rask-Madsen, C.; Li, Q.; Freund, B.; Feather, D.; Abramov, R.; Wu, I.H.; Chen, K.; Yamamoto-Hiraoka, J.; Goldenbogen, J.; Sotiropoulos, K.B.; et al. Loss of insulin signaling in vascular endothelial cells accelerates atherosclerosis in apolipoprotein E null mice. Cell Metab. 2010, 11, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Aljada, A.; Ghanim, H.; Saadeh, R.; Dandona, P. Insulin inhibits NFkappaB and MCP-1 expression in human aortic endothelial cells. J. Clin. Endocrinol. Metab. 2001, 86, 450–453. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, M.G.; Einspanier, R.; Klein, D.; Jauch, K.W. Insulin attenuates the systemic inflammatory response to thermal trauma. Mol. Med. 2002, 8, 443–450. [Google Scholar] [CrossRef]
- Diabetes Control and Complications Trial Research Group; Nathan, D.M.; Genuth, S.; Lachin, J.; Cleary, P.; Crofford, O.; Davis, M.; Rand, L.; Siebert, C. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N. Engl. J. Med. 1993, 329, 977–986. [Google Scholar] [CrossRef]
- Diabetes Control and Complications Trial (DCCT)/Epidemiology of Diabetes Interventions and Complications (EDIC) Study Research Group. Intensive Diabetes Treatment and Cardiovascular Outcomes in Type 1 Diabetes: The DCCT/EDIC Study 30-Year Follow-up. Diabetes Care 2016, 39, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Chow, E.; Bernjak, A.; Williams, S.; Fawdry, R.A.; Hibbert, S.; Freeman, J.; Sheridan, P.J.; Heller, S.R. Risk of cardiac arrhythmias during hypoglycemia in patients with type 2 diabetes and cardiovascular risk. Diabetes 2014, 63, 1738–1747. [Google Scholar] [CrossRef] [PubMed]
- Zoungas, S.; Patel, A.; Chalmers, J.; de Galan, B.E.; Li, Q.; Billot, L.; Woodward, M.; Ninomiya, T.; Neal, B.; MacMahon, S.; et al. ADVANCE Collaborative Group. Severe hypoglycemia and risks of vascular events and death. N. Engl. J. Med. 2010, 363, 1410–1418. [Google Scholar] [CrossRef] [PubMed]
- Trovati, M.; Anfossi, G.; Massucco, P.; Mattiello, L.; Costamagna, C.; Piretto, V.; Mularoni, E.; Cavalot, F.; Bosia, A.; Ghigo, D. Insulin stimulates nitric oxide synthesis in human platelets and, through nitric oxide, increases platelet concentrations of both guanosine-3′, 5′-cyclic monophosphate and adenosine-3′, 5′-cyclic monophosphate. Diabetes 1997, 46, 742–749. [Google Scholar] [CrossRef] [PubMed]
- Anfossi, G.; Russo, I.; Trovati, M. Platelet resistance to the anti-aggregating agents in the insulin resistant states. Curr. Diabetes Rev. 2006, 2, 409–430. [Google Scholar] [CrossRef] [PubMed]
- Worthley, M.I.; Holmes, A.S.; Willoughby, S.R.; Kucia, A.M.; Heresztyn, T.; Stewart, S.; Chirkov, Y.Y.; Zeitz, C.J.; Horowitz, J.D. The deleterious effects of hyperglycemia on platelet function in diabetic patients with acute coronary syndromes mediation by superoxide production, resolution with intensive insulin administration. J. Am. Coll. Cardiol. 2007, 49, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Nathan, D.M.; Lachin, J.; Cleary, P.; Orchard, T.; Brillon, D.J.; Backlund, J.Y.; O’Leary, D.H.; Genuth, S.; Diabetes Control and Complications Trial; Epidemiology of Diabetes Interventions and Complications Research Group. Intensive diabetes therapy and carotid intima-media thickness in type 1 diabetes mellitus. N. Engl. J. Med. 2003, 348, 2294–2303. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.K.; Nikooienejad, A.; Bray, R.; Cui, X.; Wilson, J.; Duffin, K.; Milicevic, Z.; Haupt, A.; Robins, D.A. Dual GIP and GLP-1 Receptor Agonist Tirzepatide Improves Beta-cell Function and Insulin Sensitivity in Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2021, 106, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Yaribeygi, H.; Sathyapalan, T.; Maleki, M.; Jamialahmadi, T.; Sahebkar, A. Molecular mechanisms by which SGLT2 inhibitors can induce insulin sensitivity in diabetic milieu: A mechanistic review. Life Sci. 2020, 240, 117090. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, H.; Zhang, R.; Xiang, Y.; Lu, J.; Xia, B.; Peng, L.; Wu, J. AdipoRon exerts opposing effects on insulin sensitivity via fibroblast growth factor 21-mediated time-dependent mechanisms. J. Biol. Chem. 2022, 298, 101641. [Google Scholar] [CrossRef]
