
The Contribution of Genetic and Epigenetic Factors: An Emerging Concept in the Assessment and Prognosis of Inflammatory Bowel Diseases

 , ,
, ,  , ,
, ,  , ,
, , 
Abstract
1. Introduction
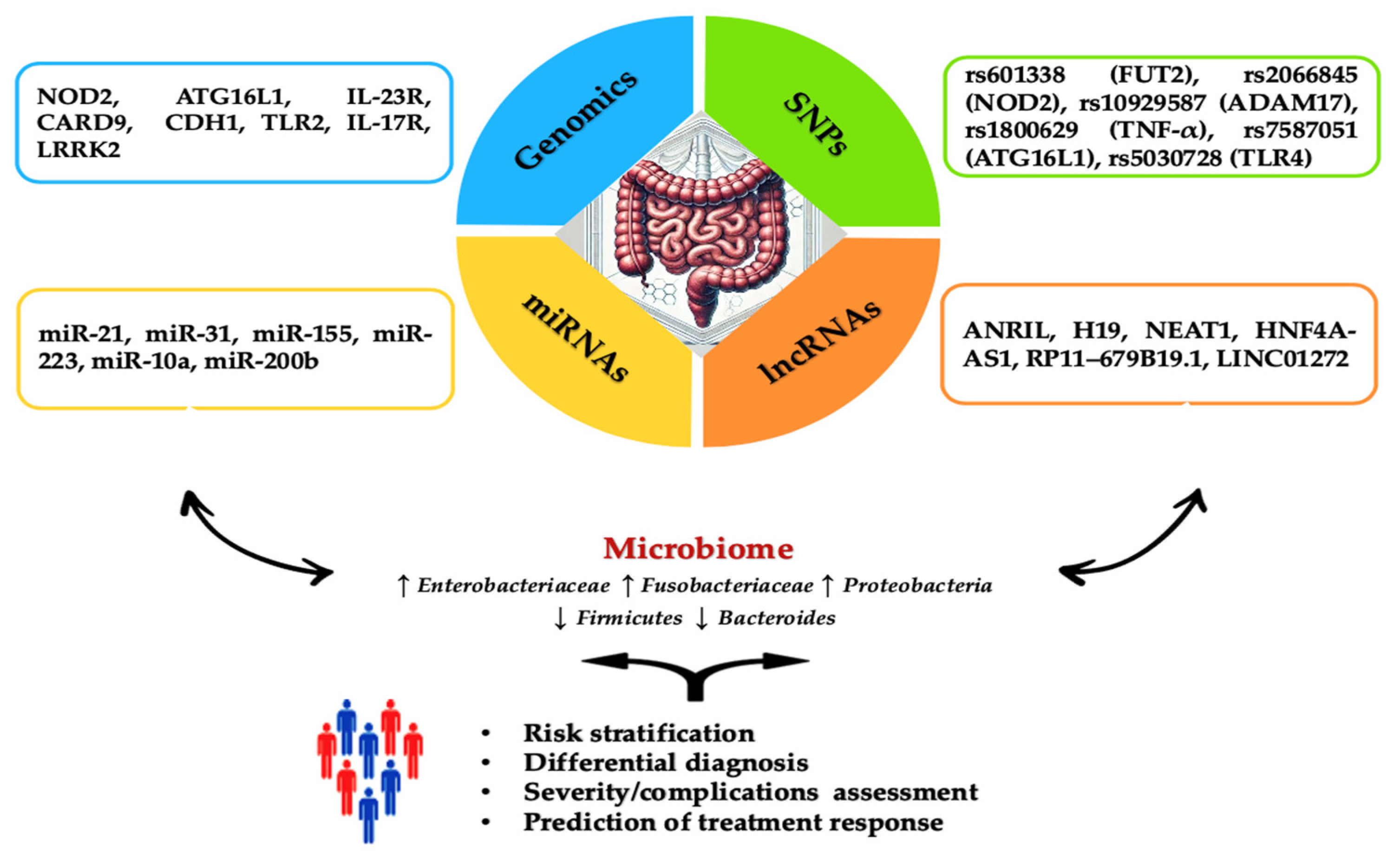
2. Genomics
2.1. The Involvement of Host Genetics in Susceptibility to IBD
2.2. Interaction between Gut Microbiota and Genomics in the IBD Pathogenesis
2.3. Prediction of the Evolution of the Disease and Treatment Response
3. The Role of Epigenetic Mechanisms on Prognosis and Therapeutic Response in IBD
3.1. The Patterns of lncRNAs in IBD
3.1.1. lncRNA as Prognostic and Diagnostic Biomarkers in IBD
3.1.2. lncRNAs as Predictors of Therapeutic Response in IBD
3.2. Role of miRNAs in IBD
3.2.1. miRNAs as Diagnostic Biomarkers of IBD

{kind=link}
| miRNAs | Disease | Sample | Main Findings | References |
|---|---|---|---|---|
| miR-21 miR-126 | UC CD | Colonic tissue | Increased levels of miR-21 (6.7-fold) in lamina propria and miR-126 (2.3-fold) in endothelial cells in UC compared to controls; miR-21 is upregulated in UC compared to CD (5.8-fold). | [157] |
| miR-31 | UC CD Controls | Colonic tissue | Hyperexpression in UC compared to CD and controls. | [159] |
| UC controls | Colonic tissue | Increased expression in active form of UC (11-fold) and inactive form (3-fold) compared to healthy controls. | [160] | |
| miR-16, miR-21, miR-223, miR-155 | CD UC controls | Blood Fecal samples | Active CD was associated with higher miR-155 expression in blood samples compared to controls (1.9-fold) and subjects (2.1-fold) in remission (AUC = 0.752; 95% CI: 0.61–0.89). | [164] |
| miR-146a, miR-155, miR-122 | CD UC controls | Colonic tissue | miR-146a and miR-155 were higher in the inflamed mucosa of children with CD and UC than in the intact mucosa. Elevated expression of miR-122 in CD compared with controls and UC. | [162] |
| CD controls | Colonic tissue | [161] | ||
| miR-125b, miR-223, miR-138, miR-155 | UC controls | Colonic tissue | Upregulation in inflamed UC tissues: miR-138 (10-fold), miR-223 (10-fold), miR-125b (2.56-fold), and miR-155 (2.33-fold). | [165] |
| miR-141, miR-200a, miR-200b, miR-200c, miR-429 | UC CD | Colonic tissue | miR-141, miR-200b, and miR-429 were downregulated in CD and UC patients compared to healthy controls. miR-141, miR-200a, miR-200b, and miR-200c were significantly downregulated in CD in comparison to UC. | [166] |
| miR-21-5p | UC controls | Blood Colonic tissue | Downregulated in UC patients compared with control and determines inhibition of the expression of IL-6, TNF-α, IL6R, STAT3, ICAM-1, NF-κB, cleaved caspase-3, cleaved caspase-9, and FasL to alleviate the inflammation and apoptosis. | [167] |
| miR-21 miR-92a | UC controls | Blood Colonic tissue | Overexpression has increased performance for differentiating UC from healthy subjects (AUC = 0.979, specificity = 1.00). | [158] |
| miR-215-5p, miR-203a-3p, miR-223-3p, miR-194-5p, miR-192-5p, miR-10b-5p, miR-10a-5p, miR-337-5p, miR-582-5p | CD controls | Colonic tissue | Downregulated in inactive CD compared to controls. Their inhibitory action on NOD2, TLR4, and IL6ST genes is involved in the innate immune response and cytokine signaling. | [168] |
| miR-29b-3p, miR-122-5p, miR-146a-3p, miR-150-5p, miR-192-5p, miR-194-5p, miR-375-3p, miR-148a-3p, miR-199a-3p | experimental model colitis mice | Blood | Hyperexpression of miR-29b-3p, -122-5p, -192-5p, -194-5p, -375-3p, -150-5p, -146a-3p, and the downregulated miR-148a-3p and -199a-3p; differentiation of UC patients from healthy controls (accuracies of 83.3%). | [169] |
| mi146b-5p | CD UC controls | Blood | The serum level (2.87- and 2.72-fold higher in patients with CD and UC than controls, respectively) is correlated with disease activity (CDEIS r = 0.579; UCIES r = 0.582) (AUC = 0.869, 95% CI: 0.764-0.940). | [170] |
3.2.2. The Role of miRNAs as Therapeutic Targets
4. Future Directions and Challenges
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Baldan-Martin, M.; Chaparro, M.; Gisbert, J.P. Tissue Proteomic Approaches to Understand the Pathogenesis of Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2021, 27, 1184–1200. [Google Scholar] [CrossRef]
- Nowak, J.K.; Kallab, R.; Satsang, J. Current and emerging biomarkers for ulcerative colitis. Expert Rev. Mol. Diagn. 2023, 23, 1107–1119. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, B.; Jin, T.; Ocansey, D.K.W.; Jiang, J.; Mao, F. Intestinal Fibrosis in Inflammatory Bowel Disease and the Prospects of Mesenchymal Stem Cell Therapy. Front. Immunol. 2022, 13, 835005. [Google Scholar]
- Li, X.; Zhang, M.; Zhou, G.; Xie, Z.; Wang, Y.; Han, J.; Li, L.; Wu, Q.; Zhang, S. Role of Rho GTPases in inflammatory bowel disease. Cell Death Discov. 2023, 9, 24. [Google Scholar] [CrossRef]
- Ma, C.; Sandborn, W.J.; D’Haens, G.R.; Zou, G.; Stitt, L.W.; Singh, S.; Ananthakrishnan, A.N.; Dulai, P.S.; Khanna, R.; Jairath, V.; et al. Discordance between Patient-Reported Outcomes and Mucosal Inflammation in Patients with Mild to Moderate Ulcerative Colitis. Clin. Gastroenterol. Hepatol. 2020, 18, 1760–1768. [Google Scholar]
- Sabino, J.; Verstockt, B.; Vermeire, S.; Ferrante, M. New biologics and small molecules in inflammatory bowel disease: An update. Therap. Adv. Gastroenterol. 2019, 12, 1756284819853208. [Google Scholar]
- Giachero, F.; Jenke, A.; Zilbauer, M. Improving prediction of disease outcome for inflammatory bowel disease: Progress through systems medicine. Expert Rev. Clin. Immunol. 2021, 17, 871–881. [Google Scholar]
- Elhag, D.A.; Kumar, M.; Saadaoui, M.; Akobeng, A.K.; Al-Mudahka, F.; Elawad, M.; Al Khodor, S. Inflammatory Bowel Disease Treatments and Predictive Biomarkers of Therapeutic Response. Int. J. Mol. Sci. 2022, 23, 6966. [Google Scholar] [CrossRef]
- Maaser, C.; Sturm, A.; Vavricka, S.R.; Kucharzik, T.; Fiorino, G.; Annese, V.; Calabrese, E.; Baumgart, D.C.; Bettenworth, D.; Borralho Nunes, P.; et al. European Crohn’s and Colitis Organisation [ECCO] and the European Society of Gastrointestinal and Abdominal Radiology [ESGAR]. ECCO-ESGAR Guideline for Diagnostic Assessment in IBD Part 1: Initial diagnosis, monitoring of known IBD, detection of complications. J. Crohn’s Colitis 2019, 13, 144–164. [Google Scholar]
- Torres, J.; Petralia, F.; Sato, T.; Wang, P.; Telesco, S.E.; Choung, R.S.; Strauss, R.; Li, X.J.; Laird, R.M.; Gutierrez, R.L.; et al. Serum Biomarkers Identify Patients Who Will Develop Inflammatory Bowel Diseases Up to 5 Years before Diagnosis. Gastroenterology 2020, 159, 96–104. [Google Scholar] [CrossRef]
- Turner, D.; Ricciuto, A.; Lewis, A.; D’Amico, F.; Dhaliwal, J.; Griffiths, A.M.; Bettenworth, D.; Sandborn, W.J.; Sands, B.E.; Reinisch, W.; et al. International Organization for the Study of IBD. STRIDE-II—An Update on the Selecting Therapeutic Targets in Inflammatory Bowel Disease (STRIDE) Initiative of the International Organization for the Study of IBD (IOIBD): Determining Therapeutic Goals for Treat-to-Target strategies in IBD. Gastroenterology 2021, 160, 1570–1583. [Google Scholar]
- Liu, X.Y.; Tang, H.; Zhou, Q.Y.; Zeng, Y.L.; Chen, D.; Xu, H.; Li, Y.; Tan, B.; Qian, J.M. Advancing the precision management of inflammatory bowel disease in the era of omics approaches and new technology. World J. Gastroenterol. 2023, 29, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Olivera, P.A.; Silverberg, M.S. Biomarkers That Predict Crohn’s Disease Outcomes. J. Can. Assoc. Gastroenterol. 2023, 7, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Hanzel, J.; Panaccione, R.; Sandborn, W.J.; D’Haens, G.R.; Ahuja, V.; Atreya, R.; Bernstein, C.N.; Bossuyt, P.; Bressler, B.; et al. CORE-IBD: A Multidisciplinary International Consensus Initiative to Develop a Core Outcome Set for Randomized Controlled Trials in Inflammatory Bowel Disease. Gastroenterology 2022, 163, 950–964. [Google Scholar] [CrossRef] [PubMed]
- Torres, J.; Halfvarson, J.; Rodríguez-Lago, I.; Hedin, C.R.H.; Jess, T.; Dubinsky, M.; Croitoru, K.; Colombel, J.F. Results of the Seventh Scientific Workshop of ECCO: Precision Medicine in IBD-Prediction and Prevention of Inflammatory Bowel Disease. J. Crohn’s Colitis 2021, 15, 1443–1454. [Google Scholar] [CrossRef]
- Aldars-García, L.; Chaparro, M.; Gisbert, J.P. Systematic Review: The Gut Microbiome and Its Potential Clinical Application in Inflammatory Bowel Disease. Microorganisms 2021, 9, 977. [Google Scholar] [CrossRef] [PubMed]
- Aldars-García, L.; Marin, A.C.; Chaparro, M.; Gisbert, J.P. The Interplay between Immune System and Microbiota in Inflammatory Bowel Disease: A Narrative Review. Int. J. Mol. Sci. 2021, 22, 3076. [Google Scholar] [CrossRef] [PubMed]
