Targeting Myeloid Differentiation Primary Response Protein 88 (MyD88) and Galectin-3 to Develop Broad-Spectrum Host-Mediated Therapeutics against SARS-CoV-2

Abstract
1. Introduction
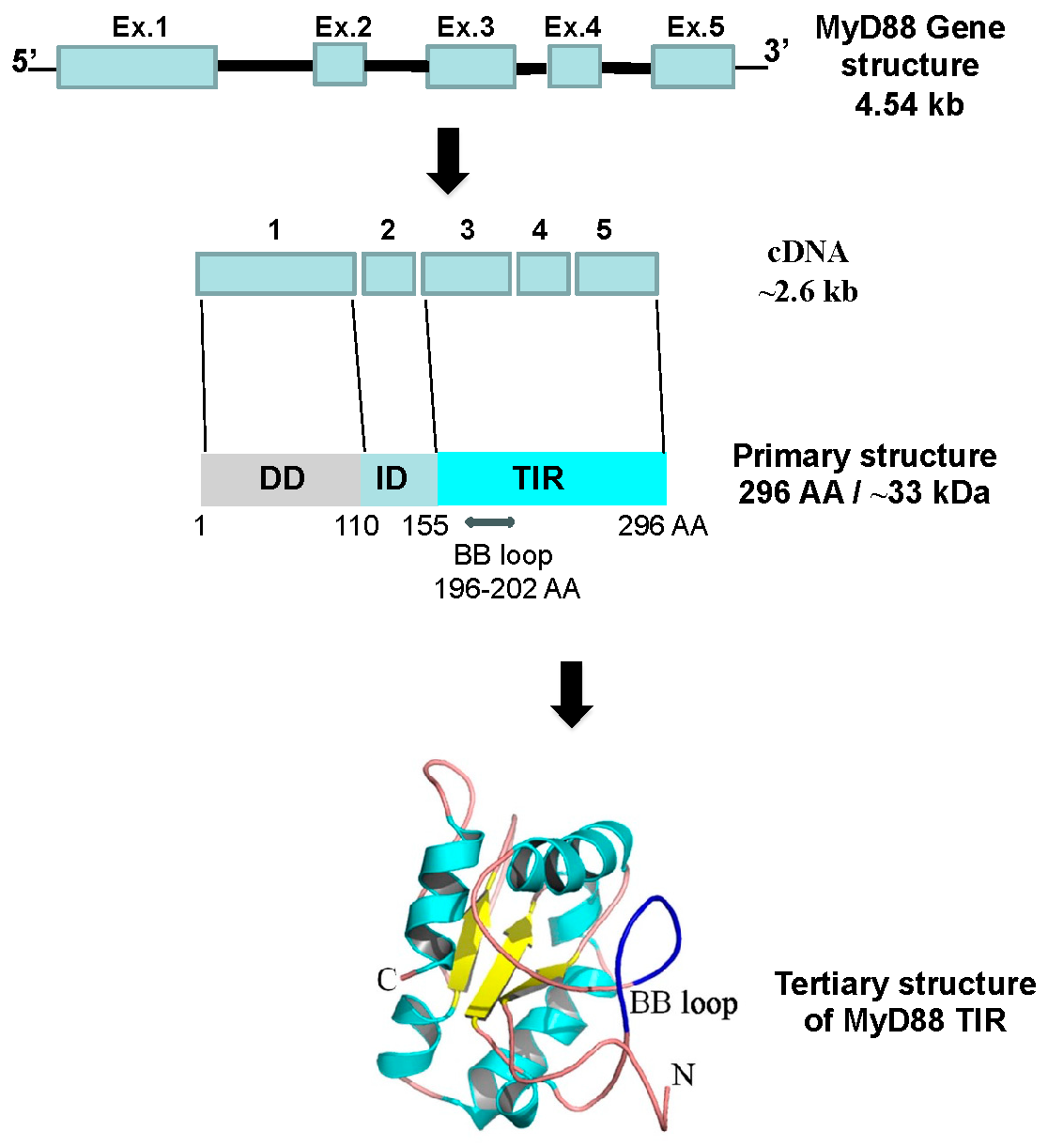
2. MyD88
2.1. Role of MyD88 in the Regulation of Host Immunity in Viral Infections—“Host Friendly” or “Hostile”?
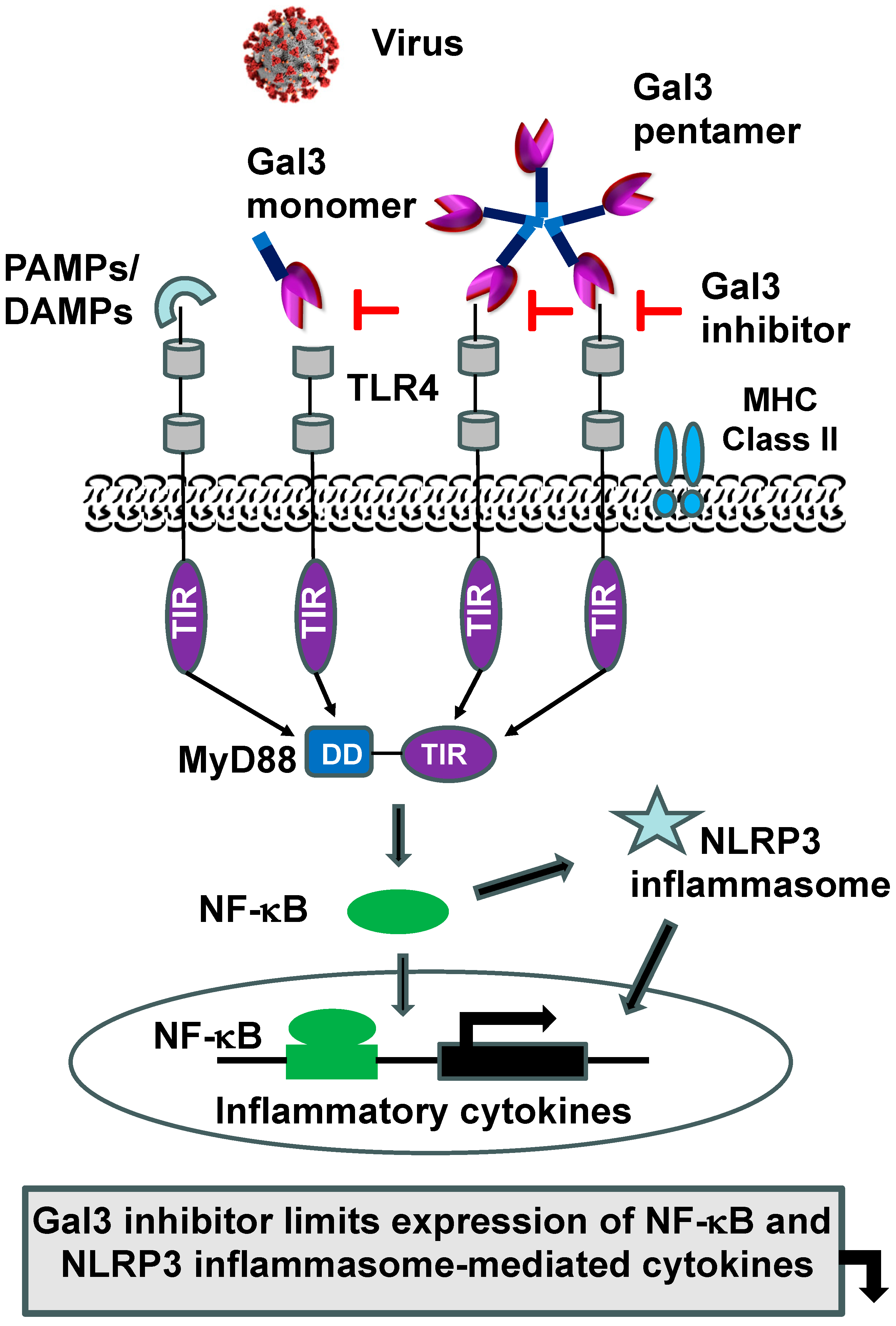
2.2. MyD88-Targeted Therapeutic Approach to Tackle Exuberant Inflammation and to Augment Antiviral IFN Response to Viral Infections, Including SARS-CoV-2
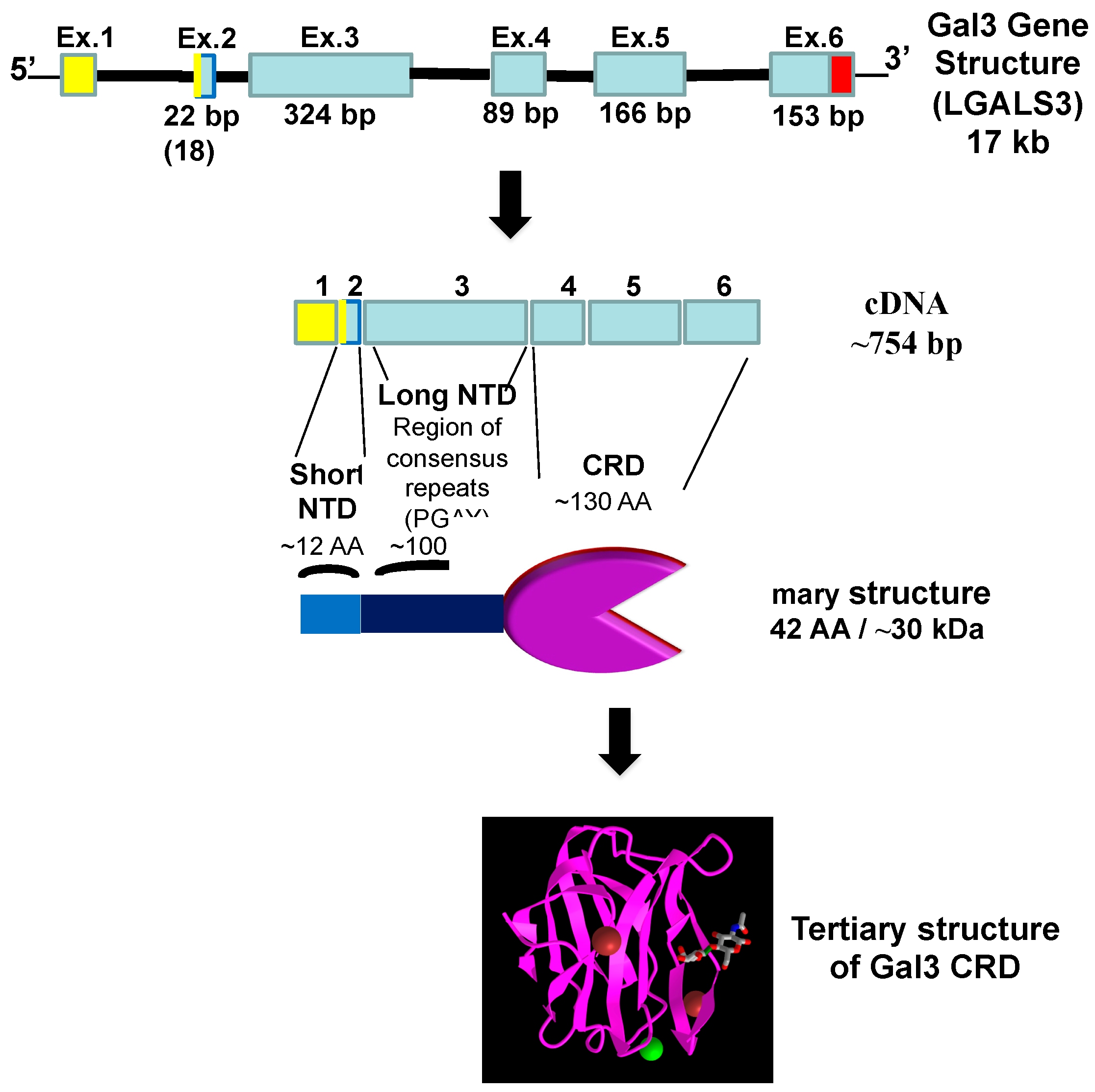
3. Galectin-3
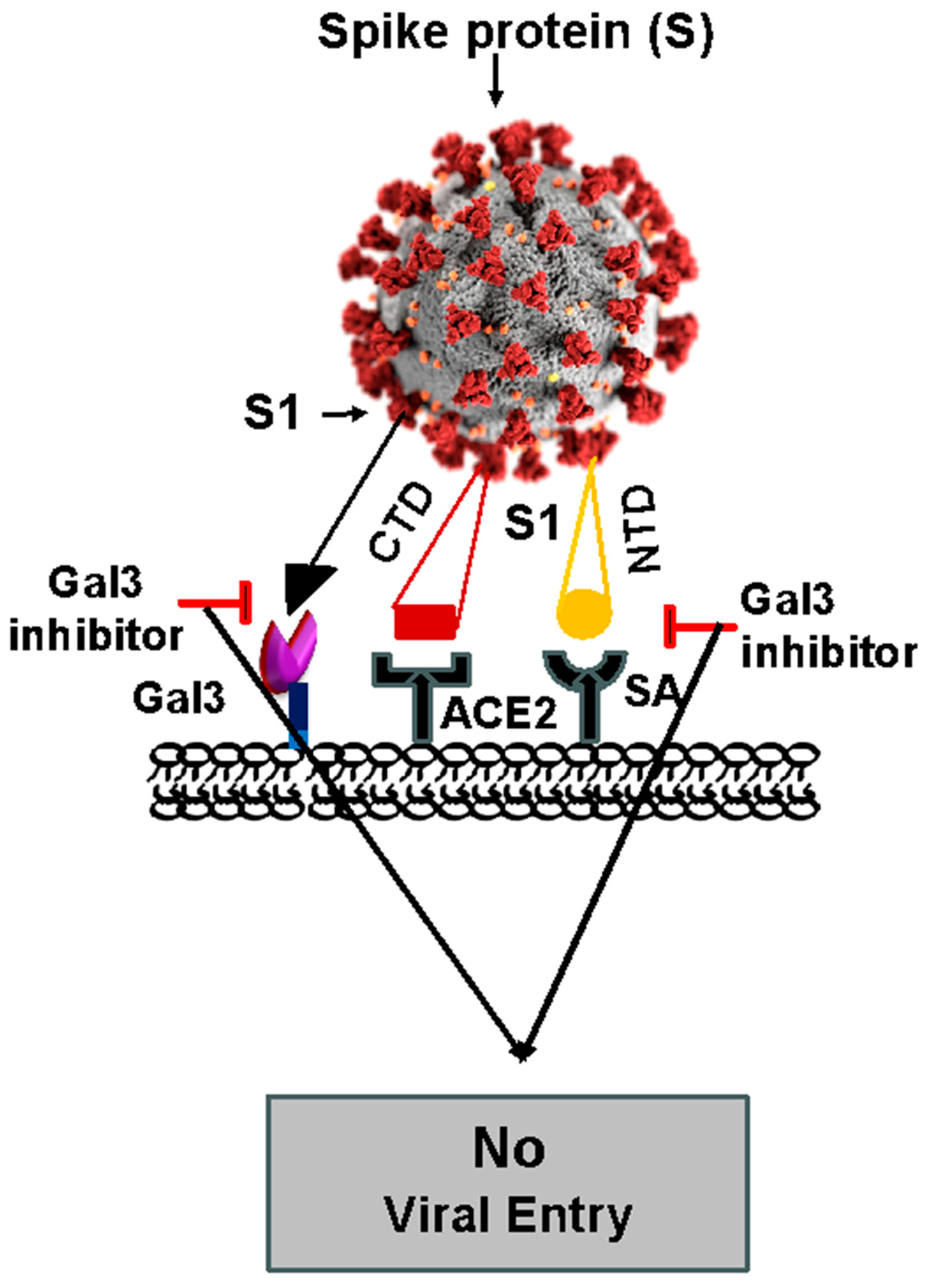
3.1. Role of Gal3 in Viral Infection, Including COVID-19
3.2. Immunomodulation of Gal3 in Viral Infection, Including COVID-19
4. Development of MyD88 and Gal3 Inhibitors to Treat COVID-19
5. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef] [PubMed]
- V’kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef]
- Lei, X.; Dong, X.; Ma, R.; Wang, W.; Xiao, X.; Tian, Z.; Wang, C.; Wang, Y.; Li, L.; Ren, L.; et al. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat. Commun. 2020, 11, 3810. [Google Scholar] [CrossRef]
- Xia, H.; Cao, Z.; Xie, X.; Zhang, X.; Chen, J.Y.; Wang, H.; Menachery, V.D.; Rajsbaum, R.; Shi, P.Y. Evasion of Type I Interferon by SARS-CoV-2. Cell Rep. 2020, 33, 108234. [Google Scholar] [CrossRef] [PubMed]
- Hatton, C.F.; Botting, R.A.; Dueñas, M.E.; Haq, I.J.; Verdon, B.; Thompson, B.J.; Spegarova, J.S.; Gothe, F.; Stephenson, E.; Gardner, A.I.; et al. Delayed induction of type I and III interferons mediates nasal epithelial cell permissiveness to SARS-CoV-2. Nat. Commun. 2021, 12, 7092. [Google Scholar] [CrossRef]
- Viox, E.G.; Hoang, T.N.; Upadhyay, A.A.; Nchioua, R.; Hirschenberger, M.; Strongin, Z.; Tharp, G.K.; Pino, M.; Nguyen, K.; Harper, J.L.; et al. Modulation of type I interferon responses potently inhibits SARS-CoV-2 replication and inflammation in rhesus macaques. Sci. Immunol. 2023, 8, eadg0033. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Taniguchi, T. IRFs: Master regulators of signaling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 2006, 6, 644–658. [Google Scholar] [CrossRef]
- Blasius, A.L.; Beutler, B. Intracellular toll-like receptors. Immunity 2010, 32, 305–315. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef]
- Takeda, K.; Kaisho, T.; Akira, S. Toll-like receptors. Ann. Rev. Immunol. 2003, 21, 335–376. [Google Scholar] [CrossRef] [PubMed]
- Saikh, K.U. MyD88 and beyond: A perspective on MyD88-targeted therapeutic approach for modulation of host immunity. Immunol. Res. 2021, 69, 117–128. [Google Scholar] [CrossRef]
- Ohnishi, H.; Tochio, H.; Kato, Z.; Orii, K.E.; Li, A.; Kimura, T.; Hiroaki, H.; Kondo, N.; Shirakawa, M. Structural basis for the multiple interactions of the MyD88 TIR domain in TLR4 signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 10260–10265. [Google Scholar] [CrossRef] [PubMed]
- Deguine, J.; Barton, G.M. MyD88: A central player in innate immune signaling. F1000Prime Rep. 2014, 6, 97. [Google Scholar] [CrossRef] [PubMed]
- Stojanovic, B.S.; Stojanovic, B.; Milovanovic, J.; Arsenijević, A.; Dimitrijevic Stojanovic, M.; Arsenijevic, N.; Milovanovic, M. The Pivotal Role of Galectin-3 in Viral Infection: A Multifaceted Player in Host-Pathogen Interactions. Int. J. Mol. Sci. 2023, 24, 9617. [Google Scholar] [CrossRef] [PubMed]
- Vasta, G.R. Roles of galectins in infection. Nat. Rev. Microbiol. 2009, 7, 424–438. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Alvarez, L.; Ortega, E. The Many Roles of Galectin-3, a Multifaceted Molecule, in Innate Immune Responses against Pathogens. Mediat. Inflamm. 2017, 2017, 9247574. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.T.; Stowell, S.R. The role of galectins in immunity and infection. Nat. Rev. Immunol. 2023, 23, 479–494. [Google Scholar] [CrossRef]
