Abstract
Inflammatory bowel disease (IBD) represents heterogeneous and relapsing intestinal conditions with a severe impact on the quality of life of individuals and a continuously increasing prevalence. In recent years, the development of sequencing technology has provided new means of exploring the complex pathogenesis of IBD. An ideal solution is represented by the approach of precision medicine that investigates multiple cellular and molecular interactions, which are tools that perform a holistic, systematic, and impartial analysis of the genomic, transcriptomic, proteomic, metabolomic, and microbiomics sets. Hence, it has led to the orientation of current research towards the identification of new biomarkers that could be successfully used in the management of IBD patients. Multi-omics explores the dimension of variation in the characteristics of these diseases, offering the advantage of understanding the cellular and molecular mechanisms that affect intestinal homeostasis for a much better prediction of disease development and choice of treatment. This review focuses on the progress made in the field of prognostic and predictive biomarkers, highlighting the limitations, challenges, and also the opportunities associated with the application of genomics and epigenomics technologies in clinical practice.
1. Introduction
Crohn’s disease (CD) and ulcerative colitis (UC) are heterogenous and relapsing intestinal inflammatory conditions that exert a severe impact on the quality of life of individuals and whose prevalence registers a continuous rise in many regions of the globe [1]. Dysfunction of the immune system and the intestinal microbiome combined with the intervention of various environmental factors, in a patient with genetic susceptibility, represent the main elements with a recognized contribution to the occurrence of IBD. Despite the numerous studies carried out in the last decades, the pathogenesis of these diseases is incompletely elucidated [2,3,4].
The clinical evolution varies between patients due to differences related to the localization and extent of intestinal inflammatory lesions, to which is added either the presence of extra-digestive manifestations or the development of severe complications [5]. The delay in establishing the diagnosis postpones the possibility of early initiation of the treatment, which substantially influences the progression of the disease and affects the health status of the patients [6]. Moreover, it has been observed that a favorable response obtained after the initiation of the administration of the therapy does not guarantee their long-term effectiveness, and the absence of a correlation between the clinical phenotype and serum or fecal biomarkers currently used to evaluate mucosal healing limits the possibility of adapting the therapeutic scheme [7].
Recently, it has been reported that various treatment options are only effective in approximately 40–60% of individuals because of molecular mechanisms involved in the modulation of intestinal inflammation [7,8]. This resistance associated with different drugs requires novel strategies by personalizing clinical management according to the new targets recommended by STRIDE II, which were defined as standardized treatment objectives to provide benefits for the patient [9,10,11]. Precision medicine, a strategy defined in 2011, recommends considering, mainly, the complexity of the enteropathogenic mechanisms—including clinical, genetic, and environmental characteristics—to improve the selection of therapeutic decisions with individual specificity [12].
Hence, biomarkers serve as straightforward and accurate, minimally intrusive instruments that could promptly steer clinical judgments and offer predictive insights into disease progression, treatment response, and postoperative recurrence [13]. A priority endorsed by the CORE-IBD initiative is the utilization of biomarkers to assess disease progression, the risk of complications, and the likelihood of adverse events linked to drug administration [14].
On the other hand, considering the heterogeneity of the phenotypes accounting for significant variations in the behavior of these diseases, it is unlikely that the determination of a single biomarker generates results that could be reliable for all patients. Additionally, it is crucial to acknowledge that focusing solely on a molecular marker followed by the search for a significant association with a clinical parameter could be an inappropriate orientation because it does not consider the complex perspective of the pathogenesis of IBD. An ideal solution is represented by the approach of multi-omics that investigates multiple cellular and molecular interactions, which are tools that perform a holistic, systematic, and impartial analysis of the genomic, transcriptomic, proteomic, metabolomic, and microbiomic sets. Moreover, the concept integrates clinical data to allow the construction of a map with individual specificity, which is useful for understanding the pathogenesis of IBD [1,15,16].
The inclusion of these techniques in the screening of the population is considered to be extremely promising for the detection of metabolic alterations in the early stage of the disease, being the support of future personalized medicine [17,18] (Figure 1).
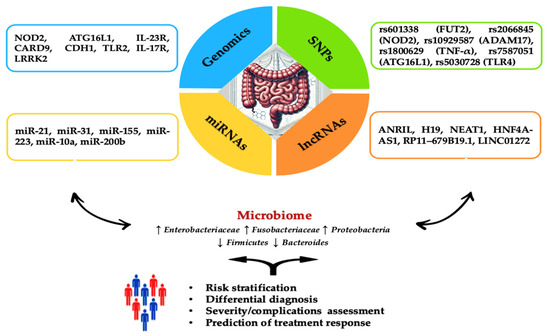
Figure 1.
Overview of genetic and epigenetic factors for IBD assessment. The diagram summarizes the specific roles of key genes, SNPs, miRNAs, and lncRNAs involved in the incidence and progression of IBD. The blue section highlights genes such as NOD2, associated with an increased risk of Crohn’s Disease (CD) development. Moreover, it has a crucial role in recognizing bacterial components and modulating the immune response. ATG16L1 increases IL-1β production, intensifying inflammatory lesions in CD. IL-23R variants improve protection against IBD by regulating the Th17 cell-mediated immune response. CARD9 is involved in inhibiting intestinal inflammation through its role in signaling pathways associated with Toll-like receptors and C-type lectin receptors. CDH1 encodes E-cadherin, essential for maintaining the integrity of the intestinal epithelial barrier, and mutations in this gene compromise barrier function, contributing to IBD pathogenesis. TLR2 is involved in recognizing pathogens and initiating immune responses, playing a role in the expression of specific receptors and maintaining immune homeostasis in the intestine. IL-17R, the receptor for interleukin-17, is associated with various phenotypes ranging from risk to protection against IBD. LRRK2 modulates autophagy, a critical process for maintaining cellular homeostasis and immune response. The green section details SNPs like rs601338 (FUT2), which is linked to an increased risk of IBD in specific populations, such as Chinese. rs2066845 (NOD2) is connected to microbiome composition in relatives of CD patients while rs10929587 (ADAM17) increases the risk of surgery in CD patients. rs1800629 (TNF-α) is associated with higher production of TNF-α. rs7587051 (ATG16L1) is linked to decreased IFX levels in the GG genotype. rs5030728 (TLR4) is associated with decreased IFX levels in GG versus GA + AA. The yellow section emphasizes the importance of miRNAs, including miR-21, which stimulates the secretion of TNF-α and macrophage inflammatory protein 2 (MIP-2), intensifying inflammatory lesions. miR-31 is overexpressed in colonic epithelial cells specific to ulcerative colitis (UC), while miR-155 induces the differentiation of M1 macrophages. miR-223 inhibits NLRP3, reducing IL-1β release and promoting healing. miR-10a downregulates NOD2 and IL-12/IL-23p40 expression in inflamed mucosa, suppressing Th1 and Th17 cell responses. miR-200b inhibits TNF-α and IL-8, preventing increased paracellular permeability by redistributing tight junction proteins claudin-1 and zonulin-1. The orange section discusses the role of lncRNAs, such as ANRIL, which has low expression in intestinal mucosa correlated with CD susceptibility and differentiates between active and remission stages of the disease. High levels of ANRIL are associated with a favorable response to infliximab treatment. H19’s overexpression affects epithelial barrier integrity by reducing the release of miR-675, which encodes zonula occludens protein (ZO-1) and E-cadherin. Stimulated by IL-22, H19 inhibits negative regulators of intestinal epithelial cell proliferation and regeneration. NEAT1 inhibition reduces intestinal permeability and improves epithelial barrier integrity, mediating the innate immune response and the secretion of inflammatory cytokines. HNF4A-AS1 is related to the presence of mucosal ulcerations, with severe prognoses in pediatric patients. RP11–679B19.1 is a long non-coding RNA associated with the WWOX gene in patients with recurrent stenotic CD. It has been strongly linked to the risk allele A, indicating a significant association with fibrostenotic complications in CD. LINC01272 is overexpressed in patients with UC and CD, and its level is significantly correlated with severe mucosal ulcerations. Abbreviations: NOD2: nucleotide-binding oligomerization domain 2; ATG16L1: autophagy-related 16-like 1; IL-1β: interleukin-1 beta; CD: Crohn’s Disease; IL-23R: interleukin-23 receptor; Th17: T helper 17; CARD9: caspase recruitment domain family member 9; CDH1: cadherin 1; TLR2: toll-like receptor 2; IL-17R: interleukin-17 receptor; LRRK2: leucine-rich repeat kinase 2; SNP: single nucleotide polymorphism; FUT2: fucosyltransferase 2; ADAM17: ADAM metallopeptidase domain 17; TNF-α: tumor necrosis factor alpha; IFX: infliximab; TLR4: toll-like receptor 4; miR: microRNA; MIP-2: macrophage inflammatory protein 2; miR-31: microRNA-31; UC: ulcerative colitis; lncRNA: long non-coding RNA; ANRIL: antisense non-coding RNA in the INK4 locus; H19: H19, imprinted maternally expressed transcript; ZO-1: zonula occludens protein 1; IL-22: interleukin-22; NEAT1: nuclear paraspeckle assembly transcript 1; HNF4A: hepatocyte nuclear factor 4 alpha; HNF4A-AS1: HNF4A antisense RNA 1; RP11–679B19.1: long non-coding RNA RP11–679B19.1; LINC01272: long intergenic non-protein coding RNA 1272.
In this review, we have assessed the progress made in recent years in the exploration of the genomic and epigenomic profile in the field of IBD, which led to the definition of promising biomarkers for the early diagnosis of complex interactions between molecular structures that have become therapeutic targets and objectives in clinical trials.
2. Genomics
2.1. The Involvement of Host Genetics in Susceptibility to IBD
The development of novel techniques led to the discovery of a significant number of genetic risk loci, characterized by a substantial heterogeneity of allele frequency and effect size in IBD, which outlined Mendelian traits within families, races, or populations [19,20]. Despite this progress, the literature provides a low rate of family history that varies between 1.5 and 28% in CD and 1.5 and 24% in UC [21]. It is estimated that hereditary factors have a stronger contribution to CD, with more than 120 genes identified, while only 22 genes have been defined for UC pathogenesis [22].
In first-degree relatives of these patients, the risk was estimated to be approximately twice as high for the development of CD (Relative Risk, RR: 7.77; 95% Confidence Interval, 95% CI: 7.05–8.56) compared to UC (RR: 4.08; 95% CI: 3.81–4.38) [23]. Moreover, it was found that the concordance rate for monozygotic and dizygotic twins reached up to 50% and 10% in CD, respectively, while in UC a lower heritability is reported (up to 15% and 4%) [24]. On the other hand, Spencer et al. reported numerous mutations in autophagy-related genes detected in the Ashkenazi Jewish population that were associated with an approximately four-fold higher prevalence of IBD and a frequent familial aggregation. However, by grouping these individuals according to the chronology of birth and age, the researchers emphasized the contribution of the common living environment that overlaps the genetic etiology for the onset of these diseases [25].
Considering these new perspectives, a genetic impact assessment has become an extremely interesting approach, both to predict IBD in the general population and perform a risk stratification, with better performance in CD (AUC = 0.75) than in UC (AUC = 0.70) [26]. Through sequencing studies, numerous independent coding variants have been identified in genes involved in modulating apoptosis and autophagy (NOD2, ATG16L1, IRGM, LRRK2) and inhibiting intestinal inflammation or infection (CARD9) [24,27]. The impact of genetic factors in modulating the expression of various receptors involved in the maintenance of intestinal barrier homeostasis is found in a broad spectrum of phenotypes ranging from risk to protection in UC and CD (Table 1).

Table 1.
Pattern of genes involved in altered epithelial barrier function in IBD.
Racial differences need to be considered when investigating genetic factors (Table 2).
Table 2.
Genes involved in IBD in different populations.
NOD2 (CARD15) is the gene associated with CD susceptibility [50]. The most common variants are R702W, G908R, and L1007fs. Patients with homozygous or compound heterozygous mutations have a 15–40-fold increased risk of CD development [54]. However, it has been observed that these alleles may occur in 0.5–2% of healthy individuals [24]. It was also found that the identification of a specific mutation (rs2066845) of this gene in first-degree relatives of patients with CD could explain the abundance of Erysipelotrichaceae in the feces. The results stress the hypothesis of the intervention of the genetic factor on the intestinal microbiome that occurs before the onset of the disease [54].
Further studies have highlighted the substantial contribution of ATG16L1, IL-23R, and fucosyltransferase 2 (FUT2) genes regarding autophagy, microbiota imbalance, and alteration of the epithelial barrier function in IBD that have been explored in different populations [44,55]. ATG16L1 deletion increases the production of IL-1β and causes the intensification of intestinal inflammatory lesions [53].
The major histocompatibility complex (HLA) genes, located on chromosome 6p21.3, encode specific proteins that initiate the T cell-mediated response and have a key role in most chronic inflammatory diseases [56]. Among these, the HLA-DRB1 and HLA-DQB1 alleles have the most consistent association with UC and CD, being involved in their severe evolution [57,58].
Gao et al. defined the risk for the development of UC depending on the presence of genes with a role in the regulation of the epithelial barrier (hepatocyte nuclear factor 4 alpha-HNF4A, laminin subunit beta 1-LAMB1, cadherin 1-CDH1, and G protein subunit alpha 12-GNA12), T cell proliferation and activation (TNFSF14), type I interferon expression (RFTN2), and Toll-like receptor stimulation (IRF5) [59]. The chromosome 1p31 is involved in the coding of a subunit of interleukin IL-23R, which has a determining role in the pathogenesis of intestinal inflammation. Loss of function of this receptor or alteration of cellular immunity associated with different IL23R variants improves protection against IBD [60]. Moreover, patients carrying different variants of TYK2 were also found to have protection from IBD through diminished T-lymphocyte response to IL-23 stimulation and reduced phosphorylation of STAT3 [61].