- Ortega Moreno, L.; Sanz-Garcia, A.; Fernández de la Fuente, M.J.; Arroyo Solera, R.; Fernández-Tomé, S.; Marin, A.C.; Mora-Gutierrez, I.; Fernández, P.; Baldan-Martin, M.; Chaparro, M.; et al. Serum adipokines as non-invasive biomarkers in Crohn’s disease. Sci. Rep. 2020, 10, 18027. [Google Scholar] [CrossRef]
- Somineni, H.K.; Nagpal, S.; Venkateswaran, S.; Cutler, D.J.; Okou, D.T.; Haritunians, T.; Simpson, C.L.; Begum, F.; Datta, L.W.; Quiros, A.J.; et al. Whole-genome sequencing of African Americans implicates differential genetic architecture in inflammatory bowel disease. Am. J. Hum. Genet. 2021, 108, 431–445. [Google Scholar] [CrossRef] [PubMed]
- Chudy-Onwugaje, K.O.; Christian, K.E.; Farraye, F.A.; Cross, R.K. A State-of-the-Art Review of New and Emerging Therapies for the Treatment of IBD. Inflamm. Bowel Dis. 2019, 25, 820–830. [Google Scholar] [CrossRef]
- Santos, M.P.C.; Gomes, C.; Torres, J. Familial and ethnic risk in inflammatory bowel disease. Ann. Gastroenterol. 2018, 31, 14–23. [Google Scholar] [CrossRef]
- Garza-Hernandez, D.; Sepulveda-Villegas, M.; Garcia-Pelaez, J.; Aguirre-Gamboa, R.; Lakatos, P.L.; Estrada, K.; Martinez-Vazquez, M.; Trevino, V. A systematic review and functional bioinformatics analysis of genes associated with Crohn’s disease identify more than 120 related genes. BMC Genom. 2022, 23, 302. [Google Scholar] [CrossRef]
- Moller, F.T.; Andersen, V.; Wohlfahrt, J.; Jess, T. Familial risk of inflammatory bowel disease: A population-based cohort study 1977-2011. Am. J. Gastroenterol. 2015, 110, 564–571. [Google Scholar] [CrossRef]
- El Hadad, J.; Schreiner, P.; Vavricka, S.R.; Greuter, T. The Genetics of Inflammatory Bowel Disease. Mol. Diagn. Ther. 2024, 28, 27–35. [Google Scholar] [CrossRef]
- Spencer, E.A.; Helmus, D.; Telesco, S.; Colombel, J.F.; Dubinsky, M.C. Road to Prevention Study Group. Inflammatory Bowel Disease Clusters within Affected Sibships in Ashkenazi Jewish Multiplex Families. Gastroenterology 2020, 159, 381–382. [Google Scholar] [CrossRef]
- Chen, G.B.; Lee, S.H.; Montgomery, G.W.; Wray, N.R.; Visscher, P.M.; Gearry, R.B.; Lawrance, I.C.; Andrews, J.M.; Bampton, P.; Mahy, G.; et al. Performance of risk prediction for inflammatory bowel disease based on genotyping platform and genomic risk score method. BMC Med. Genet. 2017, 18, 94. [Google Scholar] [CrossRef]
- Arnadottir, G.A.; Norddahl, G.L.; Gudmundsdottir, S.; Agustsdottir, A.B.; Sigurdsson, S.; Jensson, B.O.; Bjarnadottir, K.; Theodors, F.; Benonisdottir, S.; Ivarsdottir, E.V.; et al. A homozygous loss-of-function mutation leading to CYBC1 deficiency causes chronic granulomatous disease. Nat. Commun. 2018, 9, 4447. [Google Scholar] [CrossRef]
- de Ponthaud, C.; Abdalla, S.; Belot, M.P.; Shao, X.; Penna, C.; Brouquet, A.; Bougnères, P. Increased CpG methylation at the CDH1 locus in inflamed ileal mucosa of patients with Crohn disease. Clin. Epigenet. 2024, 16, 28. [Google Scholar] [CrossRef]
- Muller, M.; Hansmannel, F.; Arnone, D.; Choukour, M.; Ndiaye, N.C.; Kokten, T.; Houlgatte, R.; Peyrin-Biroulet, L. Genomic and molecular alterations in human inflammatory bowel disease-associated colorectal cancer. United Eur. Gastroenterol. J. 2020, 8, 675–684. [Google Scholar] [CrossRef]
- Yu, H.; Liu, Z. GNA12 regulates C5a-induced migration by downregulating C5aR1-PLCβ2-PI3K-AKT-ERK1/2 signaling. Biophys. Rep. 2023, 9, 33–44. [Google Scholar] [CrossRef]
- M’Koma, A.E. The Multifactorial Etiopathogeneses Interplay of Inflammatory Bowel Disease: An Overview. Gastrointest. Disord. 2019, 1, 75–105. [Google Scholar] [CrossRef]
- Canale, V.; Spalinger, M.R.; Alvarez, R.; Sayoc-Becerra, A.; Sanati, G.; Manz, S.; Chatterjee, P.; Santos, A.N.; Lei, H.; Jahng, S.; et al. PTPN2 Is a Critical Regulator of Ileal Paneth Cell Viability and Function in Mice. Cell Mol. Gastroenterol. Hepatol. 2023, 16, 39–62. [Google Scholar] [CrossRef] [PubMed]
- Niechcial, A.; Butter, M.; Manz, S.; Obialo, N.; Bäbler, K.; van der Lely, L.; Lang, S.; Gottier, C.; McCole, D.F.; Scharl, M.; et al. Presence of PTPN2 SNP rs1893217 Enhances the Anti-inflammatory Effect of Spermidine. Inflamm. Bowel Dis. 2020, 26, 1038–1049. [Google Scholar] [CrossRef] [PubMed]
- Rivas, M.A.; Graham, D.; Sulem, P.; Stevens, C.; Desch, A.N.; Goyette, P.; Gudbjartsson, D.; Jonsdottir, I.; Thorsteinsdottir, U.; Degenhardt, F.; et al. A protein-truncating R179X variant in RNF186 confers protection against ulcerative colitis. Nat. Commun. 2016, 7, 12342. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, K.; Kinoshita, M.; Tanaka, H.; Okuzaki, D.; Shimada, Y.; Kayama, H.; Okumura, R.; Furuta, Y.; Narazaki, M.; Tamura, A.; et al. Regulation of intestinal homeostasis by the ulcerative colitis-associated gene RNF186. Mucosal. Immunol. 2017, 10, 446–459. [Google Scholar] [CrossRef]
- Lu, Y.; Li, X.; Liu, S.; Zhang, Y.; Zhang, D. Toll-like Receptors and Inflammatory Bowel Disease. Front. Immunol. 2018, 9, 72. [Google Scholar] [CrossRef] [PubMed]
- Cari, L.; Rosati, L.; Leoncini, G.; Lusenti, E.; Gentili, M.; Nocentini, G.; Riccardi, C.; Migliorati, G.; Ronchetti, S. Association of GILZ with MUC2, TLR2, and TLR4 in Inflammatory Bowel Disease. Int. J. Mol. Sci. 2023, 24, 2235. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F. IL-23 in inflammatory bowel diseases and colon cancer. Cytokine Growth Factor Rev. 2019, 45, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, H.; Neurath, M.F.; Atreya, R. Role of the IL23/IL17 Pathway in Crohn’s Disease. Front. Immunol. 2021, 12, 622934. [Google Scholar] [CrossRef]
- Deng, Z.; Wang, S.; Wu, C.; Wang, C. IL-17 inhibitor-associated inflammatory bowel disease: A study based on literature and database analysis. Front. Pharmacol. 2023, 14, 1124628. [Google Scholar] [CrossRef]
- Cai, Y.; Jia, X.; Xu, L.; Chen, H.; Xie, S.; Cai, J. Interleukin-17 and inflammatory bowel disease: A 2-sample Mendelian randomization study. Front. Immunol. 2023, 14, 1238457. [Google Scholar] [CrossRef] [PubMed]
- Barnes, E.L.; Loftus, E.V., Jr.; Kappelman, M.D. Effects of Race and Ethnicity on Diagnosis and Management of Inflammatory Bowel Diseases. Gastroenterology 2021, 160, 677–689. [Google Scholar] [CrossRef]
- Yang, D.H.; Yang, S.K.; Song, K.; Hong, M.; Park, S.H.; Lee, H.S.; Kim, J.B.; Lee, H.J.; Park, S.K.; Jung, K.W.; et al. TNFSF15 is an independent predictor for the development of Crohn’s disease-related complications in Koreans. J. Crohn’s Colitis 2014, 8, 1315–1326. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Zhu, Q.; Zheng, P.F.; Feng, Y.L. Association of Fucosyltransferase 2 Gene Variant with Inflammatory Bowel Diseases: A Meta-Analysis. Med. Sci. Monit. 2019, 25, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Hu, J.; Wu, X.; Pan, J.A.; Jiao, N.; Li, Y.; Huang, Y.; Lin, X.; Zou, Y.; Chen, Y.; et al. Altered gut microbiome in FUT2 loss-of-function mutants in support of personalized medicine for inflammatory bowel diseases. J. Genet. Genom. 2021, 48, 771–780. [Google Scholar]
- Sazonovs, A.; Kennedy, N.A.; Moutsianas, L.; Heap, G.A.; Rice, D.L.; Reppell, M.; Bewshea, C.M.; Chanchlan, N.; Walker, G.J.; Perry, M.H.; et al. HLA-DQA1*05 Carriage Associated with Development of Anti-Drug Antibodies to Infliximab and Adalimumab in Patients with Crohn’s Disease. Gastroenterology 2020, 158, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Alcolado, L.; Grueso-Navarro, E.; Arias, Á.; Lucendo, A.J.; Laserna-Mendieta, E.J. Impact of HLA-DQA1∗05 Genotype in Immunogenicity and Failure to Treatment with Tumor Necrosis Factor-alpha Antagonists in Inflammatory Bowel Disease: A Systematic Review and Meta-analysis. J. Crohn’s Colitis 2024. [Google Scholar] [CrossRef] [PubMed]
- Mahdi, B.M. Role of HLA typing on Crohn’s disease pathogenesis. Ann. Med. Surg. 2015, 4, 248–253. [Google Scholar] [CrossRef]
- Ashton, J.J.; Latham, K.; Beattie, R.M.; Ennis, S. Review article: The genetics of the human leucocyte antigen region in inflammatory bowel disease. Aliment. Pharmacol. Ther. 2019, 50, 885–900. [Google Scholar] [CrossRef]
- Brant, S.R.; Okou, D.T.; Simpson, C.L.; Cutler, D.J.; Haritunians, T.; Bradfield, J.P.; Chopra, P.; Prince, J.; Begum, F.; Kumar, A.; et al. Genome-Wide Association Study Identifies African-Specific Susceptibility Loci in African Americans with Inflammatory Bowel Disease. Gastroenterology 2017, 152, 206–217. [Google Scholar] [CrossRef]
- Bergstein, S.; Spencer, E. HLA-DQA1*05 associates with immunogenicity and loss of response to anti-TNF therapy in the IBD population: A meta-analysis. Inflamm. Bowel Dis. 2023, 29, S58. [Google Scholar] [CrossRef]