- Hardiman, G.; Jenkins, N.A.; Copeland, N.G.; Gilbert, D.J.; Garcia, D.K.; Naylor, S.L.; Kastelein, R.A.; Bazan, J.F. Genetic structure and chromosomal mapping of MyD88. Genomics 1997, 45, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Alcoceba, M.; García-Álvarez, M.; Medina, A.; Maldonado, R.; González-Calle, V.; Chillón, M.C.; Sarasquete, M.E.; González, M.; García-Sanz, R.; Jiménez, C. MYD88 Mutations: Transforming the Landscape of IgM Monoclonal Gammopathies. Int. J. Mol. Sci. 2022, 23, 5570. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R.; Preston-Hurlburt, P.; Kopp, E.; Stadlen, A.; Chen, C.; Ghosh, S.; Janeway, C.A., Jr. MyD88 is an adaptor protein in the Toll/IL-1 receptor family signaling pathways. Mol. Cell 1998, 2, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Olson, M.A.; Lee, M.S.; Kissner, T.L.; Alam, S.; Waugh, D.S.; Saikh, K.U. Discovery of Small Molecule Inhibitors of MyD88-Dependent Signaling Pathways using a Computational Screen. Sci. Rep. 2015, 5, 14246. [Google Scholar] [CrossRef] [PubMed]
- Fekonja, O.; Avbelj, M.; Jerala, R. Suppression of TLR signaling by targeting TIR domain-containing proteins. Curr. Protein Pept. Sci. 2012, 13, 776–788. [Google Scholar] [CrossRef] [PubMed]
- Avbelj, M.; Horvat, S.; Roman Jerala, R. The Role of Intermediary Domain of MyD88 in Cell Activation and Therapeutic Inhibition of TLRs. J. Immunol. 2011, 187, 2394–2404. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Tao, X.; Shen, B.; Horng, T.; Medzhitov, R.; Manley, J.L.; Tong, L. Structural Basis for Signal Transduction by the Toll/interleukin-I receptor domains. Nature 2000, 408, 111–115. [Google Scholar] [CrossRef]
- Saikh, K.U.; Morazzani, E.M.; Piper, A.E.; Bakken, R.R.; Glass, P.J. A small molecule inhibitor of MyD88 exhibits broad spectrum antiviral activity by up regulation of type I interferon. Antivir. Res. 2020, 181, 104854. [Google Scholar] [CrossRef] [PubMed]
- Muzio, M.; Ni, J.; Feng, P.; Dixit, V.M. IRAK (Pelle) family member IRAK-2 and MyD88 as proximal mediators of IL-1 signaling. Science 1997, 278, 1612–1615. [Google Scholar] [CrossRef] [PubMed]
- Fuse, K.; Chan, G.; Liu, Y.; Gudgeon, P.; Husain, M.; Chen, M.; Yeh, W.C.; Akira, S.; Liu, P.P. Myeloid differentiation factor-88 plays a crucial role in the pathogenesis of Coxsackievirus B3 induced myocarditis and influences type I interferon production. Circulation 2005, 112, 2276–2285. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Maheshwari, R. Oligonucleotide array analysis of Toll-like receptors and associated signaling genes in Venezuelan equine encephalitis virus-infected mouse brain. J. Gen. Virol. 2009, 2009, 1836–1847. [Google Scholar] [CrossRef] [PubMed]
- Edwards, M.R.; Liu, G.; Mire, C.E.; Sureshchandra, S.; Luthra, P.; Yen, B.; Shabman, R.S.; Leung, D.W.; Messaoudi, I.; Geisbert, T.W.; et al. Differential regulation of interferon responses by ebola and Marburg virus VP35 proteins. Cell Rep. 2016, 14, 1632–1640. [Google Scholar] [CrossRef] [PubMed]
- Siednienko, J.; Halle, A.; Nagpal, K.; Golenbock, D.T.; Miggin, S.M. TLR3-mediated IFN-β gene induction is negatively regulated by the TLR adaptor MyD88 adaptor-like. Eur. J. Immunol. 2010, 40, 3150–3160. [Google Scholar] [CrossRef]
- Siednienko, J.; Gajanayake, T.; Fitzgerald, K.A.; Moynagh, P.; Miggin, S.M. Absence of MyD88 results in enhanced TLR3-dependent phosphorylation of IRF3 and increased IFN-β and RANTES production. J. Immunol. 2011, 186, 2514–2522. [Google Scholar] [CrossRef] [PubMed]
