


Glutathione-Dependent Pathways in Cancer Cells


Abstract
:1. Introduction
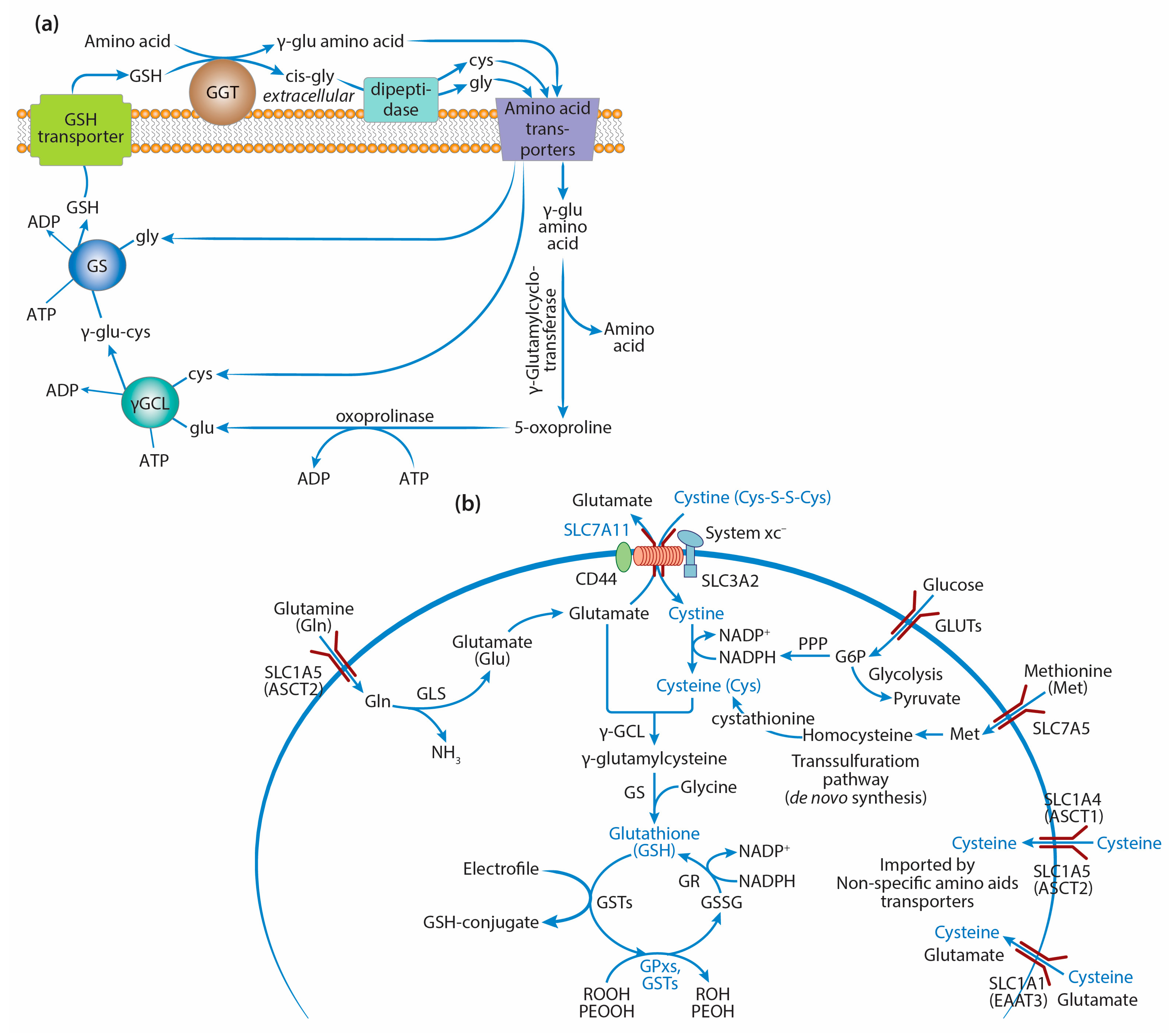
2. GSH Synthesis in Cancer Cells
2.1. γ-Glutamyl Cycle and the Key Enzymes of GSH Synthesis
2.2. GSH Synthesis and Precursor Amino Acids
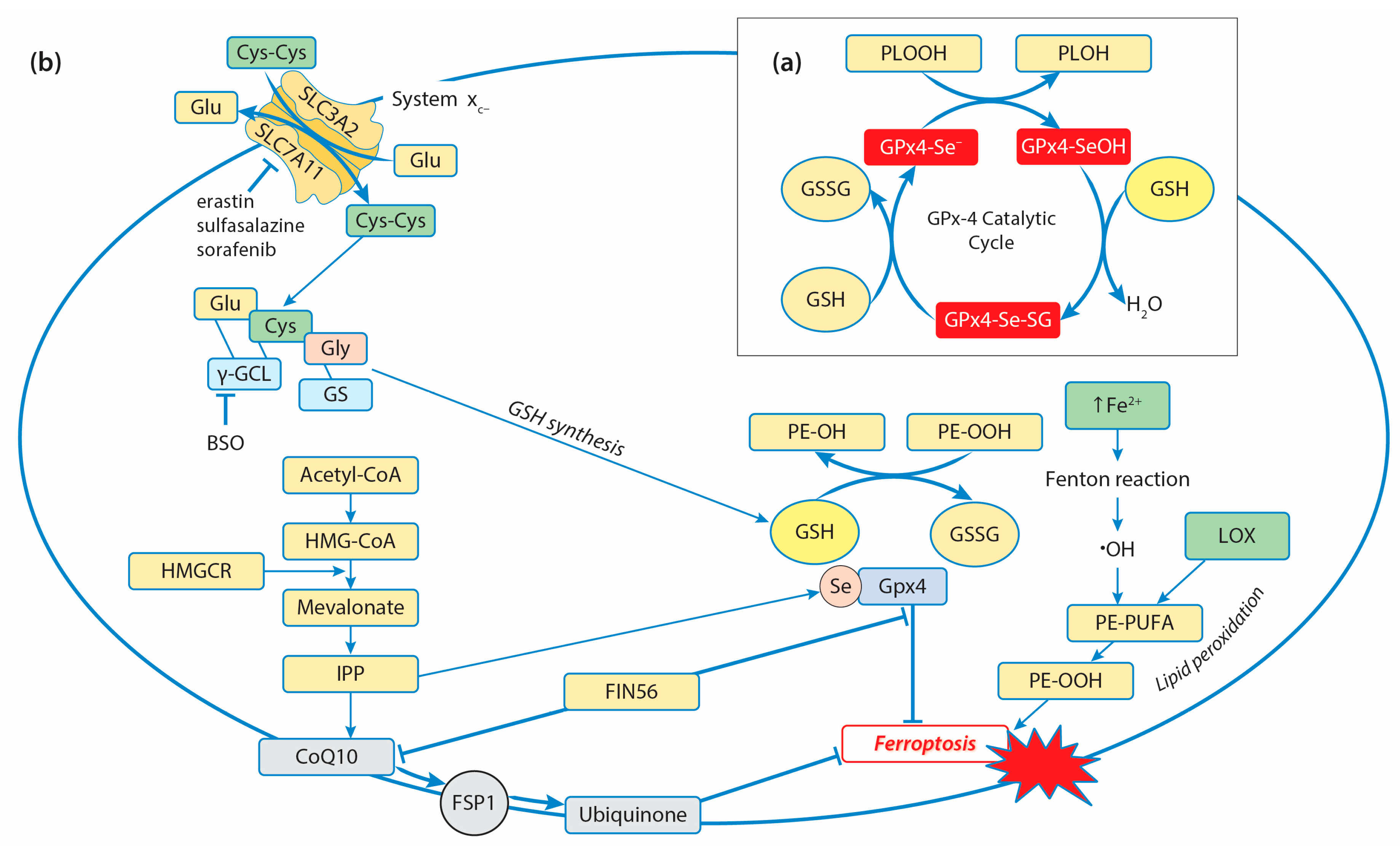