2.2. Interaction between Gut Microbiota and Genomics in the IBD Pathogenesis
An increased amount of evidence supports the role of genetic susceptibility on the homeostasis of the intestinal microbiota as an essential element for the triggering of an abnormal immune response and the alteration of the epithelial barrier [62]. Among the genes with impact in IBD, a significant association stands between Caspase recruitment domain family member 9 (CARD9), Nucleotide-binding oligomerization domain containing 2 (NOD2), Autophagy related 16 like 1 (ATG16L) and Fucosyltransferase 2 (FUT2), and the low abundance of the protective Roseburia species that switches acetate into butyrate [63]. NOD2 represents the most relevant susceptibility gene for Crohn’s disease, frequently associated with the phenotype that develops strictures, ileal damage, and presents an increased risk of surgical intervention [64]. This gene encodes an intracellular receptor that recognizes muramyl dipeptide from the microbial peptidoglycan structure to synthesize an oligomer capable of activating a pro-inflammatory signaling cascade and to stimulate autophagy through ATG16L1 [65]. Up to this moment, both experimental models in mice and studies in humans have emphasized the impact of these genes on the diversity of the microbiota. For example, in the intestinal mucosa of NOD2 knock-out mice, an increased abundance of microorganisms from the genus Firmicutes, Bacteroides, and Bacillus was found [66].
The establishment of intestinal dysbiosis alters the epithelial barrier by disrupting the tight junction proteins with a much more serious evolution of colitis compared to control groups of mice [67,68]. In CD patients, the polymorphism of these genes alters the ability of Paneth cells to recognize and eliminate pathogens, favoring the accumulation of the Actinobacteria group and the Firmicutes/Bacillus class, and a reduced presence of Ruminococcaceae family, which increases the potential for the development of intestinal inflammatory lesions [69].
The identification of a specific mutation (rs2066845) of NOD2 in first-degree relatives of patients with Crohn’s disease explained the abundance of Erysipelotrichaceae in the feces. These results stress the hypothesis of the intervention of the genetic factor on the change of the intestinal microbiome that occurs long before the onset of the disease [54]. Another gene involved in the recognition of pathogens is the caspase recruitment domain-containing protein 9 (CARD9) and causes a reduction in the response to immunoglobulin G in individuals with susceptibility loci for both CD and UC. Furthermore, biallelic loss-of-function (LOF) variants of CARD9 have been observed to be associated with increased susceptibility to Candida albicans infections [65]. Although LOF variants of the interleukin (IL)-23 receptor (IL23R) have been shown to protect against CD development, they increase susceptibility to various microbial infections. As a result, the blockade of IL12/23p40 and L23p19 through the administration of ustekinumab and risankizumab, respectively, has therapeutic efficacy in IBD [70].
2.3. Prediction of the Evolution of the Disease and Treatment Response
The investigation of the genetic background has become an important element for an individualized therapeutic strategy based on optimizing the dose to which a favorable response is obtained, without triggering side effects [71]. Although many medications are effective, a frequently encountered problem in clinical practice is the loss of long-term response. In this context, a genetic risk assessment is an attractive strategy, both to predict IBD in the general population and to stratify patients according to their response to the chosen treatment [72,73].
A significant number of patients face an unpredictable evolution that complicates the decision to select medicines. Currently, a reduced number of genetic tests could anticipate the effectiveness of the response or the probability of developing toxic reactions [24,74,75]. Even though they have distinct pharmacodynamic profiles, the administration of anti-TNF monoclonal antibodies (infliximab, adalimumab, and golimumab) act identically, reducing the extent of inflammatory lesions and the probability of relapses, but approximately one-third of patients become non-responders, with genetic factors being responsible for this evolution [76].
Verstockt et al. observed that the measurement of a low signal for triggering Receptor Expressed on Myeloid cells 1 (TREM1) at the beginning of anti-TNF therapy is predictive of a favorable response associated with endoscopic remission, with better accuracy for mucosal expression (AUC = 0.77, 95% CI: 0.616–0.919; p = 0.003) than serum expression (AUC = 0.58, p = 0.31) [77]. A multigenic signature, different from the original profile of T lymphocytes, was defined by Biasci et al. to characterize an aggressive phenotype that presents an increased risk for treatment escalation in UC (Hazard Ratio, HR: 3.1; CI 95%: 1.25–7.72, p = 0.02) and CD (HR: 2.7, CI 95%: 1.32–5.34, p = 0.01). In this study, the sensitivity and specificity for predicting multiple exacerbations in the following 18 months of therapy was 72.7% and 73.2% in CD and 100% and 48% in UC, respectively [78]. In the same context, Lee et al. identified four different loci (FOXO3, XACT, IGFBP1, MHC) related to the susceptibility of treatment escalation in CD and, in addition, proved that rs7576459 associated with IGFBP1/IGFBP3 represents a clinical phenotype with a severe prognosis (Odds Ratio, OR: 3.91; 95% CI: 3.10–5.01; p < 0.001) [79]. Although pretreatment genotyping is recommended in the European Crohn’s and Colitis Organization (ECCO) guidelines, it has been proven that it could not replace the need to monitor the occurrence of possible adverse effects during the administration of certain drugs [80].
The HLA system is considered as a predictor of the occurrence of adverse effects during the administration of thiopurine, azathioprine, or mercaptopurine. Using the GWAS approach, two haplotypes were identified—HLA-DQA1*02:01 and HLA-DRB1*07:01—most often involved in the unpredictable evolution of therapy. Additionally, the risk of developing pancreatitis three months after the initiation of immunosuppressive therapy was 9% and 17% in heterozygous and homozygous patients, respectively, in direct relation to the presence of rs2647087 (OR: 2.59, CI 95%: 2.07–3.26, p < 0.001) [81]. Several studies have reported the involvement of single-nucleotide polymorphisms (SNPs) in the response to anti-TNF therapy in IBD patients (rs1800629, rs1799724, rs767455, rs1061624, and rs976881 with an inhibitory effect, while rs4149570, rs361525, and rs3397 determine a superior result), the majority being located in the TNF 1 (TNFR1) and TNF 2 (TNFR2) receptor genes and in those that regulate innate immunity (TLR4, IL6, IL1, IL17, TLR2, TLR9), autophagy, and apoptosis (Fas-L, CASP9, and ATG16L1) [82,83,84]. Genes that encode proteins involved in the regulation of the immune response have become a new target for evaluating anti-TNF treatment efficacy. Due to involvement in the pathogenesis of IBD, the genetic information for the synthesis of Toll-Like Receptor 4 (TLR4) possesses a role in the recognition of molecular patterns specific to certain pathogens, such as CD14 for ensuring homeostasis of the intestinal barrier [85].
Based on a retrospective evaluation in the PANTS study, it was observed in CD that baseline expression of major histocompatibility complex, antigen presentation, and myeloid cell enriched receptor were significantly higher in anti-TNF responders (infliximab and adalimumab) compared to patients who did not respond [46]. A model including 10 polymorphisms located in genes involved in the regulation of the NF-κB pathway (TLR2, TLR4, and NFKBIA) and signaling of TNF-α (TNFRSF1A) or other cytokines (NLRP3, IL1RN, IL18, and JAK2) was associated with the prediction of the rate of non-response to anti-TNF treatment, much higher in patients with UC (23%) compared to those with CD (13%) (OR: 1.98; 95% CI: 1.54–2.55, p = 0.0001). On the other hand, it was found that the inflammatory lesions genetically determined by the association of TNFRSF1A, IL1RN, and IL18 benefit from the favorable effect obtained by anti-TNF. Conversely, the intervention of IL-1β or IL-18 increases the probability of ineffectiveness as these cytokines could maintain inflammation even after the completion of therapy [86]. Recently, a significant number of studies have reported important correlations between different genetic polymorphisms and the response or lack of response to biological treatments, which reflected the heterogeneity of efficacy in IBD patients (Table 3).
Table 3.
Single-nucleotide polymorphism and the response to anti-TNF therapy.
Further studies concentrated on the genes involved in apoptosis and autophagy (BAX, BCL2, CASP3, and CASP9). It was observed that the low expression of these genes in peripheral blood lymphocytes influences the immune response, with the development of inflammatory lesions of the intestinal mucosa in UC. Moreover, the BAX/BCL2 ratio was significantly correlated (r = 0.473, p = 0.0095) with the duration of the active phase of the disease. Instead, in CD patients who received biological medication, a significantly lower (BAX/BCL2) ratio was noted compared to the placebo-treated group, suggesting that apoptosis modulation could become an important therapeutic mechanism in this pathology [97]. In various studies, numerous genetic polymorphisms that impact the alteration of cytokines (IL-1p, IL-1RA, IL-6, IL-11, IL-13, IL-17, and IL-27) were investigated. An example is the interleukin 17 (IL-17) family, a cytokine secreted by Th17 cells, which includes six ligands (IL-17A to IL-17F) and five receptors (IL-17RA to IL-17RE), located mainly in Paneth cells (IL-17A) and colonic epithelium (IL-17F). In CD patients, these receptors are overexpressed and transmit signals to downstream pathways through traf3-interacting protein 2 (TRAF3IP2), which share intracellular signal transduction molecules, such as I-κB and NF-κB, with the TNF-α signaling pathway. Hence, Urabe et al. managed to confirm the genotypes G/G for IL17F (rs766748) and C/C or C/A for TRAF3IP2 (rs1883136), which were strongly associated with a favorable response obtained after one year of treatment with IFX (OR = 37.92; p = 0.0019), with the sensitivity and specificity estimate of the genetic test being 70.0% and 100%, respectively [94].
In CD patients who received ADA, a pharmacogenetic relationship was highlighted between the polymorphisms rs755622 in the macrophage migration inhibitory factor gene (MIF) and rs3740691 in the ADP Ribosylation Factor GTPase Activating Protein 2 gene (ARFGAP2). However, the most consistent association with establishing a favorable response after 30 weeks of therapy was found for the variant rs2275913 in the IL17A gene (p = 9.73 × 10−3) [98]. It was observed that IL-23 cytokine, the ligand for IL23R, along with IL-12, to which it is related through the p40 subunit, became targets for the action of neutralizing antibodies in the treatment of IBD. However, it has been shown that the anti-inflammatory effect is the result of blocking the p19 subunit included in IL-23, with the generation of selective antibodies for therapy with guselkumab, risankizumab, and mirikizumab that act either by disrupting signaling pathways (JAK-STAT, NF-κB) or causing the loss of T helper-17, CD4+, and CD8+ cell function [99]. Although genetic risk assessment is considered a useful approach for the stratification of IBD patients, with increased performance in CD compared to UC, it has not yet been implemented in medical practice due to the increased costs of obtaining minor benefits [72].
3. The Role of Epigenetic Mechanisms on Prognosis and Therapeutic Response in IBD
The description of the dynamic and reversible changes that occur in the regulation of gene expression through interaction with various environmental factors, without altering the DNA sequence, represents a new direction of multi-omics research that has proven useful for understanding the pathogenesis of IBD in order to achieve precision medicine [24,84]. In recent years, evidence has appeared regarding the contribution of epigenetic mechanisms (hypo- or hypermethylation of DNA, histone modification, chromatin remodeling, or non-coding RNA sequences) for the maintenance of homeostasis of the intestinal epithelium, cell development, and differentiation or modulation of immune responses against pathogens [100]. By applying epigenome-wide association studies (EWAS), these signatures have been found to be stable over multiple cycles of cell division and replication with the possibility of being inherited [101]. Moreover, it was observed that the different epigenetic profiles could be used as biomarkers for early diagnosis and differentiation of clinical phenotypes; simultaneously, they became new therapeutic targets that could predict the response and occurrence of adverse reactions in IBD patients [100]. One of the epigenetic mechanisms that provides a new perspective on the pathogenesis of IBD is DNA methylation in gene promoters, with a key role in reducing their expression from a transcriptional point of view [102]. The methylation characteristics proved to be important for differentiating the types and activity stages of these diseases [100].
The detection of an increased number of differentially methylated positions (DMPs) in both pediatric and adult cohorts justifies its use for clinical phenotype classification. An analysis performed on rectal biopsy samples highlighted a methylation profile in the Thyroid Hormone Receptor Associated Protein (THRAP2), Forssman Glycosyltransferase 1 (GBGT1), Tumor Necrosis Factor Ligand (TNFSF), and Fucosyltransferase 7 (FUT7) that allows the differentiation of IBD patients from healthy controls [103]. Although these epigenetic features are significant, they disappear after mucosal inflammation is reduced by treatment. This finding suggests that abnormal DNA methylation might no longer exist when disease activity is reduced [104]. Furthermore, evidence supports that local inflammation could accelerate the methylation process, favoring the disruption of cellular replication and differentiation. As a result, DMPs could be used as markers for the early identification of tumors or dysplastic lesions occurring in patients with IBD [105,106].
An important role in regulating the structure or function of chromatin, with a major impact on the dynamics of various cellular processes, belongs to the post-translational modifications of histones (H2A, H2B, H3, H4, and H1) catalyzed by specific enzymes, such as histone-acetyltransferases (HAT), histone deacetylases (HDAC), histone methyltransferases (HMT), and histone lysine transferases (KAT) [107,108,109]. The progress registered in recent years regarding the exploration of the impact of chromatin changes has offered perspectives for the approach of new therapeutic interventions in these diseases (Table 4).
Table 4.
The impact of various epigenetics mechanisms regarding the pathogenesis of IBD.
3.1. The Patterns of lncRNAs in IBD
Although they do not contain genetic information for protein synthesis, long non-coding RNAs (lncRNAs)—sequences made up of more than 200 nucleotides—have an important role in various biological processes by modulating the expression of genes involved in post-transcriptional/transcriptional regulation or by guiding chromatin-modifying complexes into specific genomic loci [128]. Recent cumulative data from GWAS have proven that lncRNAs promote inflammation and carcinogenesis, being associated with numerous diseases, which represents a promising perspective for clinical applications [129].