- Dewit, O.; Moreels, T.; Baert, F.; Peeters, H.; Reenaers, C.; de Vos, M.; Van Hootegem, P.; Muls, V.; Veereman, G.; Mana, F.; et al. Limitations of extensive TPMT genotyping in the management of azathioprine-induced myelosuppression in IBD patients. Clin. Biochem. 2011, 44, 1062–1066. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.; Ammitzboell, M.; Nys, K.; Seidelin, J.B.; Nielsen, O.H. ATG16L1: A multifunctional susceptibility factor in Crohn disease. Autophagy 2015, 11, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Turpin, W.; Bedrani, L.; Espin-Garcia, O.; Xu, W.; Silverberg, M.S.; Smith, M.I.; Garay, J.A.R.; Lee, S.H.; Guttman, D.S.; Griffiths, A.; et al. Associations of NOD2 polymorphisms with Erysipelotrichaceae in stool of in healthy first degree relatives of Crohn’s disease subjects. BMC Med. Genet. 2020, 21, 204. [Google Scholar] [CrossRef] [PubMed]
- Cotterill, L.; Payne, D.; Levinson, S.; McLaughlin, J.; Wesley, E.; Feeney, M.; Durbin, H.; Lal, S.; Makin, A.; Petryszyn, P.; et al. C3435T Polymorphism of the ABCB1 Gene in Polish Patients with Inflammatory Bowel Disease: A Case-Control and Meta-Analysis Study. Genes 2021, 12, 1419. [Google Scholar] [CrossRef]
- Rojas, M.; Restrepo-Jiménez, P.; Monsalve, D.M.; Pacheco, Y.; Acosta-Ampudia, Y.; Ramírez-Santana, C.; Leung, P.S.C.; Ansari, A.A.; Gershwin, M.E.; Anaya, J.M. Molecular mimicry and autoimmunity. J. Autoimmun. 2018, 95, 100–123. [Google Scholar] [CrossRef] [PubMed]
- Naito, T.; Okada, Y. HLA imputation and its application to genetic and molecular fine-mapping of the MHC region in autoimmune diseases. Semin. Immunopathol. 2022, 44, 15–28. [Google Scholar] [CrossRef]
- Verstockt, B.; Parkes, M.; Lee, J.C. How Do We Predict a Patient’s Disease Course and Whether They Will Respond to Specific Treatments? Gastroenterology 2022, 162, 1383–1395. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Liu, Z. The latest breakthrough on genetic characteristics of inflammatory bowel disease in Chinese and other East Asian ancestries. Precis. Clin. Med. 2023, 6, pbad017. [Google Scholar] [CrossRef]
- Sivanesan, D.; Beauchamp, C.; Quinou, C.; Lee, J.; Lesage, S.; Chemtob, S.; Rioux, J.D.; Michnick, S.W. IL23R (Interleukin 23 Receptor) Variants Protective against Inflammatory Bowel Diseases (IBD) Display Loss of Function due to Impaired Protein Stability and Intracellular Trafficking. J. Biol. Chem. 2016, 291, 8673–8685. [Google Scholar] [CrossRef]
- Momozawa, Y.; Dmitrieva, J.; Théâtre, E.; Deffontaine, V.; Rahmouni, S.; Charloteaux, B.; Crins, F.; Docampo, E.; Elansary, M.; Gori, A.S.; et al. IBD risk loci are enriched in multigenic regulatory modules encompassing putative causative genes. Nat. Commun. 2018, 9, 2427. [Google Scholar] [CrossRef] [PubMed]
- Qiu, P.; Ishimoto, T.; Fu, L.; Zhang, J.; Zhang, Z.; Liu, Y. The Gut Microbiota in Inflammatory Bowel Disease. Front. Cell Infect. Microbiol. 2022, 12, 733992. [Google Scholar] [CrossRef] [PubMed]
- Imhann, F.; Vich Vila, A.; Bonder, M.J.; Fu, J.; Gevers, D.; Visschedijk, M.C.; Spekhorst, L.M.; Alberts, R.; Franke, L.; van Dullemen, H.M.; et al. Interplay of host genetics and gut microbiota underlying the onset and clinical presentation of inflammatory bowel disease. Gut 2018, 67, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Kayali, S.; Fantasia, S.; Gaiani, F.; Cavallaro, L.G.; de’Angelis, G.L.; Laghi, L. NOD2 and Crohn’s Disease Clinical Practice: From Epidemiology to Diagnosis and Therapy, Rewired. Inflamm. Bowel Dis. 2024. [Google Scholar] [CrossRef] [PubMed]
- Noble, A.J.; Nowak, J.K.; Adams, A.T.; Uhlig, H.H.; Satsangi, J. Defining Interactions Between the Genome, Epigenome, and the Environment in Inflammatory Bowel Disease: Progress and Prospects. Gastroenterology 2023, 165, 44–60. [Google Scholar] [CrossRef] [PubMed]
- Butera, A.; Di Paola, M.; Pavarini, L.; Strati, F.; Pindo, M.; Sanchez, M.; Cavalieri, D.; Boirivant, M.; De Filippo, C. Nod2 Deficiency in mice is Associated with Microbiota Variation Favouring the Expansion of mucosal CD4+ LAP+ Regulatory Cells. Sci. Rep. 2018, 8, 14241. [Google Scholar] [CrossRef] [PubMed]
- Coyne, M.J.; Comstock, L.E. Type VI Secretion Systems and the Gut Microbiota. Microbiol. Spectr. 2019, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Wellman, A.S.; Metukuri, M.R.; Kazgan, N.; Xu, X.; Xu, Q.; Ren, N.S.X.; Czopik, A.; Shanahan, M.T.; Kang, A.; Chen, W. Intestinal Epithelial Sirtuin 1 Regulates Intestinal Inflammation During Aging in Mice by Altering the Intestinal Microbiota. Gastroenterology 2017, 153, 772–786. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Zhang, Y.; Tian, X.; Wang, X.; Gathungu, G.; Wolber, A.; Shiekh, S.S.; Sartor, R.B.; Davidson, N.O.; Ciorba, M.A.; et al. Influence of Crohn’s disease related polymorphisms in innate immune function on ileal microbiome. PLoS ONE 2019, 14, e0213108. [Google Scholar] [CrossRef]
- Moschen, A.R.; Tilg, H.; Raine, T. IL-12, IL-23 and IL-17 in IBD: Immunobiology and therapeutic targeting. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 185–196. [Google Scholar] [CrossRef]
- Borg-Bartolo, S.P.; Boyapati, R.K.; Satsangi, J.; Kalla, R. Precision medicine in inflammatory bowel disease: Concept, progress and challenges. F1000Research 2020, 9, F1000. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Etxebarria, K.; Merino, O.; Gaite-Reguero, A.; Rodrigues, P.M.; Herrarte, A.; Etxart, A.; Ellinghaus, D.; Alonso-Galan, H.; Franke, A.; Marigorta, U.M.; et al. Local genetic variation of inflammatory bowel disease in Basque population and its effect in risk prediction. Sci. Rep. 2022, 12, 3386. [Google Scholar] [CrossRef]
- Gettler, K.; Levantovsky, R.; Moscati, A.; Giri, M.; Wu, Y.; Hsu, N.Y.; Chuang, L.S.; Sazonovs, A.; Venkateswaran, S.; Korie, U.; et al. Common and Rare Variant Prediction and Penetrance of IBD in a Large, Multi-ethnic, Health System-based Biobank Cohort. Gastroenterology 2021, 160, 1546–1557. [Google Scholar] [CrossRef]
- Dickson, A.L.; Daniel, L.L.; Zanussi, J.; Dale Plummer, W.; Wei, W.Q.; Liu, G.; Reese, T.; Anandi, P.; Birdwell, K.A.; Kawai, V.; et al. TPMT and NUDT15 Variants Predict Discontinuation of Azathioprine for Myelotoxicity in Patients with Inflammatory Disease: Real-World Clinical Results. Clin. Pharmacol. Ther. 2022, 111, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Relling, M.V.; Schwab, M.; Whirl-Carrillo, M.; Suarez-Kurtz, G.; Pui, C.H.; Stein, C.M.; Moyer, A.M.; Evans, W.E.; Klein, T.E.; Antillon-Klussmann, F.G.; et al. Clinical Pharmacogenetics Implementation Consortium Guideline for Thiopurine Dosing Based on TPMT and NUDT15 Genotypes: 2018 Update. Clin. Pharmacol. Ther. 2019, 105, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Gareb, B.; Otten, A.T.; Frijlink, H.W.; Dijkstra, G.; Kosterink, J.G.W. Review: Local Tumor Necrosis Factor-α Inhibition in Inflammatory Bowel Disease. Pharmaceutics 2020, 12, 539. [Google Scholar] [CrossRef] [PubMed]
- Verstockt, B.; Verstockt, S.; Dehairs, J.; Ballet, V.; Blevi, H.; Wollants, W.J.; Breynaert, C.; Van Assche, G.; Vermeire, S.; Ferrante, M. Low TREM1 expression in whole blood predicts anti-TNF response in inflammatory bowel disease. EBioMedicine 2019, 40, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Biasci, D.; Lee, J.C.; Noor, N.M.; Pombal, D.R.; Hou, M.; Lewis, N.; Ahmad, T.; Hart, A.; Parkes, M.; McKinney, E.F.; et al. A blood-based prognostic biomarker in IBD. Gut 2019, 68, 1386–1395. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Biasci, D.; Roberts, R.; Gearry, R.B.; Mansfield, J.C.; Ahmad, T.; Prescott, N.J.; Satsangi, J.; Wilson, D.C.; Jostins, L.; et al. Genome-wide association study identifies distinct genetic contributions to prognosis and susceptibility in Crohn’s disease. Nat. Genet. 2017, 49, 262–268. [Google Scholar] [CrossRef]
- Ruemmele, F.M.; Veres, G.; Kolho, K.L.; Griffiths, A.; Levine, A.; Escher, J.C.; Amil Dias, J.; Barabino, A.; Braegger, C.P.; Bronsky, J.; et al. European Crohn’s and Colitis Organisation; European Society of Pediatric Gastroenterology, Hepatology and Nutrition. Consensus guidelines of ECCO/ESPGHAN on the medical management of pediatric Crohn’s disease. J. Crohn’s Colitis 2014, 8, 1179–1207. [Google Scholar] [CrossRef]
- Heap, G.A.; Weedon, M.N.; Bewshea, C.M.; Singh, A.; Chen, M.; Satchwell, J.B.; Vivian, J.P.; So, K.; Dubois, P.C.; Andrews, J.M.; et al. HLA-DQA1-HLA-DRB1 variants confer susceptibility to pancreatitis induced by thiopurine immunosuppressants. Nat. Genet. 2014, 46, 1131–1134. [Google Scholar] [CrossRef] [PubMed]
- Lauro, R.; Mannino, F.; Irrera, N.; Squadrito, F.; Altavilla, D.; Squadrito, G.; Pallio, G.; Bitto, A. Pharmacogenetics of Biological Agents Used in Inflammatory Bowel Disease: A Systematic Review. Biomedicines 2021, 9, 1748. [Google Scholar] [CrossRef] [PubMed]
- Bek, S.; Nielsen, J.V.; Bojesen, A.B.; Franke, A.; Bank, S.; Vogel, U.; Andersen, V. Systematic review: Genetic biomarkers associated with anti-TNF treatment response in inflammatory bowel diseases. Aliment. Pharmacol. Ther. 2016, 44, 554–567. [Google Scholar] [CrossRef]