- TenOver, B.R. The Evolution of antiviral defense systems. Cell Host Microbe 2016, 19, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Janeway, C.A., Jr.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Chen, Z.J. Innate immune sensing and signaling of cytosolic nucleic acids. Annu. Rev. Immunol. 2014, 32, 461–488. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Yanai, H. Interferon signaling network in innate defense. Cell Microbiol. 2006, 8, 907–922. [Google Scholar]
- Brierle, M.M.; Fish, E.N. IFN- alpha beta receptor interactions to biologic outcomes; understanding the circuitry. J. Interferon Cytokine Res. 2002, 22, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Lazear, H.M.; Schoggins, J.W.; Diamond, M.S. Shared and distinct functions of type I and type III interferons. Immunity 2019, 50, 907–923. [Google Scholar] [CrossRef] [PubMed]
- Hur, S. Double stranded RNA sensors and modulators of innate immunity. Annu. Rev. Immunol. 2019, 37, 349–375. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.X.; Fish, E.N. The yin and yang of viruses and Interferons. Trends Immunol. 2012, 33, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Toporovski, R.; Morrow, M.P.; Weiner, D.B. Interferons as potential adjuvants in prophylactic vaccines. Expert. Opin. Biol. Ther. 2010, 10, 1489–1500. [Google Scholar] [CrossRef] [PubMed]
- Galani, I.E.; Rovina, N.; Lampropoulou, V.; Triantafyllia, V.; Manioudaki, M.; Pavlos, E.; Koukaki, E.; Fragkou, P.C.; Panou, V.; Rapti, V.; et al. Untuned antiviral immunity in COVID-19 revealed by temporal type I/III interferon patterns and flu comparison. Nat. Immunol. 2021, 22, 32–40. [Google Scholar] [CrossRef]
- Sallard, E.; Lescure, F.-X.; Yazdanapanah, Y.; Mentre, F.; Peiffer-Smadja, N. Type 1 interferons as a potential treatment against COVID-19. Antivir. Res. 2020, 178, 10479. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, T.; Ogasawara, K.; Takaoka, A.; Tanaka, N. IRF family of transcription factors as regulators of host defense. Annu. Rev. Immunol. 2001, 19, 623–655. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Heylbroeck, C.; Pitha, P.M.; Hiscott, J. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome mediated degradation. Mol. Cell. Biol. 1998, 18, 2986–2996. [Google Scholar] [CrossRef]
- Nguyen, H.; Hiscott, J.; Pitha, P.M. The growing family of interferon regulatory factors. Cytokine Growth Factor Rev. 1997, 8, 293–312. [Google Scholar] [CrossRef] [PubMed]
- Mamane, Y.; Heylbroeck, C.; Génin, P.; Algarté, M.; Servant, M.J.; LePage, C.; DeLuca, C.; Kwon, H.; Lin, R.; Hiscott, J. Interferon regulatory factors: The next generation. Gene 1999, 237, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Yanai, H.; Negishi, H.; Asagiri, M.; Sato, M.; Mizutani, T.; Shimada, N.; Ohba, Y.; Takaoka, A.; Yoshida, N.; et al. IRF-7 is he master regulator of type-1 interferon-dependent immune responses. Nature 2005, 434, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.A.; McWhirter, S.M.; Faia, K.L.; Rowe, D.C.; Latz, E.; Golenbock, D.T.; Coyle, A.J.; Liao, S.M.; Maniatis, T. IKK epsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003, 4, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Planès, R.; Ben Haij, N.; Leghmari, K.; Serrero, M.; BenMohamed, L.; Bahraoui, E. HIV-1 Tat Protein Activates both the MyD88 and TRIF Pathways to Induce Tumor Necrosis Factor Alpha Interleukin-10 in Human Monocytes. J. Virol. 2016, 90, 5886–5898. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.V.; Cicha, M.Z.; Meyerholz, D.K.; Chapleau, M.W.; Abboud, F.M. Dual Activation of TRIF and MyD88 Adaptor Proteins by Angiotensin II Evokes Opposing Effects on Pressure, Cardiac Hypertrophy, and Inflammatory Gene Expression. Hypertension 2015, 66, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Kenny, E.F.; Talbot, S.; Gong, M.; Golenbock, D.T.; Bryant, C.E.; O’Neill, L.A. MyD88 adaptor like is not essential for TLR2 signaling and inhibits signaling by TLR3. J. Immunol. 2009, 183, 3642–3651. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.C.; Li, X.; Pearlman, E. MyD88 functions as a negative regulator of TLR3/TRIFinduced corneal inflammation by inhibiting activation of c-Jun N-terminal kinase. J. Biol. Chem. 2003, 278, 3988–39960. [Google Scholar]
- Saito, T.; Gale, M., Jr. Differential recognition of double-stranded RNA by RIG-I-like receptors in antiviral immunity. J. Exp. Med. 2008, 205, 1523–1527. [Google Scholar] [CrossRef] [PubMed]
- Ayithan, N.; Bradfute, S.B.; Anthony, S.M.; Stuthman, K.S.; Dye, J.M.; Bavari, S.; Bray, M.; Ozato, K. Ebola virus-like particles stimulate Type I interferons and pro-inflammatory cytokine expression through the toll-like receptors and interferon signaling pathways. J. Interferon Cytokine Res. 2014, 34, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Saikh, K.U.; Ranji, C.M. Cells Stimulated with More Than One Toll-Like Receptor-Ligand in the Presence of a MyD88 Inhibitor Augmented Interferon-β via MyD88-Independent Signaling Pathway. Viral Immunol. 2021, 34, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Ribero, M.S.; Jouvenet, N.; Dreux, M.; Nisole, S. Interplay between SARS-CoV-2 and type I interferon response. PLoS Pathog. 2020, 16, e1008737. [Google Scholar] [CrossRef] [PubMed]
- Totura, L.A.; Whitmore, A.; Agnihothram, S.; Schäfer, A.; Katze, M.G.; Heise, M.T.; Baric, R.S. Toll-like receptor signaling via TRIF contributes to a protective Innate Immune Response to Sever acute respiratory syndrome coronavirus infection. mBio 2015, 6, e00638-15. [Google Scholar] [CrossRef] [PubMed]
- Abou-Ismail, M.Y.; Diamond, A.; Kapoor, S.; Arafah, Y.; Nayak, L. The hypercoagulable state in COVID-19: Incidence, pathophysiology, and management. Thromb Res. 2020, 194, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Jean, K.; Lim, J.K.; Albrecht, R.A.; tenOever, B.R. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Iampietro, M.; Aurine, N.; Dhondt, K.P.; Dumont, C.; Pelissier, R.; Spanier, J.; Valive, A.; Raoul, H.; Kalinke, U.; Horvat, B. Control of nipah virus infection in mice by the host adaptors mitrochondrial antiviral signaling protein (MAVS) and myeloid differentiation primary response 88 (MyD88). J. Infect. Dis. 2019, 221 (Suppl. S4), S401–S406. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.X.; Zhang, B.C.; Sun, L. Poly (I:C) induces antiviral immune responses in Japanese flounder (Paralichthys olivaceus) that require TLR3 and MDA5 and is negatively regulated by MyD88. PLoS ONE 2014, 9, e112918. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 49. [Google Scholar] [CrossRef] [PubMed]
- Diao, B.; Wang, C.; Tan, Y.; Chen, X.; Liu, Y.; Ning, L.; Chen, L.; Li, M.; Liu, Y.; Wang, G.; et al. Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID-19). Front Immunol. 2020, 11, 827. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Gao, Y.; Wang, G.; Song, G.; Liu, S.; Sun, D.; Xu, Y.; Tian, Z. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell Mol. Immunol. 2020, 17, 533–535. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Gil, L.; Ayllon, J.; Ortigoza, M.B.; Garcia-satre, A.; Shaw, M.L.; Palese, P. Identification of small molecules with type I interferon inducing properties by High-throughput Screening. PLoS ONE 2012, 7, e49049. [Google Scholar] [CrossRef] [PubMed]