Even though their potential has not been sufficiently explored, it was discovered that they initiate and influence the evolution of inflammatory intestinal diseases, intervening in the maintenance of the integrity of the intestinal epithelium barrier, apoptosis, and the interaction of immune cells, which allowed their use as diagnostic and prognostic biomarkers [125]. Numerous studies have revealed the link between lncRNAs and the disruption of epithelial barrier function, reduction in junctional proteins, and increased intestinal permeability, which are predictive aspects for IBD relapse. LncRNA nuclear paraspeckle assembly transcript 1 (NEAT1) is a key component that mediates the innate immune response and the secretion of inflammatory cytokines. Liu et al. found that inhibition of NEAT1 expression in TNF-α and dextran sodium sulfate (DSS) reduced intestinal permeability and improved epithelial barrier integrity [130].
In various tissues during the embryonic stage, the lncRNA transcribed by the H19 gene on chromosome 11 is silenced after birth. In pathological conditions, the increased modulation of H19 induced by IL-22 inhibits the negative regulators of the proliferation and regeneration of intestinal epithelial cells (protein p53, miR-34a, and let-7), which stimulates mucosal healing [131]. However, overexpression of H19 was observed to reduce the release of miR-675 encoding zonula occludens protein (ZO-1) and the E-cadherin protein, strongly affecting epithelial barrier integrity [132].
3.1.1. lncRNA as Prognostic and Diagnostic Biomarkers in IBD
Even though numerous studies have focused on the evaluation of the genomic impact, there is evidence that lncRNAs have an important role in the regulation of cellular physiology and are differentially expressed in IBD. The monitorization of the level of these lncRNAs could be applied to clinical evaluation and prognosis. As an example, the dysfunction or upregulation of lncRNAs (DIO3OS, KIF9-AS1, and LINC01272) is considered a biomarker that has an increased performance to differentiate IBD patients from healthy controls [125]. An important aspect refers to the possibility of using lncRNAs to investigate the late stages of the disease in which various complications are present (Table 5).
Table 5.
The role of lncRNA in IBD pathogenesis.
3.1.2. lncRNAs as Predictors of Therapeutic Response in IBD
Several studies have concluded that certain lncRNAs might be considered excellent therapeutic targets. Among these, an anti-inflammatory small molecule drug (ABX464) stands out, a new antiviral drug with modulating potential for lncRNA 0599–205, which, in addition, triggers strong anti-inflammatory reactions in the dextran sulfate sodium (DSS)-induced colitis model [135].
New molecular therapies aimed at controlling intestinal permeability stimulate the increase in the level of lncRNA SPRY4-IT1, which has a protective effect on the epithelial barrier through the overexpression of tight junction proteins (claudin-1, occludin, and JAM-1) [136]. Additionally, ANRIL became a target that reflects the effectiveness of infliximab treatment for these patients. High concentrations were detected in responders compared to non-responders, who maintained a constant level of this drug [126].
Corticosteroids represent effective drugs to induce short-term emission in IBD. However, there are patients who develop resistance to this therapy due to the cytoplasmic accumulation of lncRNA (GAS5) that intervenes at the post-transcriptional level. As a result, GAS5 could be considered a biomarker with increased potential for personalizing corticosteroid therapy [137].
3.2. Role of miRNAs in IBD
In recent years, there has been increased interest in exploring the potential of a class of small molecules of endogenous origin, consisting of non-coding single-stranded RNA, with a length of 18–25 nucleotides (miRNAs), which have begun to be used as diagnostic and prognostic biomarkers of severe evolution or unfavorable therapeutic response in a variety of autoimmune, neurodegenerative, cardiovascular, or neoplastic diseases [138,139]. Moreover, it was discovered that in UC and CD, they maintain a key role in the post-transcriptional regulation of genes involved in the intestinal modulation of innate and adaptive immunity, in response to cellular changes or the action of environmental factors. This process affects autophagy, the homeostasis of the intestinal microbiome, and the integrity of the epithelial barrier [140].
In the context of complex pathogenesis, the modification of the expression profile of miRNAs due to various mechanisms, including DNA methylation, deacetylation, and keratin modification, acts on the intercellular signaling pathways, intervening in the synthesis of pro- or anti-inflammatory cytokines and regulating the permeability of the intestinal mucosa (Table 6).
Table 6.
Types of miRNAs signatures in inflammatory bowel disease.
3.2.1. miRNAs as Diagnostic Biomarkers of IBD
Different types of miRNAs have been evaluated in numerous studies. A list of potential biomarkers for diagnostic and prognostic assessment in IBD patients is included in Table 7. After comparing the expression of miRNAs in tissues, feces, and peripheral blood, a repeated presence of miR-16, miR-21, miR-155, miR-223, or miR-31 is noted, which proved a functional relevance, both for patients with UC and CD, as well as in experimental models on mice [156]. A potential diagnostic marker for UC is represented by miR-21, with a significantly higher level compared to CD, which is mainly detected in inflamed lamina propria cells and partially damaged crypts [157]. It has also been shown that the increased serum level of this miRNA accompanied by an overexpression for miR-92a has excellent performance in differentiating UC from healthy subjects [158].
Other researchers have found that the upregulation of miR-31–3p in colonic epithelial cells is specific for UC. Furthermore, intracolonic administration of a chemical inhibitor of miR-31-3p to mice was reported to exacerbate DSS-induced colitis by stimulating the release of pro-inflammatory cytokines (IL-6, IL-8, and TNF-ά) [159]. Differences in miRNA composition and expression levels have also been suggested to be influenced by the evolution of the disease. As a result, it was found that miR-31 expression in sigmoid colon biopsies is strongly regulated in favor of the active UC form (up to 11 folds) compared to healthy controls [160]. In contrast, in CD patients, there was an upregulation in the inflamed duodenal mucosa for miR-146a and miR-155 [161].
On the other hand, it was found that the active form of this disease was associated with a much higher serum level for miR-155 compared to patients in remission [162]. Although differential diagnosis between CD and UC is considered a real challenge when inflammatory lesions are limited to the colon, a report indicated that a complex panel including miR-19a, miR-21, miR-31, miR-101, miR-146a, and miR-375, with an increased expression in saliva and blood, has a superior diagnostic performance [163].
Table 7.
Correlation between IBD diagnosis and miRNAs.
Table 7.
Correlation between IBD diagnosis and miRNAs.
| miRNAs | Disease | Sample | Main Findings | References |
|---|---|---|---|---|
| miR-21 miR-126 | UC CD | Colonic tissue | Increased levels of miR-21 (6.7-fold) in lamina propria and miR-126 (2.3-fold) in endothelial cells in UC compared to controls; miR-21 is upregulated in UC compared to CD (5.8-fold). | [157] |
| miR-31 | UC CD Controls | Colonic tissue | Hyperexpression in UC compared to CD and controls. | [159] |
| UC controls | Colonic tissue | Increased expression in active form of UC (11-fold) and inactive form (3-fold) compared to healthy controls. | [160] | |
| miR-16, miR-21, miR-223, miR-155 | CD UC controls | Blood Fecal samples | Active CD was associated with higher miR-155 expression in blood samples compared to controls (1.9-fold) and subjects (2.1-fold) in remission (AUC = 0.752; 95% CI: 0.61–0.89). | [164] |
| miR-146a, miR-155, miR-122 | CD UC controls | Colonic tissue | miR-146a and miR-155 were higher in the inflamed mucosa of children with CD and UC than in the intact mucosa. Elevated expression of miR-122 in CD compared with controls and UC. | [162] |
| CD controls | Colonic tissue | [161] | ||
| miR-125b, miR-223, miR-138, miR-155 | UC controls | Colonic tissue | Upregulation in inflamed UC tissues: miR-138 (10-fold), miR-223 (10-fold), miR-125b (2.56-fold), and miR-155 (2.33-fold). | [165] |
| miR-141, miR-200a, miR-200b, miR-200c, miR-429 | UC CD | Colonic tissue | miR-141, miR-200b, and miR-429 were downregulated in CD and UC patients compared to healthy controls. miR-141, miR-200a, miR-200b, and miR-200c were significantly downregulated in CD in comparison to UC. | [166] |
| miR-21-5p | UC controls | Blood Colonic tissue | Downregulated in UC patients compared with control and determines inhibition of the expression of IL-6, TNF-α, IL6R, STAT3, ICAM-1, NF-κB, cleaved caspase-3, cleaved caspase-9, and FasL to alleviate the inflammation and apoptosis. | [167] |
| miR-21 miR-92a | UC controls | Blood Colonic tissue | Overexpression has increased performance for differentiating UC from healthy subjects (AUC = 0.979, specificity = 1.00). | [158] |
| miR-215-5p, miR-203a-3p, miR-223-3p, miR-194-5p, miR-192-5p, miR-10b-5p, miR-10a-5p, miR-337-5p, miR-582-5p | CD controls | Colonic tissue | Downregulated in inactive CD compared to controls. Their inhibitory action on NOD2, TLR4, and IL6ST genes is involved in the innate immune response and cytokine signaling. | [168] |
| miR-29b-3p, miR-122-5p, miR-146a-3p, miR-150-5p, miR-192-5p, miR-194-5p, miR-375-3p, miR-148a-3p, miR-199a-3p | experimental model colitis mice | Blood | Hyperexpression of miR-29b-3p, -122-5p, -192-5p, -194-5p, -375-3p, -150-5p, -146a-3p, and the downregulated miR-148a-3p and -199a-3p; differentiation of UC patients from healthy controls (accuracies of 83.3%). | [169] |
| mi146b-5p | CD UC controls | Blood | The serum level (2.87- and 2.72-fold higher in patients with CD and UC than controls, respectively) is correlated with disease activity (CDEIS r = 0.579; UCIES r = 0.582) (AUC = 0.869, 95% CI: 0.764-0.940). | [170] |
CD: Crohn disease, UC: ulcerative colitis, CDEIS: Crohn’s disease endoscopic index of severity, CI: Confidence Interval, miR: micro-Ribonucleic Acid, TNF-α: Tumor Necrosis Factor Alpha, NF-κB: Nuclear Factor Kappa B, IL: Interleukin.
3.2.2. The Role of miRNAs as Therapeutic Targets
The role of miRNAs in IBD therapy has gained significance due to their involvement in pathological processes. As a result, it was concluded that monitoring the immune status according to the changes of miRNA signatures in the blood circulation and at the tissue level is an effective approach in IBD for the design of an individualized therapeutic strategy, having a predictive role in identifying the loss of the favorable response. Recently, miR-31 has shown promising results in order to reduce inflammation in experimental colitis models, indicating its potential therapeutic application [171]. Similarly, miR-223 has been found to alleviate symptoms such as occult bleeding and weight loss in DSS colitis models, suggesting its efficacy in improving clinical status [172]. By targeting dendritic cells, miR-29b possesses the ability to manage immune response [173]. Moreover, the therapeutic impact of miRNA-126 and miR-20a in pediatric CD patients treated with infliximab underscores their relevance in managing immune regulation and epithelial barrier function [174]. Inflamed tissues present elevated levels of miR-146a and miR-146b, which act as inhibitors of the NF-kB signaling pathway, presenting a mechanism to reduce inflammation. Moreover, miR-320a contributes to the healing of ulcers in the colonic mucosa, highlighting restorative properties [175]. The therapeutic implications of miR-29, particularly in reducing IL-23 expression by targeting the IL-12p40 subunit, validate its role as a biomarker for ustekinumab’s therapeutic efficacy [176] (Table 8).
Table 8.
Impact of miRNAs on therapeutic response in IBD.
4. Future Directions and Challenges
One significant implication is the advancement of personalized treatment strategies. By identifying specific genetic and epigenetic markers, clinicians could tailor therapies to individual patients, potentially improving treatment efficacy and reducing adverse effects. Future research may focus on the development of predictive models that utilize genetic and epigenetic data to identify individuals at high risk of developing IBD before clinical symptoms appear. Early detection could enable preventive measures and interventions, potentially altering the disease course and improving long-term outcomes. Moreover, understanding the genetic predisposition and early epigenetic changes provides insights into the initial triggers of IBD. The concept of multi-omics provides a comprehensive understanding of the molecular mechanisms underlying the diseases. Future perspectives include the development of integrated multi-omics platforms that create a holistic view of disease processes. These platforms could facilitate the discovery of novel biomarkers and therapeutic targets, enhancing the ability to diagnose, monitor, and treat IBD with unprecedented precision. The accuracy and potential of these biomarkers for in-depth exploration of genetic variation between populations must be verified in large cohorts of IBD patients in which datasets are collected longitudinally to ensure reproducibility and validation of the results regarding disease subtype classification and therapeutic intervention.
In recent years, the development of sequencing technology has provided new means of exploring the complex pathogenesis of IBD. Despite the progress achieved in describing the architecture of the genome and understanding the mechanisms related to the heredity of susceptible individuals, it is still claimed that they pose a minor contribution to the development or influence of the prognosis of these diseases since most of the risk alleles are extremely rare. Moreover, a real assessment of the impact of genotyping on therapeutic strategies is lacking, especially in patients who face the loss of favorable response over time. A major challenge associated with epigenetic studies is the heterogeneity of nonspecific IBD cells detected in blood or tissue samples, making data interpretation difficult due to the interference of different individual epigenetic characteristics. Despite the applications presented in this review, the usefulness of approaching genomic or epigenomic profiles in clinical practice is relatively limited due to the low accessibility for assessing complex analysis systems that, in addition, involve high costs in the context of obtaining small benefits. Moreover, there are a series of limitations of GWAS that refer, above all, to the linkage disequilibrium of the human genome in which different alleles could associate randomly and, subsequently, might be transmitted simultaneously.