- Puca, P.; Capobianco, I.; Coppola, G.; Di Vincenzo, F.; Trapani, V.; Petito, V.; Laterza, L.; Pugliese, D.; Lopetuso, L.R.; Scaldaferri, F. Cellular and Molecular Determinants of Biologic Drugs Resistance and Therapeutic Failure in Inflammatory Bowel Disease. Int. J. Mol. Sci. 2024, 25, 2789. [Google Scholar] [CrossRef]
- Bai, B.Y.H.; Reppell, M.; Smaoui, N.; Waring, J.F.; Pivorunas, V.; Guay, H.; Lin, S.; Chanchlani, N.; Bewshea, C.; Goodhand, J.R.; et al. Baseline Expression of Immune Gene Modules in Blood is Associated with Primary Response to Anti-TNF Therapy in Crohn’s Disease Patients. J. Crohn’s Colitis 2024, 18, 431–445. [Google Scholar] [CrossRef]
- Bank, S.; Julsgaard, M.; Abed, O.K.; Burisch, J.; Broder Brodersen, J.; Pedersen, N.K.; Gouliaev, A.; Ajan, R.; Nytoft-Rasmussen, D.; Honore Grauslund, C.; et al. Polymorphisms in the NFkB, TNF-alpha, IL-1beta, and IL-18 pathways are associated with response to anti-TNF therapy in Danish patients with inflammatory bowel disease. Aliment. Pharmacol. Ther. 2019, 49, 890–903. [Google Scholar] [CrossRef]
- Salvador-Martín, S.; Zapata-Cobo, P.; Velasco, M.; Palomino, L.M.; Clemente, S.; Segarra, O.; Sánchez, C.; Tolín, M.; Moreno-Álvarez, A.; Fernández-Lorenzo, A.; et al. Association between HLA DNA Variants and Long-Term Response to Anti-TNF Drugs in a Spanish Pediatric Inflammatory Bowel Disease Cohort. Int. J. Mol. Sci. 2023, 24, 1797. [Google Scholar] [CrossRef]
- Lykowska-Szuber, L.; Walczak, M.; Stawczyk-Eder, K.; Krela-Kazmierczak, I.; Eder, P.; Zakerska-Banaszak, O.; Dobrowolska, A.; Skrzypczak-Zielinska, M. Variants of the CASP9 gene as candidate markers for primary response to anti-TNF therapy in Crohn’s disease patients. J. Appl. Genet. 2023, 64, 759–768. [Google Scholar] [CrossRef]
- Tang, J.; Zhang, C.B.; Lyu, K.S.; Jin, Z.M.; Guan, S.X.; You, N.; Huang, M.; Wang, X.D.; Gao, X. Association of polymorphisms in C1orf106, IL1RN, and IL10 with post-induction infliximab trough level in Crohn’s disease patients. Gastroenterol. Rep. 2019, 8, 367–373. [Google Scholar] [CrossRef]
- Salvador-Martín, S.; Pujol-Muncunill, G.; Bossacoma, F.; Navas-López, V.M.; Gallego-Fernández, C.; Segarra, O.; Clemente, S.; Muñoz-Codoceo, R.; Viada, J.; Magallares, L.; et al. Pharmacogenetics of trough serum anti-TNF levels in paediatric inflammatory bowel disease. Br. J. Clin. Pharmacol. 2021, 87, 447–457. [Google Scholar] [CrossRef]
- Salvador-Martín, S.; López-Cauce, B.; Nuñez, O.; Laserna-Mendieta, E.J.; García, M.I.; Lobato, E.; Abarca-Zabalía, J.; Sanjurjo-Saez, M.; Lucendo, A.J.; Marín-Jiménez, I.; et al. Genetic predictors of long-term response and trough levels of infliximab in Crohn’s disease. Pharmacol. Res. 2019, 149, 104478. [Google Scholar] [CrossRef]
- Laserna-Mendieta, E.J.; Salvador-Martín, S.; Arias, A.; López-Cauce, B.; Marín-Jiménez, I.; Menchén, L.A.; Marín-Rubio, L.; Ontañón Rodríguez, J.; López-Fernánde, L.A.; Lucendo, A.J. Single nucleotide polymorphisms in ADAM17, IL23R and SLCO1C1 genes protect against infliximab failure in adults with Crohn’s disease. Biomed. Pharmacother. 2023, 159, 114225. [Google Scholar] [CrossRef]
- Hoffmann, P.; Lamerz, D.; Hill, P.; Kirchner, M.; Gauss, A. Gene Polymorphisms of NOD2, IL23R, PTPN2 and ATG16L1 in Patients with Crohn’s Disease: On the Way to Personalized Medicine? Genes 2021, 12, 866. [Google Scholar] [CrossRef] [PubMed]
- Urabe, S.; Isomoto, H.; Ishida, T.; Maeda, K.; Inamine, T.; Kondo, S.; Higuchi, N.; Sato, K.; Uehara, R.; Yajima, H.; et al. Genetic Polymorphisms of IL-17F and TRAF3IP2 Could Be Predictive Factors of the Long-Term Effect of Infliximab against Crohn’s Disease. BioMed Res. Int. 2015, 2015, 416838. [Google Scholar] [CrossRef]
- Matsuoka, K.; Hamada, S.; Shimizu, M.; Nanki, K.; Mizuno, S.; Kiyohara, H.; Arai, M.; Sugimoto, S.; Iwao, Y.; Ogata, H.; et al. Factors predicting the therapeutic response to infliximab during maintenance therapy in Japanese patients with Crohn’s disease. PLoS ONE 2018, 13, e0204632. [Google Scholar] [CrossRef]
- Koder, S.; Repnik, K.; Ferkolj, I.; Pernat, C.; Skok, P.; Weersma, R.K.; Potočnik, U. Genetic polymorphism in ATG16L1 gene influences the response to adalimumab in Crohn’s disease patients. Pharmacogenomics 2015, 16, 191–204. [Google Scholar] [CrossRef]
- Dudzińska, E.; Szymona, K.; Gil-Kulik, P.; Chomik, P.; Świstowska, M.; Gryzińska, M.; Kocki, J. Imbalance of Controlled Death in Peripheral Blood Lymphocytes in Crohn’s Disease and Ulcerative Colitis. Medicina 2019, 55, 231. [Google Scholar] [CrossRef]
- Jezernik, G.; Gorenjak, M.; Potočnik, U. MIF Variant rs755622 Is Associated with Severe Crohn’s Disease and Better Response to Anti-TNF Adalimumab Therapy. Genes 2023, 14, 452. [Google Scholar] [CrossRef] [PubMed]
- Abreu, M.T.; Rowbotham, D.S.; Danese, S.; Sandborn, W.J.; Miao, Y.; Zhang, H.; Tikhonov, I.; Panaccione, R.; Hisamatsu, T.; Scherl, E.J.; et al. Efficacy and Safety of Maintenance Ustekinumab for Ulcerative Colitis Through 3 Years: UNIFI Long-term Extension. J. Crohn’s Colitis 2022, 16, 1222–1234. [Google Scholar] [CrossRef]
- Xu, J.; Xu, H.M.; Yang, M.F.; Liang, Y.J.; Peng, Q.Z.; Zhang, Y.; Tian, C.M.; Wang, L.S.; Yao, J.; Nie, Y.Q.; et al. New Insights Into the Epigenetic Regulation of Inflammatory Bowel Disease. Front. Pharmacol. 2022, 13, 813659. [Google Scholar] [CrossRef]
- Agrawal, M.; Allin, K.H.; Petralia, F.; Colombel, J.F.; Jess, T. Multiomics to elucidate inflammatory bowel disease risk factors and pathways. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 399–409. [Google Scholar] [CrossRef]
- Joustra, V.; Hageman, I.L.; Satsangi, J.; Adams, A.; Ventham, N.T.; de Jonge, W.J.; Henneman, P.; D’Haens, G.R.; Li Yim, A.Y.F. Systematic Review and Meta-analysis of Peripheral Blood DNA Methylation Studies in Inflammatory Bowel Disease. J. Crohn’s Colitis 2023, 17, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Cooke, J.; Zhang, H.; Greger, L.; Silva, A.-L.; Massey, D.; Dawson, C.; Metz, A.; Ibrahim, A.; Parkes, M. Mucosal Genome-wide Methylation Changes in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2012, 18, 2128–2137. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.A.; Nagy-Szakal, D.; Mir, S.A.; Frank, E.; Szigeti, R.; Kaplan, J.L.; Bronsky, J.; Opekun, A.; Ferry, G.D.; Winter, H.; et al. DNA methylation-associated colonic mucosal immune and defense responses in treatment-naïve pediatric ulcerative colitis. Epigenetics 2014, 9, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Wawrzyniak, M.; Scharl, M. Genetics and epigenetics of inflammatory bowel disease. Swiss. Med. Wkly. 2018, 14, w14671. [Google Scholar] [CrossRef] [PubMed]
- Azuara, D.; Aussó, S.; Rodriguez-Moranta, F.; Guardiola, J.; Sanjuan, X.; Lobaton, T.; Boadas, J.; Piqueras, M.; Monfort, D.; Guinó, E.; et al. New Methylation Biomarker Panel for Early Diagnosis of Dysplasia or Cancer in High-Risk Inflammatory Bowel Disease Patients. Inflamm. Bowel Dis. 2018, 24, 2555–2564. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lee, K.; Kim, K.; Yi, S.J. The role of histone modifications: From neurodevelopment to neurodiseases. Signal. Transduct. Target. Ther. 2022, 7, 217. [Google Scholar] [CrossRef] [PubMed]
- Millán-Zambrano, G.; Burton, A.; Bannister, A.J.; Schneider, R. Histone post-translational modifications—Cause and consequence of genome function. Nat. Rev. Genet. 2022, 3, 563–580. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Wang, S.; Li, J. Treatment of Inflammatory Bowel Disease: A Comprehensive Review. Front. Med. 2021, 8, 765474. [Google Scholar] [CrossRef]
- Kalla, R.; Adams, A.T.; Nowak, J.K.; Bergemalm, D.; Vatn, S.; Ventham, N.T.; Kennedy, N.A.; Ricanek, P.; Lindstrom, J.; Söderholm, J.; et al. Analysis of Systemic Epigenetic Alterations in Inflammatory Bowel Disease: Defining Geographical, Genetic and Immune-Inflammatory influences on the Circulating Methylome. J. Crohn’s Colitis 2023, 17, 170–184. [Google Scholar] [CrossRef]
- Ventham, N.T.; Kennedy, N.A.; Adams, A.T.; Kalla, R.; Heath, S.; O’Leary, K.R.; Drummond, H.; IBD BIOM Consortium; IBD CHARACTER Consortium; Wilson, D.C.; et al. Integrative epigenome-wide analysis demonstrates that DNA methylation may mediate genetic risk in inflammatory bowel disease. Nat. Commun. 2016, 7, 13507. [Google Scholar] [CrossRef] [PubMed]
- Li Yim, A.Y.F.; Duijvis, N.W.; Ghiboub, M.; Sharp, C.; Ferrero, E.; Mannens, M.M.A.M.; D’Haens, G.R.; de Jonge, W.J.; Te Velde, A.A.; Henneman, P. Whole-Genome DNA Methylation Profiling of CD14+ Monocytes Reveals Disease Status and Activity Differences in Crohn’s Disease Patients. J. Clin. Med. 2020, 9, 1055. [Google Scholar] [CrossRef] [PubMed]