- Suckling, C.J.; Alam, S.; Olson, M.A.; Saikh, K.U.; Harnett, M.M.; Harnett, W. Small Molecule Analogues of the parasitic worm product ES-62 interact with the TIR domain domain of MyD88 to inhibit pro-inflammatory signaling. Sci. Rep. 2018, 8, 2123. [Google Scholar] [CrossRef] [PubMed]
- Kadrofske, M.M.; Openo, K.P.; Wang, J.L. The HumanLGALS3 (Galectin-3) Gene: Determination of the Gene Structure and Functional Characterization of the Promoter. Arch. Biochem. Biophys. 1998, 349, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Gong, H.C.; Honjo, Y.; Nangia-Makker, P.; Hogan, V.; Mazurak, N.; Bresalier, R.S.; Raz, A. The NH2 terminus of galectin-3 governs cellular compartmentalization and functions in cancer cells. Cancer Res. 1999, 59, 6239–6245. [Google Scholar] [PubMed]
- Dumic, J.; Dabelic, S.; Flögel, M. Galectin-3: An open-ended story. Biochim. Biophys. Acta 2006, 1760, 616–635. [Google Scholar] [CrossRef] [PubMed]
- Elola, M.T.; Wolfenstein-Todel, C.; Troncoso, M.F.; Vasta, G.R.; Rabinovich, G.A. Galectins: Matricellular glycan-binding proteins linking cell adhesion, migration, and survival. Cell Mol. Life Sci. 2007, 64, 1679–1700. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, R.; Anam, K.; Ahmed, H. Development of Galectin-3 Targeting Drugs for Therapeutic Applications in Various Diseases. Int. J. Mol. Sci. 2023, 24, 8116. [Google Scholar] [CrossRef] [PubMed]
- Caniglia, J.L.; Asuthkar, S.; Tsung, A.J.; Guda, M.R.; Velpula, K.K. Immunopathology of galectin-3: An increasingly promising target in COVID-19. F1000Research 2020, 9, 1078. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.F.; Tsao, C.H.; Lin, Y.T.; Hsu, D.K.; Chiang, M.L.; Lo, C.H.; Chien, F.C.; Chen, P.; Arthur Chen, Y.M.; Chen, H.Y.; et al. Galectin-3 promotes HIV-1 budding via association with Alix and Gag p6. Glycobiology 2014, 24, 1022–1035. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.F.; Hung, Y.H.; Tsao, C.H.; Chiang, C.Y.; Teoh, P.G.; Chiang, M.L.; Lin, W.H.; Hsu, D.K.; Jan, H.M.; Lin, H.C.; et al. Galectin-3 facilitates cell-to-cell HIV-1 transmission by altering the composition of membrane lipid rafts in CD4 T cells. Glycobiology 2022, 32, 760–777. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, R.; Prasad, A. Exosomes Derived from HIV-1 Infected DCs Mediate Viral trans-Infection via Fibronectin and Galectin-3. Sci. Rep. 2017, 7, 14787. [Google Scholar] [CrossRef] [PubMed]
- Woodward, A.M.; Mauris, J.; Argüeso, P. Binding of transmembrane mucins to galectin-3 limits herpesvirus 1 infection of human corneal keratinocytes. J. Virol. 2013, 87, 5841–5847. [Google Scholar] [CrossRef]
- Nita-Lazar, M.; Banerjee, A.; Feng, C.; Amin, M.N.; Frieman, M.B.; Chen, W.H.; Cross, A.S.; Wang, L.-X.; Vasta, G.R. Desialylation of airway epithelial cells during influenza virus infection enhances pneumococcal adhesion via galectin binding. Mol. Immunol. 2015, 65, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-J.; Wang, S.-F.; Weng, I.-C.; Hong, M.-H.; Lo, T.-H.; Jan, J.-T.; Hsu, L.-C.; Chen, H.-Y.; Liu, F.-T. Galectin-3 enhances avian H5N1 influenza a virus-induced pulmonary inflammation by promoting NLRP3 inflammasome activation. Am. J. Pathol. 2018, 188, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215. [Google Scholar] [CrossRef] [PubMed]