However, when this imbalance is minimal, it is possible that the polymorphisms are not transmitted with the encoded gene and, as a result, they could not be detected by GWAS. On the other hand, it was found that the same gene variation might generate different clinical phenotypes through epigenetic changes or transcriptional and translational regulation.
5. Conclusions
In the context of an incompletely elucidated pathogenesis, it was discovered that immunological, genetic, and environmental factors have an important contribution to the evolution of these diseases. Genomics and epigenetics explore the dimension of variation in the characteristics of these diseases, offering the advantage of understanding the cellular and molecular mechanisms that affect intestinal homeostasis for a much better prediction of the prognosis. However, the important perspective suggested by the studies included in this review is represented by the integration of genetic or epigenetic profiles in the field of precision medicine through the pretreatment detection of risk genes, which could improve the decision to apply a personalized strategy. The findings require an intensification of research aimed at epigenetic changes that are under selection pressure associated with the microbiome or intestinal inflammation, influencing the key pathways associated with the pathology of IBD.
Author Contributions
Conceptualization, H.M., A.T., M.M. and A.-M.S.; methodology, H.M., A.-M.S., C.M. and M.M.; software, C.V.S. and S.C.; validation, S.J., M.M., I.G. and C.M.; formal analysis C.S., C.M. and A.T.; investigation, A.-M.S., I.D.M., I.G. and C.V.S.; data curation, M.M. and S.J.; writing—original draft preparation, H.M., M.M., A.T. and A.-M.S.; writing—review and editing, C.S., H.M. and A.T.; visualization, C.M. and I.D.M.; supervision, A.T. and C.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Baldan-Martin, M.; Chaparro, M.; Gisbert, J.P. Tissue Proteomic Approaches to Understand the Pathogenesis of Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2021, 27, 1184–1200. [Google Scholar] [CrossRef]
- Nowak, J.K.; Kallab, R.; Satsang, J. Current and emerging biomarkers for ulcerative colitis. Expert Rev. Mol. Diagn. 2023, 23, 1107–1119. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, B.; Jin, T.; Ocansey, D.K.W.; Jiang, J.; Mao, F. Intestinal Fibrosis in Inflammatory Bowel Disease and the Prospects of Mesenchymal Stem Cell Therapy. Front. Immunol. 2022, 13, 835005. [Google Scholar]
- Li, X.; Zhang, M.; Zhou, G.; Xie, Z.; Wang, Y.; Han, J.; Li, L.; Wu, Q.; Zhang, S. Role of Rho GTPases in inflammatory bowel disease. Cell Death Discov. 2023, 9, 24. [Google Scholar] [CrossRef]
- Ma, C.; Sandborn, W.J.; D’Haens, G.R.; Zou, G.; Stitt, L.W.; Singh, S.; Ananthakrishnan, A.N.; Dulai, P.S.; Khanna, R.; Jairath, V.; et al. Discordance between Patient-Reported Outcomes and Mucosal Inflammation in Patients with Mild to Moderate Ulcerative Colitis. Clin. Gastroenterol. Hepatol. 2020, 18, 1760–1768. [Google Scholar]
- Sabino, J.; Verstockt, B.; Vermeire, S.; Ferrante, M. New biologics and small molecules in inflammatory bowel disease: An update. Therap. Adv. Gastroenterol. 2019, 12, 1756284819853208. [Google Scholar]
- Giachero, F.; Jenke, A.; Zilbauer, M. Improving prediction of disease outcome for inflammatory bowel disease: Progress through systems medicine. Expert Rev. Clin. Immunol. 2021, 17, 871–881. [Google Scholar]
- Elhag, D.A.; Kumar, M.; Saadaoui, M.; Akobeng, A.K.; Al-Mudahka, F.; Elawad, M.; Al Khodor, S. Inflammatory Bowel Disease Treatments and Predictive Biomarkers of Therapeutic Response. Int. J. Mol. Sci. 2022, 23, 6966. [Google Scholar] [CrossRef]
- Maaser, C.; Sturm, A.; Vavricka, S.R.; Kucharzik, T.; Fiorino, G.; Annese, V.; Calabrese, E.; Baumgart, D.C.; Bettenworth, D.; Borralho Nunes, P.; et al. European Crohn’s and Colitis Organisation [ECCO] and the European Society of Gastrointestinal and Abdominal Radiology [ESGAR]. ECCO-ESGAR Guideline for Diagnostic Assessment in IBD Part 1: Initial diagnosis, monitoring of known IBD, detection of complications. J. Crohn’s Colitis 2019, 13, 144–164. [Google Scholar]
- Torres, J.; Petralia, F.; Sato, T.; Wang, P.; Telesco, S.E.; Choung, R.S.; Strauss, R.; Li, X.J.; Laird, R.M.; Gutierrez, R.L.; et al. Serum Biomarkers Identify Patients Who Will Develop Inflammatory Bowel Diseases Up to 5 Years before Diagnosis. Gastroenterology 2020, 159, 96–104. [Google Scholar] [CrossRef]
- Turner, D.; Ricciuto, A.; Lewis, A.; D’Amico, F.; Dhaliwal, J.; Griffiths, A.M.; Bettenworth, D.; Sandborn, W.J.; Sands, B.E.; Reinisch, W.; et al. International Organization for the Study of IBD. STRIDE-II—An Update on the Selecting Therapeutic Targets in Inflammatory Bowel Disease (STRIDE) Initiative of the International Organization for the Study of IBD (IOIBD): Determining Therapeutic Goals for Treat-to-Target strategies in IBD. Gastroenterology 2021, 160, 1570–1583. [Google Scholar]
- Liu, X.Y.; Tang, H.; Zhou, Q.Y.; Zeng, Y.L.; Chen, D.; Xu, H.; Li, Y.; Tan, B.; Qian, J.M. Advancing the precision management of inflammatory bowel disease in the era of omics approaches and new technology. World J. Gastroenterol. 2023, 29, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Olivera, P.A.; Silverberg, M.S. Biomarkers That Predict Crohn’s Disease Outcomes. J. Can. Assoc. Gastroenterol. 2023, 7, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Hanzel, J.; Panaccione, R.; Sandborn, W.J.; D’Haens, G.R.; Ahuja, V.; Atreya, R.; Bernstein, C.N.; Bossuyt, P.; Bressler, B.; et al. CORE-IBD: A Multidisciplinary International Consensus Initiative to Develop a Core Outcome Set for Randomized Controlled Trials in Inflammatory Bowel Disease. Gastroenterology 2022, 163, 950–964. [Google Scholar] [CrossRef] [PubMed]
- Torres, J.; Halfvarson, J.; Rodríguez-Lago, I.; Hedin, C.R.H.; Jess, T.; Dubinsky, M.; Croitoru, K.; Colombel, J.F. Results of the Seventh Scientific Workshop of ECCO: Precision Medicine in IBD-Prediction and Prevention of Inflammatory Bowel Disease. J. Crohn’s Colitis 2021, 15, 1443–1454. [Google Scholar] [CrossRef]
- Aldars-García, L.; Chaparro, M.; Gisbert, J.P. Systematic Review: The Gut Microbiome and Its Potential Clinical Application in Inflammatory Bowel Disease. Microorganisms 2021, 9, 977. [Google Scholar] [CrossRef] [PubMed]
- Aldars-García, L.; Marin, A.C.; Chaparro, M.; Gisbert, J.P. The Interplay between Immune System and Microbiota in Inflammatory Bowel Disease: A Narrative Review. Int. J. Mol. Sci. 2021, 22, 3076. [Google Scholar] [CrossRef] [PubMed]
- Ortega Moreno, L.; Sanz-Garcia, A.; Fernández de la Fuente, M.J.; Arroyo Solera, R.; Fernández-Tomé, S.; Marin, A.C.; Mora-Gutierrez, I.; Fernández, P.; Baldan-Martin, M.; Chaparro, M.; et al. Serum adipokines as non-invasive biomarkers in Crohn’s disease. Sci. Rep. 2020, 10, 18027. [Google Scholar] [CrossRef]
- Somineni, H.K.; Nagpal, S.; Venkateswaran, S.; Cutler, D.J.; Okou, D.T.; Haritunians, T.; Simpson, C.L.; Begum, F.; Datta, L.W.; Quiros, A.J.; et al. Whole-genome sequencing of African Americans implicates differential genetic architecture in inflammatory bowel disease. Am. J. Hum. Genet. 2021, 108, 431–445. [Google Scholar] [CrossRef] [PubMed]
- Chudy-Onwugaje, K.O.; Christian, K.E.; Farraye, F.A.; Cross, R.K. A State-of-the-Art Review of New and Emerging Therapies for the Treatment of IBD. Inflamm. Bowel Dis. 2019, 25, 820–830. [Google Scholar] [CrossRef]
- Santos, M.P.C.; Gomes, C.; Torres, J. Familial and ethnic risk in inflammatory bowel disease. Ann. Gastroenterol. 2018, 31, 14–23. [Google Scholar] [CrossRef]
- Garza-Hernandez, D.; Sepulveda-Villegas, M.; Garcia-Pelaez, J.; Aguirre-Gamboa, R.; Lakatos, P.L.; Estrada, K.; Martinez-Vazquez, M.; Trevino, V. A systematic review and functional bioinformatics analysis of genes associated with Crohn’s disease identify more than 120 related genes. BMC Genom. 2022, 23, 302. [Google Scholar] [CrossRef]
- Moller, F.T.; Andersen, V.; Wohlfahrt, J.; Jess, T. Familial risk of inflammatory bowel disease: A population-based cohort study 1977-2011. Am. J. Gastroenterol. 2015, 110, 564–571. [Google Scholar] [CrossRef]
- El Hadad, J.; Schreiner, P.; Vavricka, S.R.; Greuter, T. The Genetics of Inflammatory Bowel Disease. Mol. Diagn. Ther. 2024, 28, 27–35. [Google Scholar] [CrossRef]
- Spencer, E.A.; Helmus, D.; Telesco, S.; Colombel, J.F.; Dubinsky, M.C. Road to Prevention Study Group. Inflammatory Bowel Disease Clusters within Affected Sibships in Ashkenazi Jewish Multiplex Families. Gastroenterology 2020, 159, 381–382. [Google Scholar] [CrossRef]
- Chen, G.B.; Lee, S.H.; Montgomery, G.W.; Wray, N.R.; Visscher, P.M.; Gearry, R.B.; Lawrance, I.C.; Andrews, J.M.; Bampton, P.; Mahy, G.; et al. Performance of risk prediction for inflammatory bowel disease based on genotyping platform and genomic risk score method. BMC Med. Genet. 2017, 18, 94. [Google Scholar] [CrossRef]
- Arnadottir, G.A.; Norddahl, G.L.; Gudmundsdottir, S.; Agustsdottir, A.B.; Sigurdsson, S.; Jensson, B.O.; Bjarnadottir, K.; Theodors, F.; Benonisdottir, S.; Ivarsdottir, E.V.; et al. A homozygous loss-of-function mutation leading to CYBC1 deficiency causes chronic granulomatous disease. Nat. Commun. 2018, 9, 4447. [Google Scholar] [CrossRef]
- de Ponthaud, C.; Abdalla, S.; Belot, M.P.; Shao, X.; Penna, C.; Brouquet, A.; Bougnères, P. Increased CpG methylation at the CDH1 locus in inflamed ileal mucosa of patients with Crohn disease. Clin. Epigenet. 2024, 16, 28. [Google Scholar] [CrossRef]
- Muller, M.; Hansmannel, F.; Arnone, D.; Choukour, M.; Ndiaye, N.C.; Kokten, T.; Houlgatte, R.; Peyrin-Biroulet, L. Genomic and molecular alterations in human inflammatory bowel disease-associated colorectal cancer. United Eur. Gastroenterol. J. 2020, 8, 675–684. [Google Scholar] [CrossRef]
- Yu, H.; Liu, Z. GNA12 regulates C5a-induced migration by downregulating C5aR1-PLCβ2-PI3K-AKT-ERK1/2 signaling. Biophys. Rep. 2023, 9, 33–44. [Google Scholar] [CrossRef]
- M’Koma, A.E. The Multifactorial Etiopathogeneses Interplay of Inflammatory Bowel Disease: An Overview. Gastrointest. Disord. 2019, 1, 75–105. [Google Scholar] [CrossRef]