- Comi, M.; Amodio, G.; Gregori, S. Interleukin-10-Producing DC-10 Is a Unique Tool to Promote Tolerance Via Antigen-Specific T Regulatory Type 1 Cells. Front. Immunol. 2018, 9, 682. [Google Scholar] [CrossRef]
- Fernandes, P.; MacSharry, J.; Darby, T.; Fanning, A.; Shanahan, F.; Houston, A.; Brint, E. Differential expression of key regulators of Toll-like receptors in ulcerative colitis and Crohn’s disease: A role for Tollip and peroxisome proliferator-activated receptor gamma? Clin. Exp. Immunol. 2016, 183, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, K.; Alonso, S.; Miyaki, Y.; Okada, S.; Ogura, H.; Shiiya, N.; Konishi, F.; Taya, T.; Perucho, M.; Suzuki, K. Array-based identification of common DNA methylation alterations in ulcerative colitis. Int. J. Oncol. 2012, 40, 983–994. [Google Scholar] [CrossRef] [PubMed]
- Samarani, S.; Dupont-Lucas, C.; Marcil, V.; Mack, D.; Israel, D.; Deslandres, C.; Jantchou, P.; Ahmad, A.; Amre, D. CpG Methylation in TGFβ1 and IL-6 Genes as Surrogate Biomarkers for Diagnosis of IBD in Children. Inflamm. Bowel Dis. 2020, 26, 1572–1578. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, M.; Gerbeth, L.; Gerling, M.; Rosenthal, R.; Steiger, K.; Weidinger, C.; Keye, J.; Wu, H.; Schmidt, F.; Weichert, W.; et al. HDAC inhibitors promote intestinal epithelial regeneration via autocrine TGFβ1 signalling in inflammation. Mucosal. Immunol. 2019, 12, 656–667. [Google Scholar] [CrossRef]
- Chang, J.; Ji, X.; Deng, T.; Qiu, J.; Ding, Z.; Li, Z.; Ma, Y.; Hu, X.; Li, L.; Qiu, J. Setd2 determines distinct properties of intestinal ILC3 subsets to regulate intestinal immunity. Cell Rep. 2022, 38, 110530. [Google Scholar] [CrossRef]
- Ding, Z.; Cai, T.; Tang, J.; Sun, H.; Qi, X.; Zhang, Y.; Ji, Y.; Yuan, L.; Chang, H.; Ma, Y.; et al. Setd2 supports GATA3+ST2+thymic-derived Treg cells and suppresses intestinal inflammation. Nat. Commun. 2022, 13, 7468. [Google Scholar] [CrossRef]
- Eshleman, E.M.; Shao, T.Y.; Woo, V.; Rice, T.; Engleman, L.; Didriksen, B.J.; Whitt, J.; Haslam, D.B.; Way, S.S.; Alenghat, T. Intestinal epithelial HDAC3 and MHC class II coordinate microbiota-specific immunity. J. Clin. Investig. 2023, 133, e162190. [Google Scholar] [CrossRef]
- Li, C.; Chen, Y.; Zhu, H.; Zhang, X.; Han, L.; Zhao, Z.; Wang, J.; Ning, L.; Zhou, W.; Lu, C.; et al. Inhibition of Histone Deacetylation by MS-275 Alleviates Colitis by Activating the Vitamin D Receptor. J. Crohn’s Colitis 2020, 14, 1103–1118. [Google Scholar] [CrossRef]
- Kazakevych, J.; Denizot, J.; Liebert, A.; Portovedo, M.; Mosavie, M.; Jain, P.; Stellato, C.; Fraser, C.; Corrêa, R.O.; Célestine, M.; et al. Smarcad1 mediates microbiota-induced inflammation in mouse and coordinates gene expression in the intestinal epithelium. Genome Biol. 2020, 21, 64. [Google Scholar] [CrossRef]
- Južnić, L.; Peuker, K.; Strigli, A.; Brosch, M.; Herrmann, A.; Häsler, R.; Koch, M.; Matthiesen, L.; Zeissig, Y.; Löscher, B.S.; et al. SETDB1 is required for intestinal epithelial differentiation and the prevention of intestinal inflammation. Gut 2021, 70, 485–498. [Google Scholar] [CrossRef]
- Wang, R.; Li, H.; Wu, J.; Cai, Z.Y.; Li, B.; Ni, H.; Qiu, X.; Chen, H.; Liu, W.; Yang, Z.H.; et al. Gut stem cell necroptosis by genome instability triggers bowel inflammation. Nature 2020, 580, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Hou, Y.; Chen, W.; Wang, J.; Xie, W.; Zhang, X.; Zeng, L. KIF9-AS1, LINC01272 and DIO3OS lncRNAs as novel biomarkers for inflammatory bowel disease. Mol. Med. Rep. 2018, 17, 2195–2202. [Google Scholar] [CrossRef] [PubMed]
- Ge, Q.; Dong, Y.; Lin, G.; Cao, Y. Long Noncoding RNA Antisense Noncoding RNA in the INK4 Locus Correlates with Risk, Severity, Inflammation and Infliximab Efficacy in Crohn’s Disease. Am. J. Med. Sci. 2019, 357, 134–142. [Google Scholar] [CrossRef]
- Visschedijk, M.C.; Spekhors, L.M.; Cheng, S.C.; van Loo, E.S.; Jansen, B.H.D.; Blokzijl, T.; Kil, H.; de Jong, D.J.; Pierik, M.; Maljaars, J.P.W.J.; et al. Genomic and Expression Analyses Identify a Disease-Modifying Variant for Fibrostenotic Crohn’s Disease. J. Crohn’s Colitis 2018, 12, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Zhou, G.; Chen, P.; Wang, Y.; Han, J.; Chen, M.; He, Y.; Zhang, S. Which long noncoding RNAs and circular RNAs contribute to inflammatory bowel disease? Cell Death Dis. 2020, 11, 456. [Google Scholar] [CrossRef]
- Liu, R.; Tang, A.; Wang, X.; Chen, X.; Zhao, L.; Xiao, Z.; Shen, S. Inhibition of lncRNA NEAT1 suppresses the inflammatory response in IBD by modulating the intestinal epithelial barrier and by exosome-mediated polarization of macrophages. Int. J. Mol. Med. 2018, 42, 2903–2913. [Google Scholar] [CrossRef]
- Geng, H.; Bu, H.F.; Liu, F.; Wu, L.; Pfeifer, K.; Chou, P.M.; Wang, X.; Sun, J.; Lu, L.; Pandey, A.; et al. In Inflamed Intestinal Tissues and Epithelial Cells, Interleukin 22 Signaling Increases Expression of H19 Long Noncoding RNA, Which Promotes Mucosal Regeneration. Gastroenterology 2018, 155, 144–155. [Google Scholar] [CrossRef] [PubMed]
- Zou, T.; Jaladanki, S.K.; Liu, L.; Xiao, L.; Chung, H.K.; Wang, J.Y.; Xu, Y.; Gorospe, M.; Wang, J.Y. H19 Long Noncoding RNA Regulates Intestinal Epithelial Barrier Function via MicroRNA 675 by Interacting with RNA-Binding Protein HuR. Mol. Cell Biol. 2016, 36, 1332–1341. [Google Scholar] [CrossRef]
- Wu, F.; Huang, Y.; Dong, F.; Kwon, J.H. Ulcerative Colitis-Associated Long Noncoding RNA, BC012900, Regulates Intestinal Epithelial Cell Apoptosis. Inflamm. Bowel Dis. 2016, 22, 782–795. [Google Scholar] [CrossRef]
- Haberman, Y.; BenShoshan, M.; Di Segni, A.; Dexheimer, P.J.; Braun, T.; Weiss, B.; Walters, T.D.; Baldassano, R.N.; Noe, J.D.; Markowitz, J.; et al. Long ncRNA Landscape in the Ileum of Treatment-Naive Early-Onset Crohn Disease. Inflamm. Bowel Dis. 2018, 24, 346–360. [Google Scholar] [CrossRef]
- Vautrin, A.; Manchon, L.; Garcel, A.; Campos, N.; Lapasset, L.; Laaref, A.M.; Bruno, R.; Gislard, M.; Dubois, E.; Scherrer, D.; et al. Both anti-inflammatory and antiviral properties of novel drug candidate ABX464 are mediated by modulation of RNA splicing. Sci. Rep. 2019, 9, 792. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Rao, J.N.; Cao, S.; Liu, L.; Chung, H.K.; Zhang, Y.; Zhang, J.; Liu, Y.; Gorospe, M.; Wang, J.Y. Long noncoding RNA SPRY4-IT1 regulates intestinal epithelial barrier function by modulating the expression levels of tight junction proteins. Mol. Biol. Cell. 2016, 27, 617–626. [Google Scholar] [CrossRef]
- Lucafò, M.; Di Silvestre, A.; Romano, M.; Avian, A.; Antonelli, R.; Martelossi, S.; Naviglio, S.; Tommasini, A.; Stocco, G.; Ventura, A.; et al. Role of the Long Non-Coding RNA Growth Arrest-Specific 5 in Glucocorticoid Response in Children with Inflammatory Bowel Disease. Basic Clin. Pharmacol. Toxicol. 2018, 122, 87–93. [Google Scholar] [CrossRef]
- Nag, S.; Mitra, O.; Tripathi, G.; Samanta, S.; Bhattacharya, B.; Chandane, P.; Mohanto, S.; Sundararajan, V.; Malik, S.; Rustagi, S.; et al. Exploring the theranostic potentials of miRNA and epigenetic networks in autoimmune diseases: A comprehensive review. Immun. Inflamm. Dis. 2023, 11, e1121. [Google Scholar] [CrossRef]
- James, J.P.; Riis, L.B.; Malham, M.; Høgdall, E.; Langholz, E.; Nielsen, B.S. MicroRNA Biomarkers in IBD-Differential Diagnosis and Prediction of Colitis-Associated Cancer. Int. J. Mol. Sci. 2020, 21, 7893. [Google Scholar] [CrossRef] [PubMed]
- Dhuppar, S.; Murugaiyan, G. miRNA effects on gut homeostasis: Therapeutic implications for inflammatory bowel disease. Trends Immunol. 2022, 43, 917–931. [Google Scholar] [CrossRef]
- Neudecker, V.; Haneklaus, M.; Jensen, O.; Khailova, L.; Masterson, J.C.; Tye, H.; Biette, K.; Jedlicka, P.; Brodsky, K.S.; Gerich, M.E.; et al. Myeloid-derived miR-223 regulates intestinal inflammation via repression of the NLRP3 inflammasome. J. Exp. Med. 2017, 214, 1737–1752. [Google Scholar] [CrossRef] [PubMed]
- Butin-Israeli, V.; Bui, T.M.; Wiesolek, H.L.; Mascarenhas, L.; Lee, J.J.; Mehl, L.C.; Knutson, K.R.; Adam, S.A.; Goldman, R.D.; Beyder, A.; et al. Neutrophil-induced genomic instability impedes resolution of inflammation and wound healing. J. Clin. Investig. 2019, 129, 712–726. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Gao, X.; Liu, L.; Li, Z.; Wan, Z.; Dong, Y.; Chen, X.; Niu, Y.; Zhang, J.; Yang, G. Visceral Adipose Tissue Derived Exosomes Exacerbate Colitis Severity via Pro-inflammatory MiRNAs in High Fat Diet Fed Mice. ACS Nano 2020, 14, 5099–5110. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Liang, Y.; Yang, J.; Xia, Y.; Chen, H.; Han, H.; Yang, Y.; Wu, W.; Gao, R.; Qin, H. MicroRNA-21 knockout improve the survival rate in DSS induced fatal colitis through protecting against inflammation and tissue injury. PLoS ONE 2013, 8, e66814. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; He, C.; Liu, C.; Cao, A.T.; Xue, X.; Evans-Marin, H.L.; Sun, M.; Fang, L.; Yao, S.; Pinchuk, I.V.; et al. miR-10a inhibits dendritic cell activation and Th1/Th17 cell immune responses in IBD. Gut 2015, 64, 1755–1764. [Google Scholar] [CrossRef]