- Behloul, N.; Baha, S.; Shi, R.; Meng, J. Role of the GTNGTKR Motif in the N-Terminal Receptor-Binding Domain of the SARS-CoV-2 Spike Protein. Virus Res. 2020, 286, 198058. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yu, Y.; Xu, J.; Shu, H.; Liu, H.; Wu, Y.; Zhang, L.; Yu, Z.; Fang, M.; Yu, T.; et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia inWuhan, China: A single-centered, retrospective, observational study. Lancet Respir. Med. 2020, 8, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.; et al. Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.J.; Dong, X.; Cao, Y.Y.; Yuan, Y.D.; Yang, Y.B.; Yan, Y.Q.; Akdis, C.A.; Gao, Y.D. Clinical characteristics of 140 patients infected by SARS-CoV-2 in Wuhan, China. Allergy 2020, 75, 1730–1741. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Yang, J.; Zhao, F.; Zhi, L.; Wang, X.; Liu, L.; Bi, Z.; Zhao, Y. Prevalence and impact of cardiovascularmetabolic diseases on COVID-19 in China. Clin. Res. Cardiol. 2020, 109, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Seferovic, J.P.; Lalic, N.M.; Floridi, F.; Tesic, M.; Seferovic, P.M.; Giga, V.; Lalic, K.; Jotic, A.; Jovicic, S.; Colak, E.; et al. Structural myocardial alterations in diabetes and hypertension: The role of galectin-3. Clin. Chem. Lab. Med. 2014, 52, 1499–1505. [Google Scholar] [CrossRef] [PubMed]
- Pejnovic, N. Galectin-3 in obesity and type 2 diabetes. Ser. J. Exp. Clin. Res. 2015, 16, 273–280. [Google Scholar] [CrossRef]
- Yilmaz, H.; Cakmak, M.; Inan, O.; Darcin, T.; Akcay, A. Increased levels of galectin-3 were associated with prediabetes and diabetes: New risk factor? J. Endocrinol. Investig. 2015, 38, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Bobronnikova, L. Galectin-3 as a potential biomarker of metabolic disorders and cardiovascular remodeling in patients with hypertension and type 2 diabetes. Vessel. Plus 2017, 1, 61–67. [Google Scholar] [CrossRef]
- Portacci, A.; Diaferia, F.; Santomasi, C.; Dragonieri, S.; Boniello, E.; Di Serio, F.; Carpagnano, G.E. Galectin-3 as prognostic biomarker in patients with COVID-19 acute respiratory failure. Respir. Med. 2021, 187, 106556. [Google Scholar] [CrossRef] [PubMed]
- Cervantes-Alvarez, E.; la Rosa, N.L.; la Mora, M.S.; Valdez-Sandoval, P.; Palacios-Jimenez, M.; Rodriguez-Alvarez, F.; Vera-Maldonado, B.I.; Aguirre-Aguilar, E.; Escobar-Valderrama, J.M.; Alanis-Mendizabal, J.; et al. Galectin-3 as a potential prognostic biomarker of severe COVID-19 in SARS-CoV-2 infected patients. Sci. Rep. 2022, 12, 1856. [Google Scholar] [CrossRef] [PubMed]
- Karsli, E.; Anabarli Metin, D.; Canacik, O.; Sabirli, R.; Kaymaz, B.; Kurt, O.; Koseler, A. Galectin-3 as a Potential Prognostic Biomarker for COVID-19 Disease: A Case-Control Study. Cureus 2022, 14, e28805. [Google Scholar] [CrossRef] [PubMed]
- Gajovic, N.; Markovic, S.S.; Jurisevic, M.; Jovanovic, M.; Arsenijevic, N.; Mijailovic, Z.; Jovanovic, M.; Jovanovic, I. Galectin-3 as an important prognostic marker for COVID-19 severity. Sci. Rep. 2023, 13, 1460. [Google Scholar] [CrossRef] [PubMed]
- Shajahan, A.; Supekar, N.T.; Gleinich, A.S.; Azadi, P. Deducing the N- and O-glycosylation profile of the spike protein of novel coronavirus SARS-CoV-2. Glycobiology 2020, 30, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Tortorici, M.A.; Veesler, D. Structural insights into coronavirus entry. Adv. Virus Res. 2019, 105, 93–116. [Google Scholar] [PubMed]
- Finkelstein, M.T.; Mermelstein, A.G.; Parker Miller, E.; Seth, P.C.; Stancofski, E.S.D.; Fera, D. Structural Analysis of Neutralizing Epitopes of the SARS-CoV-2 Spike to Guide Therapy and Vaccine Design Strategies. Viruses 2021, 13, 134. [Google Scholar] [CrossRef] [PubMed]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Casalino, L.; Gaieb, Z.; Goldsmith, J.A.; Hjorth, C.K.; Dommer, A.C.; Harbison, A.M.; Fogarty, C.A.; Barros, E.P.; Taylor, B.C.; McLellan, J.S.; et al. Beyond Shielding: The Roles of Glycans in the SARS-CoV-2 Spike Protein. ACS Cent. Sci. 2020, 6, 1722–1734. [Google Scholar] [CrossRef] [PubMed]