- Canale, V.; Spalinger, M.R.; Alvarez, R.; Sayoc-Becerra, A.; Sanati, G.; Manz, S.; Chatterjee, P.; Santos, A.N.; Lei, H.; Jahng, S.; et al. PTPN2 Is a Critical Regulator of Ileal Paneth Cell Viability and Function in Mice. Cell Mol. Gastroenterol. Hepatol. 2023, 16, 39–62. [Google Scholar] [CrossRef] [PubMed]
- Niechcial, A.; Butter, M.; Manz, S.; Obialo, N.; Bäbler, K.; van der Lely, L.; Lang, S.; Gottier, C.; McCole, D.F.; Scharl, M.; et al. Presence of PTPN2 SNP rs1893217 Enhances the Anti-inflammatory Effect of Spermidine. Inflamm. Bowel Dis. 2020, 26, 1038–1049. [Google Scholar] [CrossRef] [PubMed]
- Rivas, M.A.; Graham, D.; Sulem, P.; Stevens, C.; Desch, A.N.; Goyette, P.; Gudbjartsson, D.; Jonsdottir, I.; Thorsteinsdottir, U.; Degenhardt, F.; et al. A protein-truncating R179X variant in RNF186 confers protection against ulcerative colitis. Nat. Commun. 2016, 7, 12342. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, K.; Kinoshita, M.; Tanaka, H.; Okuzaki, D.; Shimada, Y.; Kayama, H.; Okumura, R.; Furuta, Y.; Narazaki, M.; Tamura, A.; et al. Regulation of intestinal homeostasis by the ulcerative colitis-associated gene RNF186. Mucosal. Immunol. 2017, 10, 446–459. [Google Scholar] [CrossRef]
- Lu, Y.; Li, X.; Liu, S.; Zhang, Y.; Zhang, D. Toll-like Receptors and Inflammatory Bowel Disease. Front. Immunol. 2018, 9, 72. [Google Scholar] [CrossRef] [PubMed]
- Cari, L.; Rosati, L.; Leoncini, G.; Lusenti, E.; Gentili, M.; Nocentini, G.; Riccardi, C.; Migliorati, G.; Ronchetti, S. Association of GILZ with MUC2, TLR2, and TLR4 in Inflammatory Bowel Disease. Int. J. Mol. Sci. 2023, 24, 2235. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F. IL-23 in inflammatory bowel diseases and colon cancer. Cytokine Growth Factor Rev. 2019, 45, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, H.; Neurath, M.F.; Atreya, R. Role of the IL23/IL17 Pathway in Crohn’s Disease. Front. Immunol. 2021, 12, 622934. [Google Scholar] [CrossRef]
- Deng, Z.; Wang, S.; Wu, C.; Wang, C. IL-17 inhibitor-associated inflammatory bowel disease: A study based on literature and database analysis. Front. Pharmacol. 2023, 14, 1124628. [Google Scholar] [CrossRef]
- Cai, Y.; Jia, X.; Xu, L.; Chen, H.; Xie, S.; Cai, J. Interleukin-17 and inflammatory bowel disease: A 2-sample Mendelian randomization study. Front. Immunol. 2023, 14, 1238457. [Google Scholar] [CrossRef] [PubMed]
- Barnes, E.L.; Loftus, E.V., Jr.; Kappelman, M.D. Effects of Race and Ethnicity on Diagnosis and Management of Inflammatory Bowel Diseases. Gastroenterology 2021, 160, 677–689. [Google Scholar] [CrossRef]
- Yang, D.H.; Yang, S.K.; Song, K.; Hong, M.; Park, S.H.; Lee, H.S.; Kim, J.B.; Lee, H.J.; Park, S.K.; Jung, K.W.; et al. TNFSF15 is an independent predictor for the development of Crohn’s disease-related complications in Koreans. J. Crohn’s Colitis 2014, 8, 1315–1326. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Zhu, Q.; Zheng, P.F.; Feng, Y.L. Association of Fucosyltransferase 2 Gene Variant with Inflammatory Bowel Diseases: A Meta-Analysis. Med. Sci. Monit. 2019, 25, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Hu, J.; Wu, X.; Pan, J.A.; Jiao, N.; Li, Y.; Huang, Y.; Lin, X.; Zou, Y.; Chen, Y.; et al. Altered gut microbiome in FUT2 loss-of-function mutants in support of personalized medicine for inflammatory bowel diseases. J. Genet. Genom. 2021, 48, 771–780. [Google Scholar]
- Sazonovs, A.; Kennedy, N.A.; Moutsianas, L.; Heap, G.A.; Rice, D.L.; Reppell, M.; Bewshea, C.M.; Chanchlan, N.; Walker, G.J.; Perry, M.H.; et al. HLA-DQA1*05 Carriage Associated with Development of Anti-Drug Antibodies to Infliximab and Adalimumab in Patients with Crohn’s Disease. Gastroenterology 2020, 158, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Alcolado, L.; Grueso-Navarro, E.; Arias, Á.; Lucendo, A.J.; Laserna-Mendieta, E.J. Impact of HLA-DQA1∗05 Genotype in Immunogenicity and Failure to Treatment with Tumor Necrosis Factor-alpha Antagonists in Inflammatory Bowel Disease: A Systematic Review and Meta-analysis. J. Crohn’s Colitis 2024. [Google Scholar] [CrossRef] [PubMed]
- Mahdi, B.M. Role of HLA typing on Crohn’s disease pathogenesis. Ann. Med. Surg. 2015, 4, 248–253. [Google Scholar] [CrossRef]
- Ashton, J.J.; Latham, K.; Beattie, R.M.; Ennis, S. Review article: The genetics of the human leucocyte antigen region in inflammatory bowel disease. Aliment. Pharmacol. Ther. 2019, 50, 885–900. [Google Scholar] [CrossRef]
- Brant, S.R.; Okou, D.T.; Simpson, C.L.; Cutler, D.J.; Haritunians, T.; Bradfield, J.P.; Chopra, P.; Prince, J.; Begum, F.; Kumar, A.; et al. Genome-Wide Association Study Identifies African-Specific Susceptibility Loci in African Americans with Inflammatory Bowel Disease. Gastroenterology 2017, 152, 206–217. [Google Scholar] [CrossRef]
- Bergstein, S.; Spencer, E. HLA-DQA1*05 associates with immunogenicity and loss of response to anti-TNF therapy in the IBD population: A meta-analysis. Inflamm. Bowel Dis. 2023, 29, S58. [Google Scholar] [CrossRef]
- Dewit, O.; Moreels, T.; Baert, F.; Peeters, H.; Reenaers, C.; de Vos, M.; Van Hootegem, P.; Muls, V.; Veereman, G.; Mana, F.; et al. Limitations of extensive TPMT genotyping in the management of azathioprine-induced myelosuppression in IBD patients. Clin. Biochem. 2011, 44, 1062–1066. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.; Ammitzboell, M.; Nys, K.; Seidelin, J.B.; Nielsen, O.H. ATG16L1: A multifunctional susceptibility factor in Crohn disease. Autophagy 2015, 11, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Turpin, W.; Bedrani, L.; Espin-Garcia, O.; Xu, W.; Silverberg, M.S.; Smith, M.I.; Garay, J.A.R.; Lee, S.H.; Guttman, D.S.; Griffiths, A.; et al. Associations of NOD2 polymorphisms with Erysipelotrichaceae in stool of in healthy first degree relatives of Crohn’s disease subjects. BMC Med. Genet. 2020, 21, 204. [Google Scholar] [CrossRef] [PubMed]
- Cotterill, L.; Payne, D.; Levinson, S.; McLaughlin, J.; Wesley, E.; Feeney, M.; Durbin, H.; Lal, S.; Makin, A.; Petryszyn, P.; et al. C3435T Polymorphism of the ABCB1 Gene in Polish Patients with Inflammatory Bowel Disease: A Case-Control and Meta-Analysis Study. Genes 2021, 12, 1419. [Google Scholar] [CrossRef]
- Rojas, M.; Restrepo-Jiménez, P.; Monsalve, D.M.; Pacheco, Y.; Acosta-Ampudia, Y.; Ramírez-Santana, C.; Leung, P.S.C.; Ansari, A.A.; Gershwin, M.E.; Anaya, J.M. Molecular mimicry and autoimmunity. J. Autoimmun. 2018, 95, 100–123. [Google Scholar] [CrossRef] [PubMed]
- Naito, T.; Okada, Y. HLA imputation and its application to genetic and molecular fine-mapping of the MHC region in autoimmune diseases. Semin. Immunopathol. 2022, 44, 15–28. [Google Scholar] [CrossRef]
- Verstockt, B.; Parkes, M.; Lee, J.C. How Do We Predict a Patient’s Disease Course and Whether They Will Respond to Specific Treatments? Gastroenterology 2022, 162, 1383–1395. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Liu, Z. The latest breakthrough on genetic characteristics of inflammatory bowel disease in Chinese and other East Asian ancestries. Precis. Clin. Med. 2023, 6, pbad017. [Google Scholar] [CrossRef]
- Sivanesan, D.; Beauchamp, C.; Quinou, C.; Lee, J.; Lesage, S.; Chemtob, S.; Rioux, J.D.; Michnick, S.W. IL23R (Interleukin 23 Receptor) Variants Protective against Inflammatory Bowel Diseases (IBD) Display Loss of Function due to Impaired Protein Stability and Intracellular Trafficking. J. Biol. Chem. 2016, 291, 8673–8685. [Google Scholar] [CrossRef]
- Momozawa, Y.; Dmitrieva, J.; Théâtre, E.; Deffontaine, V.; Rahmouni, S.; Charloteaux, B.; Crins, F.; Docampo, E.; Elansary, M.; Gori, A.S.; et al. IBD risk loci are enriched in multigenic regulatory modules encompassing putative causative genes. Nat. Commun. 2018, 9, 2427. [Google Scholar] [CrossRef] [PubMed]
- Qiu, P.; Ishimoto, T.; Fu, L.; Zhang, J.; Zhang, Z.; Liu, Y. The Gut Microbiota in Inflammatory Bowel Disease. Front. Cell Infect. Microbiol. 2022, 12, 733992. [Google Scholar] [CrossRef] [PubMed]
- Imhann, F.; Vich Vila, A.; Bonder, M.J.; Fu, J.; Gevers, D.; Visschedijk, M.C.; Spekhorst, L.M.; Alberts, R.; Franke, L.; van Dullemen, H.M.; et al. Interplay of host genetics and gut microbiota underlying the onset and clinical presentation of inflammatory bowel disease. Gut 2018, 67, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Kayali, S.; Fantasia, S.; Gaiani, F.; Cavallaro, L.G.; de’Angelis, G.L.; Laghi, L. NOD2 and Crohn’s Disease Clinical Practice: From Epidemiology to Diagnosis and Therapy, Rewired. Inflamm. Bowel Dis. 2024. [Google Scholar] [CrossRef] [PubMed]
- Noble, A.J.; Nowak, J.K.; Adams, A.T.; Uhlig, H.H.; Satsangi, J. Defining Interactions Between the Genome, Epigenome, and the Environment in Inflammatory Bowel Disease: Progress and Prospects. Gastroenterology 2023, 165, 44–60. [Google Scholar] [CrossRef] [PubMed]
- Butera, A.; Di Paola, M.; Pavarini, L.; Strati, F.; Pindo, M.; Sanchez, M.; Cavalieri, D.; Boirivant, M.; De Filippo, C. Nod2 Deficiency in mice is Associated with Microbiota Variation Favouring the Expansion of mucosal CD4+ LAP+ Regulatory Cells. Sci. Rep. 2018, 8, 14241. [Google Scholar] [CrossRef] [PubMed]
- Coyne, M.J.; Comstock, L.E. Type VI Secretion Systems and the Gut Microbiota. Microbiol. Spectr. 2019, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Wellman, A.S.; Metukuri, M.R.; Kazgan, N.; Xu, X.; Xu, Q.; Ren, N.S.X.; Czopik, A.; Shanahan, M.T.; Kang, A.; Chen, W. Intestinal Epithelial Sirtuin 1 Regulates Intestinal Inflammation During Aging in Mice by Altering the Intestinal Microbiota. Gastroenterology 2017, 153, 772–786. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Zhang, Y.; Tian, X.; Wang, X.; Gathungu, G.; Wolber, A.; Shiekh, S.S.; Sartor, R.B.; Davidson, N.O.; Ciorba, M.A.; et al. Influence of Crohn’s disease related polymorphisms in innate immune function on ileal microbiome. PLoS ONE 2019, 14, e0213108. [Google Scholar] [CrossRef]
- Moschen, A.R.; Tilg, H.; Raine, T. IL-12, IL-23 and IL-17 in IBD: Immunobiology and therapeutic targeting. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 185–196. [Google Scholar] [CrossRef]