- Shen, Y.; Zhou, M.; Yan, J.; Gong, Z.; Xiao, Y.; Zhang, C.; Du, P.; Chen, Y. MiR-200b inhibits TNF-α-induced IL-8 secretion and tight junction disruption of intestinal epithelial cells in vitro. Am. J. Physiol.-Gastrointest. Liver Physiol. 2017, 312, G123–G132. [Google Scholar] [CrossRef]
- Zhang, B.; Tian, Y.; Jiang, P.; Jiang, Y.; Li, C.; Liu, T.; Zhou, R.; Yang, N.; Zhou, X.; Liu, Z. MicroRNA-122a Regulates Zonulin by Targeting EGFR in Intestinal Epithelial Dysfunction. Cell Physiol. Biochem. 2017, 42, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Shen, J.; Cheng, J.; Fan, X. MicroRNA-21 regulates intestinal epithelial tight junction permeability. Cell Biochem. Funct. 2015, 33, 235–240. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, R.; Chen, J.; Wu, Q.; Kuang, Z. Baicalin Protects against TNF-α-Induced Injury by Down-Regulating miR-191a That Targets the Tight Junction Protein ZO-1 in IEC-6 Cells. Biol. Pharm. Bull. 2017, 40, 435–443. [Google Scholar] [CrossRef]
- Li, M.; Zhang, S.; Qiu, Y.; He, Y.; Chen, B.; Mao, R.; Cui, Y.; Zeng, Z.; Chen, M. Upregulation of miR-665 promotes apoptosis and colitis in inflammatory bowel disease by repressing the endoplasmic reticulum stress components XBP1 and ORMDL3. Cell Death Dis. 2017, 8, e2699. [Google Scholar] [CrossRef]
- Liu, L.; He, J.; Wei, X.; Wan, G.; Lao, Y.; Xu, W.; Li, Z.; Hu, H.; Hu, Z.; Luo, X.; et al. MicroRNA-20a-mediated loss of autophagy contributes to breast tumorigenesis by promoting genomic damage and instability. Oncogene 2017, 36, 5874–5884. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Miao, Y.; Shan, Y.; Liu, B.; Li, Y.; Zhao, L.; Jia, L. MiR-106b and miR-93 regulate cell progression by suppression of PTEN via PI3K/Akt pathway in breast cancer. Cell Death Dis. 2017, 8, e2796. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Shan, T.; Qu, H.; Chen, Y.; Wang, N.; Xia, J. Inhibition of miR-16 Ameliorates Inflammatory Bowel Disease by Modulating Bcl-2 in Mouse Models. J. Surg. Res. 2020, 253, 185–192. [Google Scholar] [CrossRef]
- Lin, X.T.; Zheng, X.B.; Fan, D.J.; Yao, Q.Q.; Hu, J.C.; Lian, L.; Wu, X.J.; Lan, P.; He, X.S. MicroRNA-143 Targets ATG2B to Inhibit Autophagy and Increase Inflammatory Responses in Crohn’s Disease. Inflamm. Bowel Dis. 2018, 24, 781–791. [Google Scholar] [CrossRef]
- Kim, H.Y.; Kwon, H.Y.; Ha Thi, H.T.; Lee, H.J.; Kim, G.I.; Hahm, K.B.; Hong, S. MicroRNA-132 and microRNA-223 control positive feedback circuit by regulating FOXO3a in inflammatory bowel disease. J. Gastroenterol. Hepatol. 2016, 31, 1727–1735. [Google Scholar] [CrossRef] [PubMed]
- Masi, L.; Capobianco, I.; Magrì, C.; Marafini, I.; Petito, V.; Scaldaferri, F. MicroRNAs as Innovative Biomarkers for Inflammatory Bowel Disease and Prediction of Colorectal Cancer. Int. J. Mol. Sci. 2022, 23, 7991. [Google Scholar] [CrossRef] [PubMed]
- Thorlacius-Ussing, G.; Schnack Nielsen, B.; Andersen, V.; Holmstrøm, K.; Pedersen, A.E. Expression and Localization of miR-21 and miR-126 in Mucosal Tissue from Patients with Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2017, 23, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Ahmed Hassan, E.; El-Din Abd El-Rehim, A.S.; Mohammed Kholef, E.F.; Abd-Elgwad Elsewify, W. Potential role of plasma miR-21 and miR-92a in distinguishing between irritable bowel syndrome, ulcerative colitis, and colorectal cancer. Gastroenterol. Hepatol. Bed Bench. 2020, 13, 147–154. [Google Scholar] [PubMed]
- Fang, K.; Law, I.K.M.; Padua, D.; Sideri, A.; Huang, V.; Kevil, C.G.; Iliopoulos, D.; Pothoulakis, C. MicroRNA-31-3p Is Involved in Substance P (SP)-Associated Inflammation in Human Colonic Epithelial Cells and Experimental Colitis. Am. J. Pathol. 2018, 188, 586–599. [Google Scholar] [CrossRef]
- Whiteoak, S.R.; Claridge, A.; Balendran, C.A.; Harris, R.J.; Gwiggner, M.; Bondanese, V.P.; Erlandsson, F.; Hansen, M.B.; Cummings, J.R.F.; Sanchez-Elsner, T. MicroRNA-31 Targets Thymic Stromal Lymphopoietin in Mucosal Infiltrated CD4+ T Cells: A Role in Achieving Mucosal Healing in Ulcerative Colitis? Inflamm. Bowel Dis. 2018, 24, 2377–2385. [Google Scholar] [CrossRef]
- Szűcs, D.; Béres, N.J.; Rokonay, R.; Boros, K.; Borka, K.; Kiss, Z.; Arató, A.; Szabó, A.J.; Vannay, Á.; Sziksz, E.; et al. Increased duodenal expression of miR-146a and -155 in pediatric Crohn’s disease. World J. Gastroenterol. 2016, 22, 6027–6035. [Google Scholar] [CrossRef] [PubMed]
- Béres, N.J.; Szabó, D.; Kocsis, D.; Szűcs, D.; Kiss, Z.; Müller, K.E.; Lendvai, G.; Kiss, A.; Arató, A.; Sziksz, E.; et al. Role of Altered Expression of miR-146a, miR-155, and miR-122 in Pediatric Patients with Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2016, 22, 327–335. [Google Scholar] [CrossRef]
- Schaefer, J.S.; Attumi, T.; Opekun, A.R.; Abraham, B.; Hou, J.; Shelby, H.; Graham, D.Y.; Streckfus, C.; Klein, J.R. MicroRNA signatures differentiate Crohn’s disease from ulcerative colitis. BMC Immunol. 2015, 16, 5. [Google Scholar] [CrossRef] [PubMed]
- Schönauen, K.; Le, N.; von Arnim, U.; Schulz, C.; Malfertheiner, P.; Link, A. Circulating and Fecal microRNAs as Biomarkers for Inflammatory Bowel Diseases. Inflamm. Bowel Dis. 2018, 24, 1547–1557. [Google Scholar] [CrossRef] [PubMed]
- Valmiki, S.; Ahuja, V.; Paul, J. MicroRNA exhibit altered expression in the inflamed colonic mucosa of ulcerative colitis patients. World J. Gastroenterol. 2017, 23, 5324–5332. [Google Scholar] [CrossRef]
- Zidar, N.; Boštjančič, E.; Jerala, M.; Kojc, N.; Drobne, D.; Štabuc, B.; Glavač, D. Down-regulation of microRNAs of the miR-200 family and up-regulation of Snail and Slug in inflammatory bowel diseases—Hallmark of epithelial-mesenchymal transition. J. Cell Mol. Med. 2016, 20, 1813–1820. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Yu, Y.; Tan, S. The role of the miR-21-5p-mediated inflammatory pathway in ulcerative colitis. Exp. Ther. Med. 2020, 19, 981–989. [Google Scholar] [CrossRef]
- Mohammadi, A.; Kelly, O.B.; Smith, M.I.; Kabakchiev, B.; Silverberg, M.S. Differential miRNA Expression in Ileal and Colonic Tissues Reveals an Altered Immunoregulatory Molecular Profile in Individuals with Crohn’s Disease versus Healthy Subjects. J. Crohn’s Colitis 2019, 13, 1459–1469. [Google Scholar] [CrossRef]
- Viennois, E.; Zhao, Y.; Han, M.K.; Xiao, B.; Zhang, M.; Prasad, M.; Wang, L.; Merlin, D. Serum miRNA signature diagnoses and discriminates murine colitis subtypes and predicts ulcerative colitis in humans. Sci. Rep. 2017, 7, 2520. [Google Scholar] [CrossRef]
- Chen, P.; Li, Y.; Li, L.; Yu, Q.; Chao, K.; Zhou, G.; Qiu, Y.; Feng, R.; Huang, S.; He, Y.; et al. Circulating microRNA146b-5p is superior to C-reactive protein as a novel biomarker for monitoring inflammatory bowel disease. Aliment. Pharmacol. Ther. 2019, 49, 733–743. [Google Scholar] [CrossRef]
- Tian, Y.; Xu, J.; Li, Y.; Zhao, R.; Du, S.; Lv, C.; Wu, W.; Liu, R.; Sheng, X.; Song, Y.; et al. MicroRNA-31 Reduces Inflammatory Signaling and Promotes Regeneration in Colon Epithelium, and Delivery of Mimics in Microspheres Reduces Colitis in Mice. Gastroenterology 2019, 156, 2281–2296.e6. [Google Scholar] [CrossRef] [PubMed]
- Fukata, T.; Mizushima, T.; Nishimura, J.; Okuzaki, D.; Wu, X.; Hirose, H.; Yokoyama, Y.; Kubota, Y.; Nagata, K.; Tsujimura, N.; et al. The Supercarbonate Apatite-MicroRNA Complex Inhibits Dextran Sodium Sulfate-Induced Colitis. Mol. Ther.-Nucleic Acids 2018, 12, 658–671. [Google Scholar] [CrossRef] [PubMed]
- Casertano, M.; Trotta, M.C.; Cenni, S.; Creoli, M.; Miele, E.; Martinelli, M.; Lepre, C.C.; Russo, M.; Alfano, R.; D’Amico, M.; et al. Infliximab therapy decreases the expression of serum and faecal miR-126 and miR-20a in paediatric Crohn’s disease: A pilot study. Acta Paediatr. 2024, 113, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Batra, S.K.; Heier, C.R.; Diaz-Calderon, L.; Tully, C.B.; Fiorillo, A.A.; van den Anker, J.; Conklin, L.S. Serum miRNAs Are Pharmacodynamic Biomarkers Associated with Therapeutic Response in Pediatric Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2020, 26, 1597–1606. [Google Scholar] [CrossRef]
- Chapman, C.G.; Pekow, J. The emerging role of miRNAs in inflammatory bowel disease: A review. Therap. Adv. Gastroenterol. 2015, 8, 4–22. [Google Scholar] [CrossRef]
- Morilla, I.; Uzzan, M.; Laharie, D.; Cazals-Hatem, D.; Denost, Q.; Daniel, F.; Belleannee, G.; Bouhnik, Y.; Wainrib, G.; Panis, Y.; et al. Colonic MicroRNA Profiles, Identified by a Deep Learning Algorithm, that Predict Responses to Therapy of Patients with Acute Severe Ulcerative Colitis. Clin. Gastroenterol. Hepatol. 2019, 17, 905–913. [Google Scholar] [CrossRef]