- Caniglia, J.L.; Guda, M.R.; Asuthkar, S.; Tsung, A.J.; Velpula, K.K. A potential role for Galectin-3 inhibitors in the treatment of COVID-19. PeerJ 2020, 8, e9392. [Google Scholar] [CrossRef]
- Montazersaheb, S.; Hosseiniyan Khatibi, S.M.; Hejazi, M.S.; Tarhriz, V.; Farjami, A.; Ghasemian Sorbeni, F.; Farahzadi, R.; Ghasemnejad, T. COVID-19 infection: An overview on cytokine storm and related interventions. Virol. J. 2022, 19, 92. [Google Scholar] [CrossRef] [PubMed]
- Odun-Ayo, F.; Reddy, L. Potential Roles of Modified Pectin Targeting Galectin-3 against Severe Acute Respiratory Syndrome Coronavirus-2. J 2021, 4, 824–837. [Google Scholar] [CrossRef]
- Zhou, W.; Chen, X.; Hu, Q.; Chen, X.; Chen, Y.; Huang, L. Galectin-3 activates TLR4/NF- B signaling to promote lung adenocarcinoma cell proliferation through activating lncRNA-NEAT1 expression. BMC Cancer 2018, 18, 580. [Google Scholar] [CrossRef] [PubMed]
- Darif, D.; Hammi, I.; Kihel, A.; El Idrissi Saik, I.; Guessous, F.; Akarid, K. The pro-inflammatory cytokines in COVID-19 pathogenesis: What goes wrong? Microb. Pathog. 2021, 153, 104799. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Revilla, J.; Deierborg, T.; Venero, J.L.; Boza-Serrano, A. Hyperinflammation and Fibrosis in Severe COVID-19 Patients: Galectin-3, a Target Molecule to Consider. Front. Immunol. 2020, 11, 2069. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Yang, Z.S.; Wang, W.H.; Urbina, A.N.; Lin, Y.T.; Huang, J.C.; Liu, F.T.; Wang, S.F. The Antiviral Role of Galectins toward Influenza A Virus Infection-An Alternative Strategy for Influenza Therapy. Pharmaceuticals 2021, 14, 490. [Google Scholar] [CrossRef] [PubMed]
- Arsenijevic, A.; Stojanovic, B.; Milovanovic, J.; Arsenijevic, D.; Arsenijevic, N.; Milovanovic, M. Galectin-3 in Inflammasome Activation and Primary Biliary Cholangitis Development. Int. J. Mol. Sci. 2020, 21, 5097. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Su, G.; Sun, J.; Zhang, Y. Activation of the TLR4/MyD88 signaling pathway contributes to the development of human hepatocellular carcinoma via upregulation of IL-23 and IL-17A. Oncol. Lett. 2018, 15, 9647–9654. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A. The role of MyD88-like adapters in Toll-like receptor signal transduction. Biochem. Soc. Trans. 2003, 31, 643–647. [Google Scholar] [CrossRef] [PubMed]
- Park, A.; Iwasaki, A. Type I and Type III Interferons—Induction, Signaling, Evasion, and Application to Combat COVID-19. Cell Host Microbe 2020, 27, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Acharya, D.; Liu, G.; Gack, M.U. Dysregulation of type I interferon responses in COVID-19. Nat. Rev. Immunol. 2020, 20, 397–398. [Google Scholar] [CrossRef] [PubMed]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Péré, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020, 369, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Bartfai, T.; Behrens, M.M.; Gaidarova, S.; Pemberton, J.; Shivanyuk, A.; Rebek, J., Jr. A low molecular weight mimic of the TIR-domain inhibits interleukin 1 receptor-mediated responses. Proc. Natl. Acad. Sci. USA 2003, 100, 7971–7976. [Google Scholar] [CrossRef] [PubMed]
- Kissner, T.L.; Moisan, L.; Mann, E.; Ulrich, R.G.; Ping, S.; Waugh, D.S.; Rebek, M.; Rebek, J.; Saikh, K.U. A small molecule that mimics the BB-loop in Toll/IL-1 receptor domain of MyD88 attenuate Staphylococcal enterotoxin induced cytokine production and toxic shock in mice. J. Biol. Chem. 2011, 286, 31385–31396. [Google Scholar] [CrossRef] [PubMed]