- Borg-Bartolo, S.P.; Boyapati, R.K.; Satsangi, J.; Kalla, R. Precision medicine in inflammatory bowel disease: Concept, progress and challenges. F1000Research 2020, 9, F1000. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Etxebarria, K.; Merino, O.; Gaite-Reguero, A.; Rodrigues, P.M.; Herrarte, A.; Etxart, A.; Ellinghaus, D.; Alonso-Galan, H.; Franke, A.; Marigorta, U.M.; et al. Local genetic variation of inflammatory bowel disease in Basque population and its effect in risk prediction. Sci. Rep. 2022, 12, 3386. [Google Scholar] [CrossRef]
- Gettler, K.; Levantovsky, R.; Moscati, A.; Giri, M.; Wu, Y.; Hsu, N.Y.; Chuang, L.S.; Sazonovs, A.; Venkateswaran, S.; Korie, U.; et al. Common and Rare Variant Prediction and Penetrance of IBD in a Large, Multi-ethnic, Health System-based Biobank Cohort. Gastroenterology 2021, 160, 1546–1557. [Google Scholar] [CrossRef]
- Dickson, A.L.; Daniel, L.L.; Zanussi, J.; Dale Plummer, W.; Wei, W.Q.; Liu, G.; Reese, T.; Anandi, P.; Birdwell, K.A.; Kawai, V.; et al. TPMT and NUDT15 Variants Predict Discontinuation of Azathioprine for Myelotoxicity in Patients with Inflammatory Disease: Real-World Clinical Results. Clin. Pharmacol. Ther. 2022, 111, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Relling, M.V.; Schwab, M.; Whirl-Carrillo, M.; Suarez-Kurtz, G.; Pui, C.H.; Stein, C.M.; Moyer, A.M.; Evans, W.E.; Klein, T.E.; Antillon-Klussmann, F.G.; et al. Clinical Pharmacogenetics Implementation Consortium Guideline for Thiopurine Dosing Based on TPMT and NUDT15 Genotypes: 2018 Update. Clin. Pharmacol. Ther. 2019, 105, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Gareb, B.; Otten, A.T.; Frijlink, H.W.; Dijkstra, G.; Kosterink, J.G.W. Review: Local Tumor Necrosis Factor-α Inhibition in Inflammatory Bowel Disease. Pharmaceutics 2020, 12, 539. [Google Scholar] [CrossRef] [PubMed]
- Verstockt, B.; Verstockt, S.; Dehairs, J.; Ballet, V.; Blevi, H.; Wollants, W.J.; Breynaert, C.; Van Assche, G.; Vermeire, S.; Ferrante, M. Low TREM1 expression in whole blood predicts anti-TNF response in inflammatory bowel disease. EBioMedicine 2019, 40, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Biasci, D.; Lee, J.C.; Noor, N.M.; Pombal, D.R.; Hou, M.; Lewis, N.; Ahmad, T.; Hart, A.; Parkes, M.; McKinney, E.F.; et al. A blood-based prognostic biomarker in IBD. Gut 2019, 68, 1386–1395. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Biasci, D.; Roberts, R.; Gearry, R.B.; Mansfield, J.C.; Ahmad, T.; Prescott, N.J.; Satsangi, J.; Wilson, D.C.; Jostins, L.; et al. Genome-wide association study identifies distinct genetic contributions to prognosis and susceptibility in Crohn’s disease. Nat. Genet. 2017, 49, 262–268. [Google Scholar] [CrossRef]
- Ruemmele, F.M.; Veres, G.; Kolho, K.L.; Griffiths, A.; Levine, A.; Escher, J.C.; Amil Dias, J.; Barabino, A.; Braegger, C.P.; Bronsky, J.; et al. European Crohn’s and Colitis Organisation; European Society of Pediatric Gastroenterology, Hepatology and Nutrition. Consensus guidelines of ECCO/ESPGHAN on the medical management of pediatric Crohn’s disease. J. Crohn’s Colitis 2014, 8, 1179–1207. [Google Scholar] [CrossRef]
- Heap, G.A.; Weedon, M.N.; Bewshea, C.M.; Singh, A.; Chen, M.; Satchwell, J.B.; Vivian, J.P.; So, K.; Dubois, P.C.; Andrews, J.M.; et al. HLA-DQA1-HLA-DRB1 variants confer susceptibility to pancreatitis induced by thiopurine immunosuppressants. Nat. Genet. 2014, 46, 1131–1134. [Google Scholar] [CrossRef] [PubMed]
- Lauro, R.; Mannino, F.; Irrera, N.; Squadrito, F.; Altavilla, D.; Squadrito, G.; Pallio, G.; Bitto, A. Pharmacogenetics of Biological Agents Used in Inflammatory Bowel Disease: A Systematic Review. Biomedicines 2021, 9, 1748. [Google Scholar] [CrossRef] [PubMed]
- Bek, S.; Nielsen, J.V.; Bojesen, A.B.; Franke, A.; Bank, S.; Vogel, U.; Andersen, V. Systematic review: Genetic biomarkers associated with anti-TNF treatment response in inflammatory bowel diseases. Aliment. Pharmacol. Ther. 2016, 44, 554–567. [Google Scholar] [CrossRef]
- Puca, P.; Capobianco, I.; Coppola, G.; Di Vincenzo, F.; Trapani, V.; Petito, V.; Laterza, L.; Pugliese, D.; Lopetuso, L.R.; Scaldaferri, F. Cellular and Molecular Determinants of Biologic Drugs Resistance and Therapeutic Failure in Inflammatory Bowel Disease. Int. J. Mol. Sci. 2024, 25, 2789. [Google Scholar] [CrossRef]
- Bai, B.Y.H.; Reppell, M.; Smaoui, N.; Waring, J.F.; Pivorunas, V.; Guay, H.; Lin, S.; Chanchlani, N.; Bewshea, C.; Goodhand, J.R.; et al. Baseline Expression of Immune Gene Modules in Blood is Associated with Primary Response to Anti-TNF Therapy in Crohn’s Disease Patients. J. Crohn’s Colitis 2024, 18, 431–445. [Google Scholar] [CrossRef]
- Bank, S.; Julsgaard, M.; Abed, O.K.; Burisch, J.; Broder Brodersen, J.; Pedersen, N.K.; Gouliaev, A.; Ajan, R.; Nytoft-Rasmussen, D.; Honore Grauslund, C.; et al. Polymorphisms in the NFkB, TNF-alpha, IL-1beta, and IL-18 pathways are associated with response to anti-TNF therapy in Danish patients with inflammatory bowel disease. Aliment. Pharmacol. Ther. 2019, 49, 890–903. [Google Scholar] [CrossRef]
- Salvador-Martín, S.; Zapata-Cobo, P.; Velasco, M.; Palomino, L.M.; Clemente, S.; Segarra, O.; Sánchez, C.; Tolín, M.; Moreno-Álvarez, A.; Fernández-Lorenzo, A.; et al. Association between HLA DNA Variants and Long-Term Response to Anti-TNF Drugs in a Spanish Pediatric Inflammatory Bowel Disease Cohort. Int. J. Mol. Sci. 2023, 24, 1797. [Google Scholar] [CrossRef]
- Lykowska-Szuber, L.; Walczak, M.; Stawczyk-Eder, K.; Krela-Kazmierczak, I.; Eder, P.; Zakerska-Banaszak, O.; Dobrowolska, A.; Skrzypczak-Zielinska, M. Variants of the CASP9 gene as candidate markers for primary response to anti-TNF therapy in Crohn’s disease patients. J. Appl. Genet. 2023, 64, 759–768. [Google Scholar] [CrossRef]
- Tang, J.; Zhang, C.B.; Lyu, K.S.; Jin, Z.M.; Guan, S.X.; You, N.; Huang, M.; Wang, X.D.; Gao, X. Association of polymorphisms in C1orf106, IL1RN, and IL10 with post-induction infliximab trough level in Crohn’s disease patients. Gastroenterol. Rep. 2019, 8, 367–373. [Google Scholar] [CrossRef]
- Salvador-Martín, S.; Pujol-Muncunill, G.; Bossacoma, F.; Navas-López, V.M.; Gallego-Fernández, C.; Segarra, O.; Clemente, S.; Muñoz-Codoceo, R.; Viada, J.; Magallares, L.; et al. Pharmacogenetics of trough serum anti-TNF levels in paediatric inflammatory bowel disease. Br. J. Clin. Pharmacol. 2021, 87, 447–457. [Google Scholar] [CrossRef]
- Salvador-Martín, S.; López-Cauce, B.; Nuñez, O.; Laserna-Mendieta, E.J.; García, M.I.; Lobato, E.; Abarca-Zabalía, J.; Sanjurjo-Saez, M.; Lucendo, A.J.; Marín-Jiménez, I.; et al. Genetic predictors of long-term response and trough levels of infliximab in Crohn’s disease. Pharmacol. Res. 2019, 149, 104478. [Google Scholar] [CrossRef]
- Laserna-Mendieta, E.J.; Salvador-Martín, S.; Arias, A.; López-Cauce, B.; Marín-Jiménez, I.; Menchén, L.A.; Marín-Rubio, L.; Ontañón Rodríguez, J.; López-Fernánde, L.A.; Lucendo, A.J. Single nucleotide polymorphisms in ADAM17, IL23R and SLCO1C1 genes protect against infliximab failure in adults with Crohn’s disease. Biomed. Pharmacother. 2023, 159, 114225. [Google Scholar] [CrossRef]
- Hoffmann, P.; Lamerz, D.; Hill, P.; Kirchner, M.; Gauss, A. Gene Polymorphisms of NOD2, IL23R, PTPN2 and ATG16L1 in Patients with Crohn’s Disease: On the Way to Personalized Medicine? Genes 2021, 12, 866. [Google Scholar] [CrossRef] [PubMed]
- Urabe, S.; Isomoto, H.; Ishida, T.; Maeda, K.; Inamine, T.; Kondo, S.; Higuchi, N.; Sato, K.; Uehara, R.; Yajima, H.; et al. Genetic Polymorphisms of IL-17F and TRAF3IP2 Could Be Predictive Factors of the Long-Term Effect of Infliximab against Crohn’s Disease. BioMed Res. Int. 2015, 2015, 416838. [Google Scholar] [CrossRef]
- Matsuoka, K.; Hamada, S.; Shimizu, M.; Nanki, K.; Mizuno, S.; Kiyohara, H.; Arai, M.; Sugimoto, S.; Iwao, Y.; Ogata, H.; et al. Factors predicting the therapeutic response to infliximab during maintenance therapy in Japanese patients with Crohn’s disease. PLoS ONE 2018, 13, e0204632. [Google Scholar] [CrossRef]
- Koder, S.; Repnik, K.; Ferkolj, I.; Pernat, C.; Skok, P.; Weersma, R.K.; Potočnik, U. Genetic polymorphism in ATG16L1 gene influences the response to adalimumab in Crohn’s disease patients. Pharmacogenomics 2015, 16, 191–204. [Google Scholar] [CrossRef]
- Dudzińska, E.; Szymona, K.; Gil-Kulik, P.; Chomik, P.; Świstowska, M.; Gryzińska, M.; Kocki, J. Imbalance of Controlled Death in Peripheral Blood Lymphocytes in Crohn’s Disease and Ulcerative Colitis. Medicina 2019, 55, 231. [Google Scholar] [CrossRef]
- Jezernik, G.; Gorenjak, M.; Potočnik, U. MIF Variant rs755622 Is Associated with Severe Crohn’s Disease and Better Response to Anti-TNF Adalimumab Therapy. Genes 2023, 14, 452. [Google Scholar] [CrossRef] [PubMed]
- Abreu, M.T.; Rowbotham, D.S.; Danese, S.; Sandborn, W.J.; Miao, Y.; Zhang, H.; Tikhonov, I.; Panaccione, R.; Hisamatsu, T.; Scherl, E.J.; et al. Efficacy and Safety of Maintenance Ustekinumab for Ulcerative Colitis Through 3 Years: UNIFI Long-term Extension. J. Crohn’s Colitis 2022, 16, 1222–1234. [Google Scholar] [CrossRef]
- Xu, J.; Xu, H.M.; Yang, M.F.; Liang, Y.J.; Peng, Q.Z.; Zhang, Y.; Tian, C.M.; Wang, L.S.; Yao, J.; Nie, Y.Q.; et al. New Insights Into the Epigenetic Regulation of Inflammatory Bowel Disease. Front. Pharmacol. 2022, 13, 813659. [Google Scholar] [CrossRef]
- Agrawal, M.; Allin, K.H.; Petralia, F.; Colombel, J.F.; Jess, T. Multiomics to elucidate inflammatory bowel disease risk factors and pathways. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 399–409. [Google Scholar] [CrossRef]
- Joustra, V.; Hageman, I.L.; Satsangi, J.; Adams, A.; Ventham, N.T.; de Jonge, W.J.; Henneman, P.; D’Haens, G.R.; Li Yim, A.Y.F. Systematic Review and Meta-analysis of Peripheral Blood DNA Methylation Studies in Inflammatory Bowel Disease. J. Crohn’s Colitis 2023, 17, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Cooke, J.; Zhang, H.; Greger, L.; Silva, A.-L.; Massey, D.; Dawson, C.; Metz, A.; Ibrahim, A.; Parkes, M. Mucosal Genome-wide Methylation Changes in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2012, 18, 2128–2137. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.A.; Nagy-Szakal, D.; Mir, S.A.; Frank, E.; Szigeti, R.; Kaplan, J.L.; Bronsky, J.; Opekun, A.; Ferry, G.D.; Winter, H.; et al. DNA methylation-associated colonic mucosal immune and defense responses in treatment-naïve pediatric ulcerative colitis. Epigenetics 2014, 9, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Wawrzyniak, M.; Scharl, M. Genetics and epigenetics of inflammatory bowel disease. Swiss. Med. Wkly. 2018, 14, w14671. [Google Scholar] [CrossRef] [PubMed]