| Gene | Main Findings | References |
|---|---|---|
| CDH1 | Responsible for E-cadherin synthesis, a transmembrane glycoprotein expressed in the intestinal epithelium, involved in cell adhesion and maintenance of mucosal barrier balance. Methylation of several CpGs at the CDH1 locus was increased in the inflamed ileal mucosa. Risk marker for gastric and colorectal cancer. | [28,29] |
| GNA12 | Encodes the G protein Gα12, which causes destabilization of tight junctions and alteration of the intestinal barrier by phosphorylating zonulin ZO-1 and ZO-2. Anti-inflammatory factor that inhibits the excessive chemotactic migration of macrophages. | [30,31] |
| PTPN2 | Negative regulator of pro-inflammatory cytokine signaling and counteracts IFN-γ-induced epithelial damage. Maintenance of intestinal barrier function. Loss of PTPN2 causes reduced autophagy and promotes the development of intestinal inflammation. | [32,33] |
| HNF4-α | Increases intestinal permeability by modulating the expression of RNF186 that alters barrier integrity at tight junctions. A protein-truncating R179X variant in RNF186 confers protection against UC. | [34,35] |
| TRL-2 | Regulates intestinal permeability and tight junction translocation. Suppressing immune response and maintained mucosal integrity. Increased expression in the ileum of patients with active UC compared to inactive UC and healthy controls. | [36,37] |
| IL-23R | Inhibition of IL-23 or IL-23R function reduced intestinal mucosal inflammation and confers protection against IBD. Stimulation of IL-23R inhibits Th17 cells by IL-22 and IL 17-mediated reduction of commensal microbiota and maintains barrier function. | [38,39] |
| IL-17R | Interacts with IL-17R receptor to stimulate inflammatory reactions through the action of mitogen-activated protein kinase MAPK, NF-Κb, and JAK/PI3K signaling. Inhibiting IL-17RA reduces inflammation and restores intestinal barrier function. | [40,41] |
| Gene | Population Group | Disease | Main Findings | References |
|---|---|---|---|---|
| NUDT15 | Asian | IBD | Anticipates thiopurine-induced myelosuppression. Predicts development of stenosis or fistulas. | [42] |
| TNFSF15 | Asian | CD | Predictive for the development of complications (strictures, penetrating disease, and perianal fistula). | [43] |
| FUT2 | Asian | IBD | The FUT2 loss-of-function mutation alters the gut microbiota by decreasing the abundance of adherent bacteria and stimulating CD8+ and Th17 cells that produce inflammatory lesions. Found in about 20–60% of Asian and Caucasian populations. | [44,45] |
| HLA-DQA1*05 | European | CD | Increased risk of immunogenicity to anti-TNF agents. Carried by approximately 40% of Europeans. | [46,47] |
| HLA-DRB1*0103 | European | CD | Associated with extra-intestinal manifestation. | [48] |
| HLA-DRB1*1501 | European | CD | Has been involved in extensive ulcerative colitis with severe evolution. Encountered in 6–25% of people of European ancestry. | [49] |
| HLA-DRB1*07 | European and African American | CD | Frequently associated with ileal disease. This allele varies between 5% and 29% in Europeans and North Americans but is less than 1% in Japanese. | [24] |
| NOD2 (CARD15) | European and African American | CD | Regulation of inflammation and cell apoptosis. This gene is linked to early onset, severe evolution, and stenotic complications requiring surgical interventions. | [50] |
| HLA-DQA1*05 | European and African American | IBD | Associated with immunogenicity and loss of response to anti-TNF agents. | [51] |
| TPMT | European | IBD | Anticipates thiopurine-induced myelosuppression. About 25% of patients with European ancestry carry specific variants. | [52] |
| ATG16L1 | European and African American | CD | Destabilizes caspase-3 in Paneth cells. Reduces autophagy and intracellular bacterial clearance. Stimulates the release of proinflammatory cytokines. | [53] |
| Biologic Treatment | Disease | Techniques Used/Samples | Studied Genes | Treatment Effectiveness | References |
|---|---|---|---|---|---|
| IFX ADA | IBD pediatric cohort | RT-PCR Serum | rs2097432 HLA-DQA1*05 | Decreased response:
| [87] |
| IFX | rs2395185 HLA-DRB9 |
| |||
| IFX ADA | CD | RT-PCR Colonic tissue | rs1052571 rs4645978 CASP9 | Increased response:
| [88] |
| |||||
| IFX | CD | MassArray Analyzer System Serum | rs442905 C1orf106 | Decreased IFX levels in GA carriers (β = −0.949; p = 0.025). | [89] |
| rs7587051 ATG16L1 | Decreased IFX levels in GG genotype. | ||||
| rs3213448 IL1RN | Increased IFX levels in GA carriers (OR: 2.14; 95% CI: 0.99–4.77). | ||||
| IFX | IBD pediatric cohort | RT-PCR Serum | rs5030728 TLR4 | Decreased IFX levels in GG’ vs. GA + AA (OR: 3.434; 95% CI: 1.35–8.71; p = 0.020). | [90] |
| IFX | CD | RT-PCR Serum | rs3024505 IL-10 | Increased IFX levels. | [91] |
| IFX | CD | RT-PCR Serum | rs10489629 IL-23R | Protective effect against failure to respond to IFX; the lack of response to IFX was 75% lower. | [92] |
| rs10929587 ADAM17 rs3794271 SLCO1C1 | Increased risk of CD-related surgery (OR = 2.8; 95% CI = 1.1–7.3; p = 0.025). | ||||
| IFX ADA | CD | RT-PCR Serum | rs11209026 IL-23R | Increased risk of CD-related surgery. | [93] |
| IFX | IBD | RT-PCR Serum | rs5030728 TLR4 | Increased response G allele in rs5030728 in CD (OR: 2.23; 95% CI: 1.24–4.01). | [86] |
| rs10499563 IL-6 | Decreased response C allele in rs10499563 (OR: 1.31; 95% CI: 0.99–1.71). | ||||
| IFX | CD | RT-PCR Serum | rs766748 IL-17 | Increased response: G/G genotype (OR: 5.123; 95% CI: 1.261–27.77, p = 0.0213). | [94] |
| IFX | CD | RT-PCR Serum | rs1799724 TNF-α | Decreased response 857T allele higher production of TNF-α in the intestine. | [95] |
| ADA | CD | RT-PCR Serum | rs10210302 ATG16L1 | Increased response C/T and T/T genotype (OR: 9.44; 95% CI: 2.49–35.83). | [96] |
| Biomarker | Disease | Sample | Main Findings | References |
|---|---|---|---|---|
| DNA methylation analysis | ||||
| TAP1 TESPA1 RPTOR | IBD | Blood | Hypomethylation model with 3 DMPs was correlated with treatment escalation to biological agents or surgery in IBD (HR 5.19; CI 95%: 2.14–12.56). | [110] |
| RPS6KA2 VMP1 TNSF10 | CD UC | Blood | The paired hypomethylation probe biomarkers RPS6KA2/VMP1 and RPS6KA2/TNFSF10 accurately discriminate between controls and CD (AUC = 0.84/0.81) or UC (AUC = 0.73/0.71); VMP1/microRNA-21 methylation related with IBD susceptibility. | [111] |
| FKBP5 | CD UC | Blood | The polymorphism (rs4713916) in the putative promoter region of FKBP5—associated with glucocorticoid resistance only to CD (genotype GC vs. GA/AA, OR: 3.81; CI 95%: 1.66–8.75). | [112] |
| BCL3 | UC CD | Colonic tissue | Influences the severity of intestinal ulcerations. Hypomethylation of BCL3—strongly elevated in active UC and active or inactive CD. | [113,114] |
| EYA4 SLIT2 FLI1 USP44 SND1 | IBD | Colonic tissue | Early detection of colorectal cancer and dysplastic lesions in IBD patients compared to healthy controls (92% vs. 57%; OR: 8.63; p = 0.001). | [106] |
| CND1 OL4A2 HDAC2 GLI2 AXIN2 ABL1 | UC | Colonic tissue | Strong concordance of methylation alterations (hypermethylation and hypomethylation) in the cancer cells and the UC samples (OR: 22.15; CI 95%: 15.48–31.56, p < 0.001). | [115] |
| TGFβ1 | CD UC | Blood | Panels of methylation marks enable accurate diagnosis of CD (14 TGFβ1 + 4 IL-6 CpG sites, AUC = 0.95), UC (9 TGFβ1 CpG sites; AUC = 0.99 and 4 IL-6 CpG sites, AUC = 0.89), and discrimination between CD and UC (3 TGFβ1 CpG sites, AUC = 0.81). | [116] |
| Histone-modifying enzymes | ||||
| HDAC | IBD | Colonic tissue | HDAC inhibitors influences intestinal homeostasis by maintaining the integrity of the epithelial barrier. They modulate the expression of tight junction proteins (claudin-1, claudin-2, and occludin) and increase the synthesis of TGF-β1 and IL-8 to stimulate the healing of inflammatory lesions. | [117] |
| Setd2 | Experimental model colitis on mice | Cell culture Colonic tissue | Deletion of Setd2 stimulates the generation of NKp46 + ILC3 with enhanced cytotoxic signatures and tumor suppressive capacity. | [118] |
| Modulates the expression of Treg cells that inhibit the development of intestinal inflammatory lesions. | [119] | |||
| HDAC3 | Experimental model colitis on mice | Cell culture Colonic tissue | Influences the intestinal immunity (reduces the number of CD4+ T cells and stimulates the major histocompatibility complex class II (MHC II), with a role in the control of inflammatory lesions. | [120] |
| Therapeutic target: inhibit NF-κB signaling, downregulation of tight-junction proteins and stimulation of intraepithelial lymphocytes during infection. | [121] | |||