- Kissner, T.L.; Ruthel, G.; Alam, S.; Mann, E.; Ajami, D.E.; Rebek, M.; Larkin, E.S.; Fernandez, S.; Ulrich, R.G.; Ping, S.; et al. Therapeutic inhibition of pro-infammatory signaling and toxicity to staphylococcal enterotoxin B by a synthetic dimeric BB-loop mimetic of MyD88. PLoS ONE 2012, 7, e40773. [Google Scholar] [CrossRef]
- Alam, S.; Javor, S.; Degardin, M.; Ajami, D.; Rebek, M.; Kissner, T.L.; Waag, D.M.; Rebek, J., Jr.; Saikh, K.U. Structure-based design and synthesis of a small molecule that exhibits anti-infammatory activity by inhibition of MyD88- mediated signaling to bacterial toxin exposure. Chem. Biol. Drug Des. 2015, 86, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Saikh, K.U.; Rebek, J.M.; Glass, P.J. Small Molecule Inhibitor of MyD88 for Therapeutic Treatment against Alphavirus and Staphylococcal Enterotoxin Infections and Toxin Exposure. US Patent 9,833,437 B2, 5 December 2017. [Google Scholar]
- Zetterberg, F.R.; MacKinnon, A.; Brimert, T.; Gravelle, L.; Johnsson, R.E.; Kahl-Knutson, B.; Leffler, H.; Nilsson, U.J.; Pedersen, A.; Peterson, K.; et al. Discovery and Optimization of the First Highly Effective and Orally Available Galectin-3 Inhibitors for Treatment of Fibrotic Disease. J. Med. Chem. 2022, 65, 12626–12638. [Google Scholar] [CrossRef] [PubMed]
- Gaughan, E.; Sethi, T.; Quinn, T.; Hirani, N.; Mills, A. GB0139, an inhaled small molecule inhibitor of galectin-3, in COVID-19 pneumonitis: A randomised, controlled, open-label, phase 2a experimental medicine trial of safety, pharmacokinetics, and potential therapeutic value. medRxiv 2021. [Google Scholar] [CrossRef]
- Sigamani, A.; Mayo, K.H.; Chen-Walden, H.; Reddy, S.; Platt, D. Galectin approach to lower covid transmission—Drug development for clinical use. medRxiv 2022. [Google Scholar] [CrossRef]
- Guha, P.; Kaptan, E.; Bandyopadhyaya, G.; Kaczanowska, S.; Davila, E.; Thompson, K.; Martin, S.S.; Kalvakolanu, D.V.; Vasta, G.R.; Ahmed, H. Cod glycopeptide with picomolar affinity to galectin-3 suppresses T-cell apoptosis and prostate cancer metastasis. Proc. Natl. Acad. Sci. USA 2013, 110, 5052–5057. [Google Scholar] [CrossRef] [PubMed]
- Padova, F.D.; Quesniaux, V.F.J.; Ryfel, B. MyD88 as a therapeutic target for inflammatory diseases. Expert. Opin. Ther. Targets 2018, 25, 401–408. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saikh, K.U.; Anam, K.; Sultana, H.; Ahmed, R.; Kumar, S.; Srinivasan, S.; Ahmed, H. Targeting Myeloid Differentiation Primary Response Protein 88 (MyD88) and Galectin-3 to Develop Broad-Spectrum Host-Mediated Therapeutics against SARS-CoV-2. Int. J. Mol. Sci. 2024, 25, 8421. https://doi.org/10.3390/ijms25158421
Saikh KU, Anam K, Sultana H, Ahmed R, Kumar S, Srinivasan S, Ahmed H. Targeting Myeloid Differentiation Primary Response Protein 88 (MyD88) and Galectin-3 to Develop Broad-Spectrum Host-Mediated Therapeutics against SARS-CoV-2. International Journal of Molecular Sciences. 2024; 25(15):8421. https://doi.org/10.3390/ijms25158421
Chicago/Turabian StyleSaikh, Kamal U., Khairul Anam, Halima Sultana, Rakin Ahmed, Simran Kumar, Sanjay Srinivasan, and Hafiz Ahmed. 2024. "Targeting Myeloid Differentiation Primary Response Protein 88 (MyD88) and Galectin-3 to Develop Broad-Spectrum Host-Mediated Therapeutics against SARS-CoV-2" International Journal of Molecular Sciences 25, no. 15: 8421. https://doi.org/10.3390/ijms25158421
APA StyleSaikh, K. U., Anam, K., Sultana, H., Ahmed, R., Kumar, S., Srinivasan, S., & Ahmed, H. (2024). Targeting Myeloid Differentiation Primary Response Protein 88 (MyD88) and Galectin-3 to Develop Broad-Spectrum Host-Mediated Therapeutics against SARS-CoV-2. International Journal of Molecular Sciences, 25(15), 8421. https://doi.org/10.3390/ijms25158421