- Jamaluddin, M.S.; Weakley, S.M.; Yao, Q.; Chen, C. Resistin: Functional roles and therapeutic considerations for cardiovascular disease. Br. J. Pharmacol. 2012, 165, 622–632. [Google Scholar] [CrossRef]
- Galiero, R.; Caturano, A.; Vetrano, E.; Monda, M.; Marfella, R.; Sardu, C.; Salvatore, T.; Rinaldi, L.; Sasso, F.C. Precision Medicine in Type 2 Diabetes Mellitus: Utility and Limitations. Diabetes Metab. Syndr. Obes. 2023, 16, 3669–3689. [Google Scholar] [CrossRef] [PubMed]
- Sasso, F.C.; Simeon, V.; Galiero, R.; Caturano, A.; De Nicola, L.; Chiodini, P.; Rinaldi, L.; Salvatore, T.; Lettieri, M.; Nevola, R.; et al. NID-2 study group Investigators. The number of risk factors not at target is associated with cardiovascular risk in a type 2 diabetic population with albuminuria in primary cardiovascular prevention. Post-hoc analysis of the NID-2 trial. Cardiovasc. Diabetol. 2022, 21, 235. [Google Scholar] [CrossRef] [PubMed]
- Sasso, F.C.; Pafundi, P.C.; Simeon, V.; De Nicola, L.; Chiodini, P.; Galiero, R.; Rinaldi, L.; Nevola, R.; Salvatore, T.; Sardu, C.; et al. NID-2 Study Group Investigators. Efficacy and durability of multifactorial intervention on mortality and MACEs: A randomized clinical trial in type-2 diabetic kidney disease. Cardiovasc. Diabetol. 2021, 20, 145. [Google Scholar] [CrossRef] [PubMed]
- Sasso, F.C.; Pafundi, P.C.; Caturano, A.; Galiero, R.; Vetrano, E.; Nevola, R.; Petta, S.; Fracanzani, A.L.; Coppola, C.; Di Marco, V.; et al. Impact of direct acting antivirals (DAAs) on cardiovascular events in HCV cohort with pre-diabetes. Nutr. Metab. Cardiovasc. Dis. 2021, 31, 2345–2353. [Google Scholar] [CrossRef]
- Prochazkova, M.; Budinska, E.; Kuzma, M.; Pelantova, H.; Hradecky, J.; Heczkova, M.; Daskova, N.; Bratova, M.; Modos, I.; Videnska, P.; et al. Vegan Diet Is Associated with Favorable Effects on the Metabolic Performance of Intestinal Microbiota: A Cross-Sectional Multi-Omics Study. Front. Nutr. 2022, 8, 783302. [Google Scholar] [CrossRef]
- Luo, S.; Zheng, X.; Bao, W.; Nie, S.; Ding, Y.; Yue, T.; Zhou, Y.; Hu, Y.; Li, H.; Yang, Q.; et al. Real-world effectiveness of early insulin therapy on the incidence of cardiovascular events in newly diagnosed type 2 diabetes. Signal Transduct. Target. Ther. 2024, 9, 154. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caturano, A.; Galiero, R.; Vetrano, E.; Sardu, C.; Rinaldi, L.; Russo, V.; Monda, M.; Marfella, R.; Sasso, F.C. Insulin–Heart Axis: Bridging Physiology to Insulin Resistance. Int. J. Mol. Sci. 2024, 25, 8369. https://doi.org/10.3390/ijms25158369
Caturano A, Galiero R, Vetrano E, Sardu C, Rinaldi L, Russo V, Monda M, Marfella R, Sasso FC. Insulin–Heart Axis: Bridging Physiology to Insulin Resistance. International Journal of Molecular Sciences. 2024; 25(15):8369. https://doi.org/10.3390/ijms25158369
Chicago/Turabian StyleCaturano, Alfredo, Raffaele Galiero, Erica Vetrano, Celestino Sardu, Luca Rinaldi, Vincenzo Russo, Marcellino Monda, Raffaele Marfella, and Ferdinando Carlo Sasso. 2024. "Insulin–Heart Axis: Bridging Physiology to Insulin Resistance" International Journal of Molecular Sciences 25, no. 15: 8369. https://doi.org/10.3390/ijms25158369