| HDAC1 HDAC5 | Experimental model colitis on mice | Cell culture Colonic tissue | Impact on intestinal permeability and reduce inflammation by promoting mucosal colonization with Enterobacteriaceae. | [122] |
| Therapeutic target HDAC1—negative regulator of STAT signaling, NF-κB signaling, and acute phase response; stimulation of IL-1β-dependent cytokine production. | [121] | |||
| SETDB1 | Experimental model colitis on mice | Blood Feces Colonic tissue | It acts on endogenous retroviruses to inhibit DNA damage. The deletion is accompanied by damage to the integrity of the intestinal barrier, epithelial differentiation disorders, and exacerbation of inflammatory lesions with a severe prognosis. | [123] |
| SETDB1 | IBD | Cell culture Colonic tissue | SETDB1 deletion causes intestinal stem cell instability, with the release of endogenous retroviruses and necroptosis dependent on ZBP1 that influences inflammatory lesions. | [124] |
| lncRNAs | ||||
| KIF9-AS1 | CD UC | Colonic tissue Blood | Expression levels were significantly higher in patients with UC and CD compared with the healthy controls (AUC = 0.872 and 0.811, respectively, p < 0.0001). | [125] |
| LINC01272 | Expression levels were significantly higher in patients with UC and CD compared with the healthy controls (AUC = 0.777 and 0.887, respectively, p < 0.001). | |||
| DIO3OS | Expression levels were significantly lower in patients with UC and CD compared with the healthy controls (AUC = 0.653 and 0.794, respectively, p < 0.0001). | |||
| ANRIL | CD | Colonic tissue | The low expression in the intestinal mucosa differentiates CD from healthy controls (AUC = 0.803, 95% CI: 0.733–0.874, sensitivity = 0.861, specificity = 0.642) and the active remission stage of the disease (AUC = 0.839, 95% CI: 0.760–0.918, sensitivity = 0.857, specificity = 0.712). | [126] |
| An increased level is associated with favorable response to infliximab. | ||||
| RP11–679B19.1 | CD | Ileal tissue | Strong association between increased expression and the WWOX gene in patients with recurrent fibrostenotic CD carrying the risk allele A (OR = 4.13, p < 0.01). | [127] |
| lncRNA | Main Findings | References |
|---|---|---|
| ANRIL | Low expression in intestinal mucosa; increased secretion of proinflammatory cytokines; differentiates between active and remission stages of CD. | [126] |
| RP11–679B19.1 | Strong association with WWOX gene in recurrent stenotic CD (OR = 4.13, p < 0.01). | [127] |
| BC012900 | Elevated in patients with active UC; its overexpression in intestinal epithelium increases susceptibility to apoptosis. | [133] |
| HNF4A-AS1 | Related to severe mucosal ulcerations in pediatric patients; affects expression of HNF4A gene, regulating epithelial barrier function. | [134] |
| LINC01272 | Expressed in monocytes and neutrophils in intestinal epithelium; related to severe mucosal ulcerations; increased level correlated with fecal calprotectin (r = 0.9, p < 0.01). | [134] |
| DIO3OS | Biomarker with high performance to differentiate IBD patients from healthy controls. | [125] |
| KIF9-AS1 | Elevated levels in UC and CD patients; potential as a biomarker for disease activity and progression. | [125] |
| miRNAs | Target | Main Findings | References |
|---|---|---|---|
| Intestinal inflammation | |||
| miR-223 | NLRP3 | Inhibits NLRP3, which decreases the release of IL-1β and favors the healing process. | [141] |
| miR-23a miR-155 | Neutrophils | Reduces the infiltration of the intestinal tissue with activated neutrophils; prevents further injuries and the development of neoplasia. | [142] |
| miR-155 | Macrophages | Release at the intestinal level induces the differentiation of M1 macrophages, which increases the inflammatory lesions. | [143] |
| miR-21 | TNF-α MIP-2 | Stimulates the secretion of TNF-α and macrophage inflammatory protein 2 (MIP-2), which intensifies inflammatory lesions. | [144] |
| miR-10a | NOD2 IL-12/ IL-23p40 | Downregulates the expression of NOD2 and IL-12/IL-23p40 in the inflamed mucosa and suppresses T helper (Th)1 and Th17 cell responses. | [145] |
| Epithelial barrier function | |||
| miR-200b | claudin-1 zonulin-1 | Inhibits TNF-α and IL-8 and prevents the increase in paracellular permeability by redistributing proteins of intercellular tight junctions (claudin-1 and zonulin-1). | [146] |
| miR-675 | zonulin-1 cadherin E | Increases intestinal permeability by altering zonulin 1 and E-cadherin at the level of intercellular tight junctions. | [132] |
| miR-122a | EGFR | Increases the expression of zonulin-1, which is correlated with the dysfunction of the intestinal epithelium. | [147] |
| miR-21 | PTEN p-Ak pathway | Hyperproduction suppresses tensin (PTEN) expression and increases phospho-Akt (p-Akt) levels to improve paracellular permeability. | [148] |
| miR-191a | zonulin-1 | TNF-α stimulation increased miR-191a expression, leading to the decline in zonulin-1 and increased intestinal permeability. | [149] |
| Autophagy | |||
| miR-665 | XBP1 ORMDL3 | Promotes apoptosis by repressing the endoplasmic reticulum stress components XBP1 and ORMDL3. | [150] |
| miR-20a | ECN1 ATG16L1 SQSTM1 | High levels were associated with downregulation of ECN1, ATG16L1, and SQSTM1, which inhibit autophagy. | [151] |
| miR-106b miR-93 | PTEN ATG16L1 Akt pathway | Suppression of ATG16L1 expression and PTEN activity inhibits autophagy and stimulates the development of neoplastic lesions. | [152] |
| miR-16 | Bcl-2 | Downregulates the expression of Bcl-2 to disrupt colonic epithelium and inhibit autophagy. | [153] |
| miR-143 | ATG2B | Inhibits autophagy and increases inflammatory responses. | [154] |
| miR-132 miR-223 | FOXO3 | Downregulate FOXO3, which enhances NF-κB signaling and inhibits autophagy. | [155] |
| miRNAs | Role/Therapy | References |
|---|---|---|
| miR-31 | Significant reduction in colonic inflammation after capsule administration in mice with experimental colitis. | [171] |
| miR-223 | Decrease in occult bleeding, weight loss, and edema after intracolonic application of a nanoparticle emulsion in mice with DSS colitis. | [141] |
| miR-29b | Reduction in inflammatory response and inhibition of immune response through subcutaneous injection in mice with DSS colitis. | [172] |
| miR-126, miR-20a | Significantly decreased levels in serum and feces after infliximab administration in pediatric CD patients. | [173] |
| miR-126, miR-146a, miR-146b, miR-320a, let-7c, miR-636, miR-193b | Clinical and therapeutic response in pediatric patients with IBD is associated with overexpression of miR-636 and miR-193b, and decreased levels of miR-320a, miR-126, let-7c, miR-146a, and miR-146b. miR-320a involved in ulcer healing in intestinal mucosa. | [174] |
| miR-29 | Reduces IL-23 expression by targeting the IL-12p40 subunit, considered a parameter of ustekinumab therapy efficacy. | [175] |
| miR-3934, miR-100, miR-718, miR-193b, miR-3150a-5p, miR-1260b, miR-938, miR-518b, miR-1468. | Pharmacodynamic biomarkers used to stratify responders versus nonresponders to infliximab therapy (84% accuracy, AUC = 0.82). | [176] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Minea, H.; Singeap, A.-M.; Minea, M.; Juncu, S.; Muzica, C.; Sfarti, C.V.; Girleanu, I.; Chiriac, S.; Miftode, I.D.; Stanciu, C.; et al. The Contribution of Genetic and Epigenetic Factors: An Emerging Concept in the Assessment and Prognosis of Inflammatory Bowel Diseases. Int. J. Mol. Sci. 2024, 25, 8420. https://doi.org/10.3390/ijms25158420
Minea H, Singeap A-M, Minea M, Juncu S, Muzica C, Sfarti CV, Girleanu I, Chiriac S, Miftode ID, Stanciu C, et al. The Contribution of Genetic and Epigenetic Factors: An Emerging Concept in the Assessment and Prognosis of Inflammatory Bowel Diseases. International Journal of Molecular Sciences. 2024; 25(15):8420. https://doi.org/10.3390/ijms25158420
Chicago/Turabian StyleMinea, Horia, Ana-Maria Singeap, Manuela Minea, Simona Juncu, Cristina Muzica, Catalin Victor Sfarti, Irina Girleanu, Stefan Chiriac, Ioana Diandra Miftode, Carol Stanciu, and et al. 2024. "The Contribution of Genetic and Epigenetic Factors: An Emerging Concept in the Assessment and Prognosis of Inflammatory Bowel Diseases" International Journal of Molecular Sciences 25, no. 15: 8420. https://doi.org/10.3390/ijms25158420
APA StyleMinea, H., Singeap, A.-M., Minea, M., Juncu, S., Muzica, C., Sfarti, C. V., Girleanu, I., Chiriac, S., Miftode, I. D., Stanciu, C., & Trifan, A. (2024). The Contribution of Genetic and Epigenetic Factors: An Emerging Concept in the Assessment and Prognosis of Inflammatory Bowel Diseases. International Journal of Molecular Sciences, 25(15), 8420. https://doi.org/10.3390/ijms25158420