- Azuara, D.; Aussó, S.; Rodriguez-Moranta, F.; Guardiola, J.; Sanjuan, X.; Lobaton, T.; Boadas, J.; Piqueras, M.; Monfort, D.; Guinó, E.; et al. New Methylation Biomarker Panel for Early Diagnosis of Dysplasia or Cancer in High-Risk Inflammatory Bowel Disease Patients. Inflamm. Bowel Dis. 2018, 24, 2555–2564. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lee, K.; Kim, K.; Yi, S.J. The role of histone modifications: From neurodevelopment to neurodiseases. Signal. Transduct. Target. Ther. 2022, 7, 217. [Google Scholar] [CrossRef] [PubMed]
- Millán-Zambrano, G.; Burton, A.; Bannister, A.J.; Schneider, R. Histone post-translational modifications—Cause and consequence of genome function. Nat. Rev. Genet. 2022, 3, 563–580. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Wang, S.; Li, J. Treatment of Inflammatory Bowel Disease: A Comprehensive Review. Front. Med. 2021, 8, 765474. [Google Scholar] [CrossRef]
- Kalla, R.; Adams, A.T.; Nowak, J.K.; Bergemalm, D.; Vatn, S.; Ventham, N.T.; Kennedy, N.A.; Ricanek, P.; Lindstrom, J.; Söderholm, J.; et al. Analysis of Systemic Epigenetic Alterations in Inflammatory Bowel Disease: Defining Geographical, Genetic and Immune-Inflammatory influences on the Circulating Methylome. J. Crohn’s Colitis 2023, 17, 170–184. [Google Scholar] [CrossRef]
- Ventham, N.T.; Kennedy, N.A.; Adams, A.T.; Kalla, R.; Heath, S.; O’Leary, K.R.; Drummond, H.; IBD BIOM Consortium; IBD CHARACTER Consortium; Wilson, D.C.; et al. Integrative epigenome-wide analysis demonstrates that DNA methylation may mediate genetic risk in inflammatory bowel disease. Nat. Commun. 2016, 7, 13507. [Google Scholar] [CrossRef] [PubMed]
- Li Yim, A.Y.F.; Duijvis, N.W.; Ghiboub, M.; Sharp, C.; Ferrero, E.; Mannens, M.M.A.M.; D’Haens, G.R.; de Jonge, W.J.; Te Velde, A.A.; Henneman, P. Whole-Genome DNA Methylation Profiling of CD14+ Monocytes Reveals Disease Status and Activity Differences in Crohn’s Disease Patients. J. Clin. Med. 2020, 9, 1055. [Google Scholar] [CrossRef] [PubMed]
- Comi, M.; Amodio, G.; Gregori, S. Interleukin-10-Producing DC-10 Is a Unique Tool to Promote Tolerance Via Antigen-Specific T Regulatory Type 1 Cells. Front. Immunol. 2018, 9, 682. [Google Scholar] [CrossRef]
- Fernandes, P.; MacSharry, J.; Darby, T.; Fanning, A.; Shanahan, F.; Houston, A.; Brint, E. Differential expression of key regulators of Toll-like receptors in ulcerative colitis and Crohn’s disease: A role for Tollip and peroxisome proliferator-activated receptor gamma? Clin. Exp. Immunol. 2016, 183, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, K.; Alonso, S.; Miyaki, Y.; Okada, S.; Ogura, H.; Shiiya, N.; Konishi, F.; Taya, T.; Perucho, M.; Suzuki, K. Array-based identification of common DNA methylation alterations in ulcerative colitis. Int. J. Oncol. 2012, 40, 983–994. [Google Scholar] [CrossRef] [PubMed]
- Samarani, S.; Dupont-Lucas, C.; Marcil, V.; Mack, D.; Israel, D.; Deslandres, C.; Jantchou, P.; Ahmad, A.; Amre, D. CpG Methylation in TGFβ1 and IL-6 Genes as Surrogate Biomarkers for Diagnosis of IBD in Children. Inflamm. Bowel Dis. 2020, 26, 1572–1578. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, M.; Gerbeth, L.; Gerling, M.; Rosenthal, R.; Steiger, K.; Weidinger, C.; Keye, J.; Wu, H.; Schmidt, F.; Weichert, W.; et al. HDAC inhibitors promote intestinal epithelial regeneration via autocrine TGFβ1 signalling in inflammation. Mucosal. Immunol. 2019, 12, 656–667. [Google Scholar] [CrossRef]
- Chang, J.; Ji, X.; Deng, T.; Qiu, J.; Ding, Z.; Li, Z.; Ma, Y.; Hu, X.; Li, L.; Qiu, J. Setd2 determines distinct properties of intestinal ILC3 subsets to regulate intestinal immunity. Cell Rep. 2022, 38, 110530. [Google Scholar] [CrossRef]
- Ding, Z.; Cai, T.; Tang, J.; Sun, H.; Qi, X.; Zhang, Y.; Ji, Y.; Yuan, L.; Chang, H.; Ma, Y.; et al. Setd2 supports GATA3+ST2+thymic-derived Treg cells and suppresses intestinal inflammation. Nat. Commun. 2022, 13, 7468. [Google Scholar] [CrossRef]
- Eshleman, E.M.; Shao, T.Y.; Woo, V.; Rice, T.; Engleman, L.; Didriksen, B.J.; Whitt, J.; Haslam, D.B.; Way, S.S.; Alenghat, T. Intestinal epithelial HDAC3 and MHC class II coordinate microbiota-specific immunity. J. Clin. Investig. 2023, 133, e162190. [Google Scholar] [CrossRef]
- Li, C.; Chen, Y.; Zhu, H.; Zhang, X.; Han, L.; Zhao, Z.; Wang, J.; Ning, L.; Zhou, W.; Lu, C.; et al. Inhibition of Histone Deacetylation by MS-275 Alleviates Colitis by Activating the Vitamin D Receptor. J. Crohn’s Colitis 2020, 14, 1103–1118. [Google Scholar] [CrossRef]
- Kazakevych, J.; Denizot, J.; Liebert, A.; Portovedo, M.; Mosavie, M.; Jain, P.; Stellato, C.; Fraser, C.; Corrêa, R.O.; Célestine, M.; et al. Smarcad1 mediates microbiota-induced inflammation in mouse and coordinates gene expression in the intestinal epithelium. Genome Biol. 2020, 21, 64. [Google Scholar] [CrossRef]
- Južnić, L.; Peuker, K.; Strigli, A.; Brosch, M.; Herrmann, A.; Häsler, R.; Koch, M.; Matthiesen, L.; Zeissig, Y.; Löscher, B.S.; et al. SETDB1 is required for intestinal epithelial differentiation and the prevention of intestinal inflammation. Gut 2021, 70, 485–498. [Google Scholar] [CrossRef]
- Wang, R.; Li, H.; Wu, J.; Cai, Z.Y.; Li, B.; Ni, H.; Qiu, X.; Chen, H.; Liu, W.; Yang, Z.H.; et al. Gut stem cell necroptosis by genome instability triggers bowel inflammation. Nature 2020, 580, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Hou, Y.; Chen, W.; Wang, J.; Xie, W.; Zhang, X.; Zeng, L. KIF9-AS1, LINC01272 and DIO3OS lncRNAs as novel biomarkers for inflammatory bowel disease. Mol. Med. Rep. 2018, 17, 2195–2202. [Google Scholar] [CrossRef] [PubMed]
- Ge, Q.; Dong, Y.; Lin, G.; Cao, Y. Long Noncoding RNA Antisense Noncoding RNA in the INK4 Locus Correlates with Risk, Severity, Inflammation and Infliximab Efficacy in Crohn’s Disease. Am. J. Med. Sci. 2019, 357, 134–142. [Google Scholar] [CrossRef]
- Visschedijk, M.C.; Spekhors, L.M.; Cheng, S.C.; van Loo, E.S.; Jansen, B.H.D.; Blokzijl, T.; Kil, H.; de Jong, D.J.; Pierik, M.; Maljaars, J.P.W.J.; et al. Genomic and Expression Analyses Identify a Disease-Modifying Variant for Fibrostenotic Crohn’s Disease. J. Crohn’s Colitis 2018, 12, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Zhou, G.; Chen, P.; Wang, Y.; Han, J.; Chen, M.; He, Y.; Zhang, S. Which long noncoding RNAs and circular RNAs contribute to inflammatory bowel disease? Cell Death Dis. 2020, 11, 456. [Google Scholar] [CrossRef]
- Liu, R.; Tang, A.; Wang, X.; Chen, X.; Zhao, L.; Xiao, Z.; Shen, S. Inhibition of lncRNA NEAT1 suppresses the inflammatory response in IBD by modulating the intestinal epithelial barrier and by exosome-mediated polarization of macrophages. Int. J. Mol. Med. 2018, 42, 2903–2913. [Google Scholar] [CrossRef]
- Geng, H.; Bu, H.F.; Liu, F.; Wu, L.; Pfeifer, K.; Chou, P.M.; Wang, X.; Sun, J.; Lu, L.; Pandey, A.; et al. In Inflamed Intestinal Tissues and Epithelial Cells, Interleukin 22 Signaling Increases Expression of H19 Long Noncoding RNA, Which Promotes Mucosal Regeneration. Gastroenterology 2018, 155, 144–155. [Google Scholar] [CrossRef] [PubMed]
- Zou, T.; Jaladanki, S.K.; Liu, L.; Xiao, L.; Chung, H.K.; Wang, J.Y.; Xu, Y.; Gorospe, M.; Wang, J.Y. H19 Long Noncoding RNA Regulates Intestinal Epithelial Barrier Function via MicroRNA 675 by Interacting with RNA-Binding Protein HuR. Mol. Cell Biol. 2016, 36, 1332–1341. [Google Scholar] [CrossRef]
- Wu, F.; Huang, Y.; Dong, F.; Kwon, J.H. Ulcerative Colitis-Associated Long Noncoding RNA, BC012900, Regulates Intestinal Epithelial Cell Apoptosis. Inflamm. Bowel Dis. 2016, 22, 782–795. [Google Scholar] [CrossRef]
- Haberman, Y.; BenShoshan, M.; Di Segni, A.; Dexheimer, P.J.; Braun, T.; Weiss, B.; Walters, T.D.; Baldassano, R.N.; Noe, J.D.; Markowitz, J.; et al. Long ncRNA Landscape in the Ileum of Treatment-Naive Early-Onset Crohn Disease. Inflamm. Bowel Dis. 2018, 24, 346–360. [Google Scholar] [CrossRef]
- Vautrin, A.; Manchon, L.; Garcel, A.; Campos, N.; Lapasset, L.; Laaref, A.M.; Bruno, R.; Gislard, M.; Dubois, E.; Scherrer, D.; et al. Both anti-inflammatory and antiviral properties of novel drug candidate ABX464 are mediated by modulation of RNA splicing. Sci. Rep. 2019, 9, 792. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Rao, J.N.; Cao, S.; Liu, L.; Chung, H.K.; Zhang, Y.; Zhang, J.; Liu, Y.; Gorospe, M.; Wang, J.Y. Long noncoding RNA SPRY4-IT1 regulates intestinal epithelial barrier function by modulating the expression levels of tight junction proteins. Mol. Biol. Cell. 2016, 27, 617–626. [Google Scholar] [CrossRef]
- Lucafò, M.; Di Silvestre, A.; Romano, M.; Avian, A.; Antonelli, R.; Martelossi, S.; Naviglio, S.; Tommasini, A.; Stocco, G.; Ventura, A.; et al. Role of the Long Non-Coding RNA Growth Arrest-Specific 5 in Glucocorticoid Response in Children with Inflammatory Bowel Disease. Basic Clin. Pharmacol. Toxicol. 2018, 122, 87–93. [Google Scholar] [CrossRef]
- Nag, S.; Mitra, O.; Tripathi, G.; Samanta, S.; Bhattacharya, B.; Chandane, P.; Mohanto, S.; Sundararajan, V.; Malik, S.; Rustagi, S.; et al. Exploring the theranostic potentials of miRNA and epigenetic networks in autoimmune diseases: A comprehensive review. Immun. Inflamm. Dis. 2023, 11, e1121. [Google Scholar] [CrossRef]
- James, J.P.; Riis, L.B.; Malham, M.; Høgdall, E.; Langholz, E.; Nielsen, B.S. MicroRNA Biomarkers in IBD-Differential Diagnosis and Prediction of Colitis-Associated Cancer. Int. J. Mol. Sci. 2020, 21, 7893. [Google Scholar] [CrossRef] [PubMed]
- Dhuppar, S.; Murugaiyan, G. miRNA effects on gut homeostasis: Therapeutic implications for inflammatory bowel disease. Trends Immunol. 2022, 43, 917–931. [Google Scholar] [CrossRef]
- Neudecker, V.; Haneklaus, M.; Jensen, O.; Khailova, L.; Masterson, J.C.; Tye, H.; Biette, K.; Jedlicka, P.; Brodsky, K.S.; Gerich, M.E.; et al. Myeloid-derived miR-223 regulates intestinal inflammation via repression of the NLRP3 inflammasome. J. Exp. Med. 2017, 214, 1737–1752. [Google Scholar] [CrossRef] [PubMed]
- Butin-Israeli, V.; Bui, T.M.; Wiesolek, H.L.; Mascarenhas, L.; Lee, J.J.; Mehl, L.C.; Knutson, K.R.; Adam, S.A.; Goldman, R.D.; Beyder, A.; et al. Neutrophil-induced genomic instability impedes resolution of inflammation and wound healing. J. Clin. Investig. 2019, 129, 712–726. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Gao, X.; Liu, L.; Li, Z.; Wan, Z.; Dong, Y.; Chen, X.; Niu, Y.; Zhang, J.; Yang, G. Visceral Adipose Tissue Derived Exosomes Exacerbate Colitis Severity via Pro-inflammatory MiRNAs in High Fat Diet Fed Mice. ACS Nano 2020, 14, 5099–5110. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Liang, Y.; Yang, J.; Xia, Y.; Chen, H.; Han, H.; Yang, Y.; Wu, W.; Gao, R.; Qin, H. MicroRNA-21 knockout improve the survival rate in DSS induced fatal colitis through protecting against inflammation and tissue injury. PLoS ONE 2013, 8, e66814. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; He, C.; Liu, C.; Cao, A.T.; Xue, X.; Evans-Marin, H.L.; Sun, M.; Fang, L.; Yao, S.; Pinchuk, I.V.; et al. miR-10a inhibits dendritic cell activation and Th1/Th17 cell immune responses in IBD. Gut 2015, 64, 1755–1764. [Google Scholar] [CrossRef]
- Shen, Y.; Zhou, M.; Yan, J.; Gong, Z.; Xiao, Y.; Zhang, C.; Du, P.; Chen, Y. MiR-200b inhibits TNF-α-induced IL-8 secretion and tight junction disruption of intestinal epithelial cells in vitro. Am. J. Physiol.-Gastrointest. Liver Physiol. 2017, 312, G123–G132. [Google Scholar] [CrossRef]
- Zhang, B.; Tian, Y.; Jiang, P.; Jiang, Y.; Li, C.; Liu, T.; Zhou, R.; Yang, N.; Zhou, X.; Liu, Z. MicroRNA-122a Regulates Zonulin by Targeting EGFR in Intestinal Epithelial Dysfunction. Cell Physiol. Biochem. 2017, 42, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Shen, J.; Cheng, J.; Fan, X. MicroRNA-21 regulates intestinal epithelial tight junction permeability. Cell Biochem. Funct. 2015, 33, 235–240. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, R.; Chen, J.; Wu, Q.; Kuang, Z. Baicalin Protects against TNF-α-Induced Injury by Down-Regulating miR-191a That Targets the Tight Junction Protein ZO-1 in IEC-6 Cells. Biol. Pharm. Bull. 2017, 40, 435–443. [Google Scholar] [CrossRef]
- Li, M.; Zhang, S.; Qiu, Y.; He, Y.; Chen, B.; Mao, R.; Cui, Y.; Zeng, Z.; Chen, M. Upregulation of miR-665 promotes apoptosis and colitis in inflammatory bowel disease by repressing the endoplasmic reticulum stress components XBP1 and ORMDL3. Cell Death Dis. 2017, 8, e2699. [Google Scholar] [CrossRef]
- Liu, L.; He, J.; Wei, X.; Wan, G.; Lao, Y.; Xu, W.; Li, Z.; Hu, H.; Hu, Z.; Luo, X.; et al. MicroRNA-20a-mediated loss of autophagy contributes to breast tumorigenesis by promoting genomic damage and instability. Oncogene 2017, 36, 5874–5884. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Miao, Y.; Shan, Y.; Liu, B.; Li, Y.; Zhao, L.; Jia, L. MiR-106b and miR-93 regulate cell progression by suppression of PTEN via PI3K/Akt pathway in breast cancer. Cell Death Dis. 2017, 8, e2796. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Shan, T.; Qu, H.; Chen, Y.; Wang, N.; Xia, J. Inhibition of miR-16 Ameliorates Inflammatory Bowel Disease by Modulating Bcl-2 in Mouse Models. J. Surg. Res. 2020, 253, 185–192. [Google Scholar] [CrossRef]
- Lin, X.T.; Zheng, X.B.; Fan, D.J.; Yao, Q.Q.; Hu, J.C.; Lian, L.; Wu, X.J.; Lan, P.; He, X.S. MicroRNA-143 Targets ATG2B to Inhibit Autophagy and Increase Inflammatory Responses in Crohn’s Disease. Inflamm. Bowel Dis. 2018, 24, 781–791. [Google Scholar] [CrossRef]
- Kim, H.Y.; Kwon, H.Y.; Ha Thi, H.T.; Lee, H.J.; Kim, G.I.; Hahm, K.B.; Hong, S. MicroRNA-132 and microRNA-223 control positive feedback circuit by regulating FOXO3a in inflammatory bowel disease. J. Gastroenterol. Hepatol. 2016, 31, 1727–1735. [Google Scholar] [CrossRef] [PubMed]
- Masi, L.; Capobianco, I.; Magrì, C.; Marafini, I.; Petito, V.; Scaldaferri, F. MicroRNAs as Innovative Biomarkers for Inflammatory Bowel Disease and Prediction of Colorectal Cancer. Int. J. Mol. Sci. 2022, 23, 7991. [Google Scholar] [CrossRef] [PubMed]
- Thorlacius-Ussing, G.; Schnack Nielsen, B.; Andersen, V.; Holmstrøm, K.; Pedersen, A.E. Expression and Localization of miR-21 and miR-126 in Mucosal Tissue from Patients with Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2017, 23, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Ahmed Hassan, E.; El-Din Abd El-Rehim, A.S.; Mohammed Kholef, E.F.; Abd-Elgwad Elsewify, W. Potential role of plasma miR-21 and miR-92a in distinguishing between irritable bowel syndrome, ulcerative colitis, and colorectal cancer. Gastroenterol. Hepatol. Bed Bench. 2020, 13, 147–154. [Google Scholar] [PubMed]
- Fang, K.; Law, I.K.M.; Padua, D.; Sideri, A.; Huang, V.; Kevil, C.G.; Iliopoulos, D.; Pothoulakis, C. MicroRNA-31-3p Is Involved in Substance P (SP)-Associated Inflammation in Human Colonic Epithelial Cells and Experimental Colitis. Am. J. Pathol. 2018, 188, 586–599. [Google Scholar] [CrossRef]
- Whiteoak, S.R.; Claridge, A.; Balendran, C.A.; Harris, R.J.; Gwiggner, M.; Bondanese, V.P.; Erlandsson, F.; Hansen, M.B.; Cummings, J.R.F.; Sanchez-Elsner, T. MicroRNA-31 Targets Thymic Stromal Lymphopoietin in Mucosal Infiltrated CD4+ T Cells: A Role in Achieving Mucosal Healing in Ulcerative Colitis? Inflamm. Bowel Dis. 2018, 24, 2377–2385. [Google Scholar] [CrossRef]
- Szűcs, D.; Béres, N.J.; Rokonay, R.; Boros, K.; Borka, K.; Kiss, Z.; Arató, A.; Szabó, A.J.; Vannay, Á.; Sziksz, E.; et al. Increased duodenal expression of miR-146a and -155 in pediatric Crohn’s disease. World J. Gastroenterol. 2016, 22, 6027–6035. [Google Scholar] [CrossRef] [PubMed]
- Béres, N.J.; Szabó, D.; Kocsis, D.; Szűcs, D.; Kiss, Z.; Müller, K.E.; Lendvai, G.; Kiss, A.; Arató, A.; Sziksz, E.; et al. Role of Altered Expression of miR-146a, miR-155, and miR-122 in Pediatric Patients with Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2016, 22, 327–335. [Google Scholar] [CrossRef]
- Schaefer, J.S.; Attumi, T.; Opekun, A.R.; Abraham, B.; Hou, J.; Shelby, H.; Graham, D.Y.; Streckfus, C.; Klein, J.R. MicroRNA signatures differentiate Crohn’s disease from ulcerative colitis. BMC Immunol. 2015, 16, 5. [Google Scholar] [CrossRef] [PubMed]
- Schönauen, K.; Le, N.; von Arnim, U.; Schulz, C.; Malfertheiner, P.; Link, A. Circulating and Fecal microRNAs as Biomarkers for Inflammatory Bowel Diseases. Inflamm. Bowel Dis. 2018, 24, 1547–1557. [Google Scholar] [CrossRef] [PubMed]
- Valmiki, S.; Ahuja, V.; Paul, J. MicroRNA exhibit altered expression in the inflamed colonic mucosa of ulcerative colitis patients. World J. Gastroenterol. 2017, 23, 5324–5332. [Google Scholar] [CrossRef]
- Zidar, N.; Boštjančič, E.; Jerala, M.; Kojc, N.; Drobne, D.; Štabuc, B.; Glavač, D. Down-regulation of microRNAs of the miR-200 family and up-regulation of Snail and Slug in inflammatory bowel diseases—Hallmark of epithelial-mesenchymal transition. J. Cell Mol. Med. 2016, 20, 1813–1820. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Yu, Y.; Tan, S. The role of the miR-21-5p-mediated inflammatory pathway in ulcerative colitis. Exp. Ther. Med. 2020, 19, 981–989. [Google Scholar] [CrossRef]
- Mohammadi, A.; Kelly, O.B.; Smith, M.I.; Kabakchiev, B.; Silverberg, M.S. Differential miRNA Expression in Ileal and Colonic Tissues Reveals an Altered Immunoregulatory Molecular Profile in Individuals with Crohn’s Disease versus Healthy Subjects. J. Crohn’s Colitis 2019, 13, 1459–1469. [Google Scholar] [CrossRef]
- Viennois, E.; Zhao, Y.; Han, M.K.; Xiao, B.; Zhang, M.; Prasad, M.; Wang, L.; Merlin, D. Serum miRNA signature diagnoses and discriminates murine colitis subtypes and predicts ulcerative colitis in humans. Sci. Rep. 2017, 7, 2520. [Google Scholar] [CrossRef]
- Chen, P.; Li, Y.; Li, L.; Yu, Q.; Chao, K.; Zhou, G.; Qiu, Y.; Feng, R.; Huang, S.; He, Y.; et al. Circulating microRNA146b-5p is superior to C-reactive protein as a novel biomarker for monitoring inflammatory bowel disease. Aliment. Pharmacol. Ther. 2019, 49, 733–743. [Google Scholar] [CrossRef]
- Tian, Y.; Xu, J.; Li, Y.; Zhao, R.; Du, S.; Lv, C.; Wu, W.; Liu, R.; Sheng, X.; Song, Y.; et al. MicroRNA-31 Reduces Inflammatory Signaling and Promotes Regeneration in Colon Epithelium, and Delivery of Mimics in Microspheres Reduces Colitis in Mice. Gastroenterology 2019, 156, 2281–2296.e6. [Google Scholar] [CrossRef] [PubMed]
- Fukata, T.; Mizushima, T.; Nishimura, J.; Okuzaki, D.; Wu, X.; Hirose, H.; Yokoyama, Y.; Kubota, Y.; Nagata, K.; Tsujimura, N.; et al. The Supercarbonate Apatite-MicroRNA Complex Inhibits Dextran Sodium Sulfate-Induced Colitis. Mol. Ther.-Nucleic Acids 2018, 12, 658–671. [Google Scholar] [CrossRef] [PubMed]
- Casertano, M.; Trotta, M.C.; Cenni, S.; Creoli, M.; Miele, E.; Martinelli, M.; Lepre, C.C.; Russo, M.; Alfano, R.; D’Amico, M.; et al. Infliximab therapy decreases the expression of serum and faecal miR-126 and miR-20a in paediatric Crohn’s disease: A pilot study. Acta Paediatr. 2024, 113, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Batra, S.K.; Heier, C.R.; Diaz-Calderon, L.; Tully, C.B.; Fiorillo, A.A.; van den Anker, J.; Conklin, L.S. Serum miRNAs Are Pharmacodynamic Biomarkers Associated with Therapeutic Response in Pediatric Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2020, 26, 1597–1606. [Google Scholar] [CrossRef]
- Chapman, C.G.; Pekow, J. The emerging role of miRNAs in inflammatory bowel disease: A review. Therap. Adv. Gastroenterol. 2015, 8, 4–22. [Google Scholar] [CrossRef]
- Morilla, I.; Uzzan, M.; Laharie, D.; Cazals-Hatem, D.; Denost, Q.; Daniel, F.; Belleannee, G.; Bouhnik, Y.; Wainrib, G.; Panis, Y.; et al. Colonic MicroRNA Profiles, Identified by a Deep Learning Algorithm, that Predict Responses to Therapy of Patients with Acute Severe Ulcerative Colitis. Clin. Gastroenterol. Hepatol. 2019, 17, 905–913. [Google Scholar] [CrossRef]
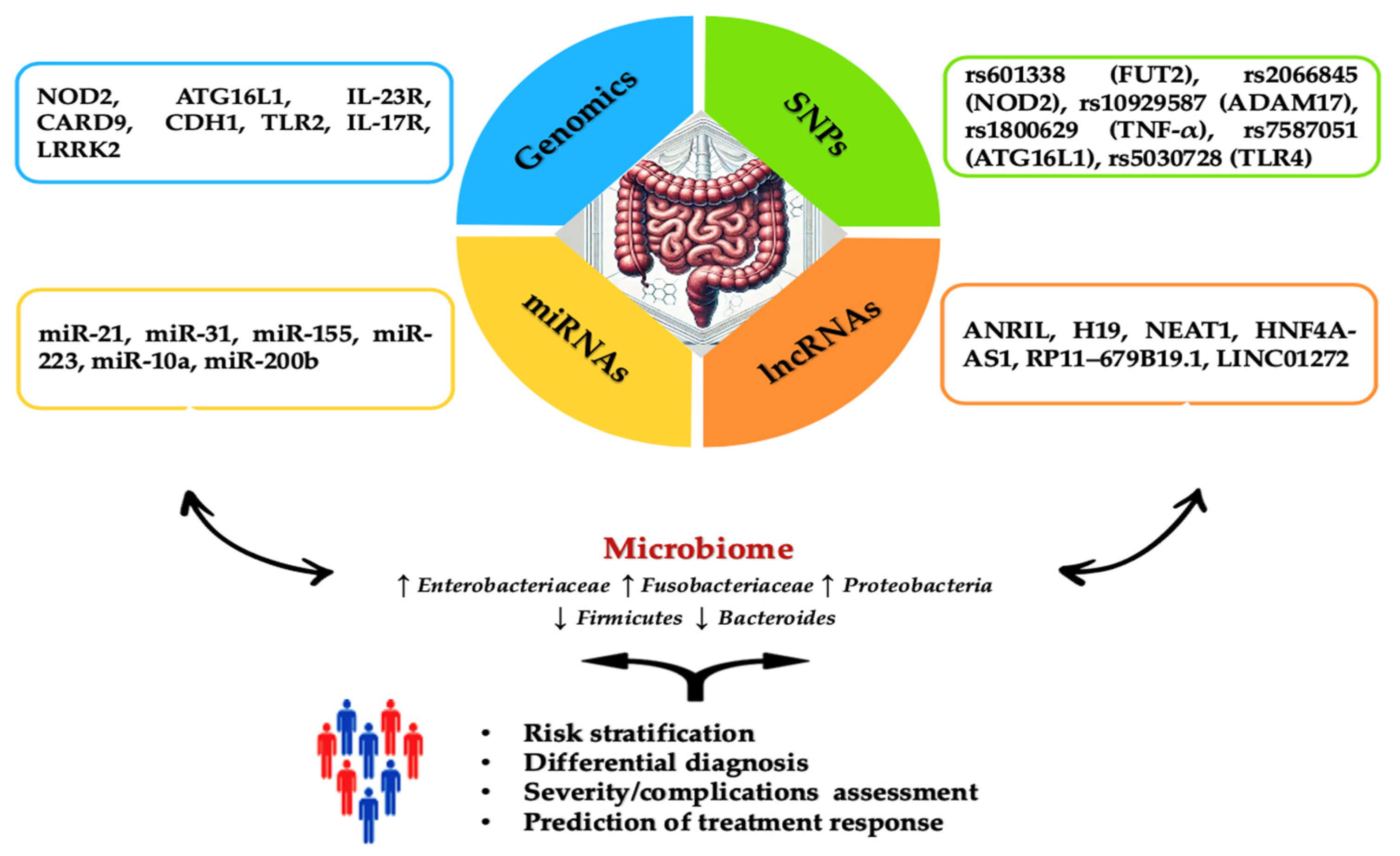
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).