3. Glutathione Peroxidases and Antioxidant Defense in Cancer Cells
3.1. Glutathione Peroxidases and Decomposition of Hydroperoxides
3.2. Mammalian Glutathione Peroxidases
3.2.1. GPx1
3.2.2. GPx2
3.2.3. GPx3
3.2.4. GPx4
3.2.5. GPx5 and GPx6
3.2.6. GPx7 and GPx8
4. Glutathione Transferases and Tumorigenesis
4.1. GST Family and Conjugation of Electrophiles to GSH
4.2. GST Polymorphisms in Cancer
4.3. GSTs’ Chaperone Function in the Regulation of Stress-Induced Signaling Pathways
4.4. GST Inhibitors and Their Antitumor Action
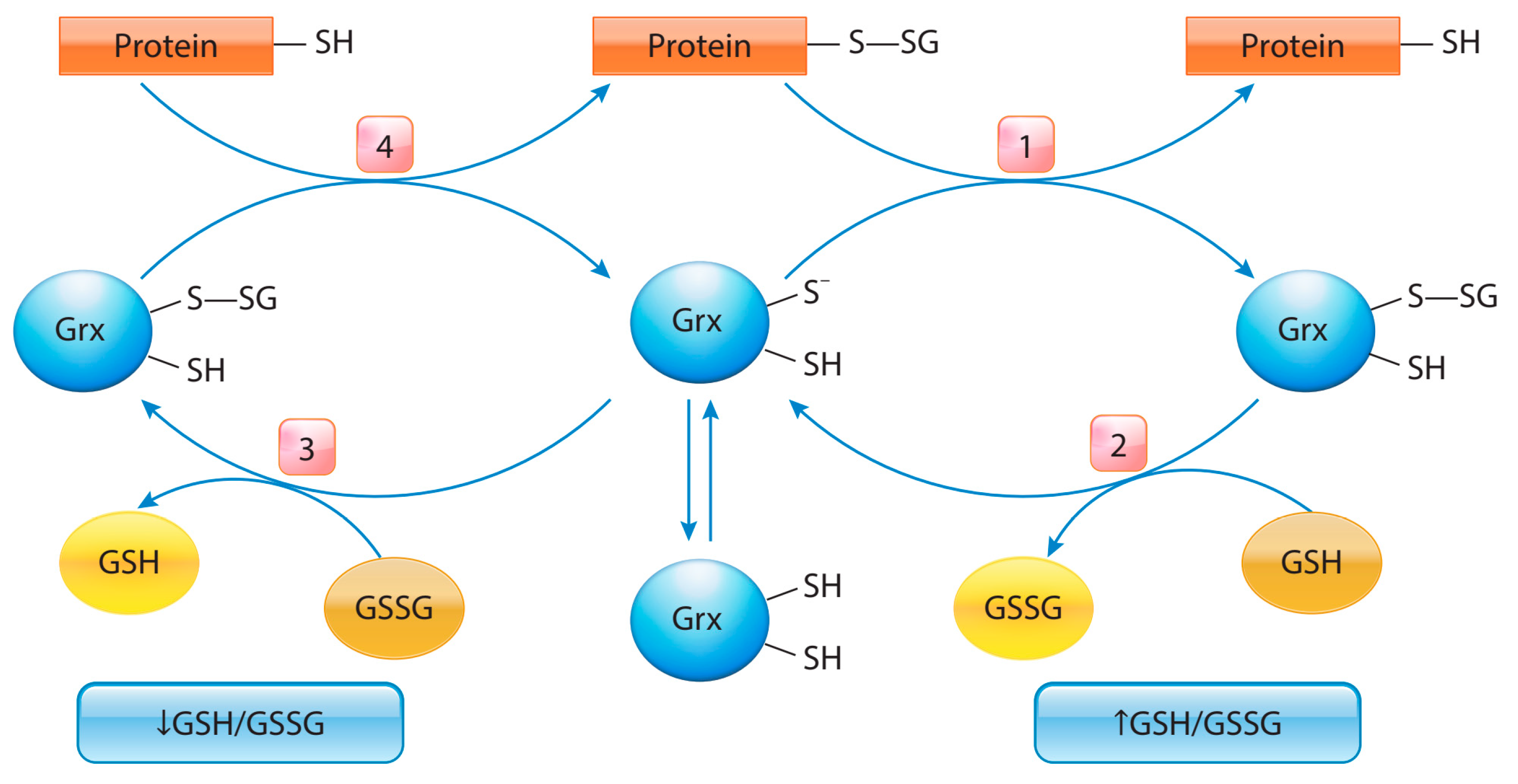
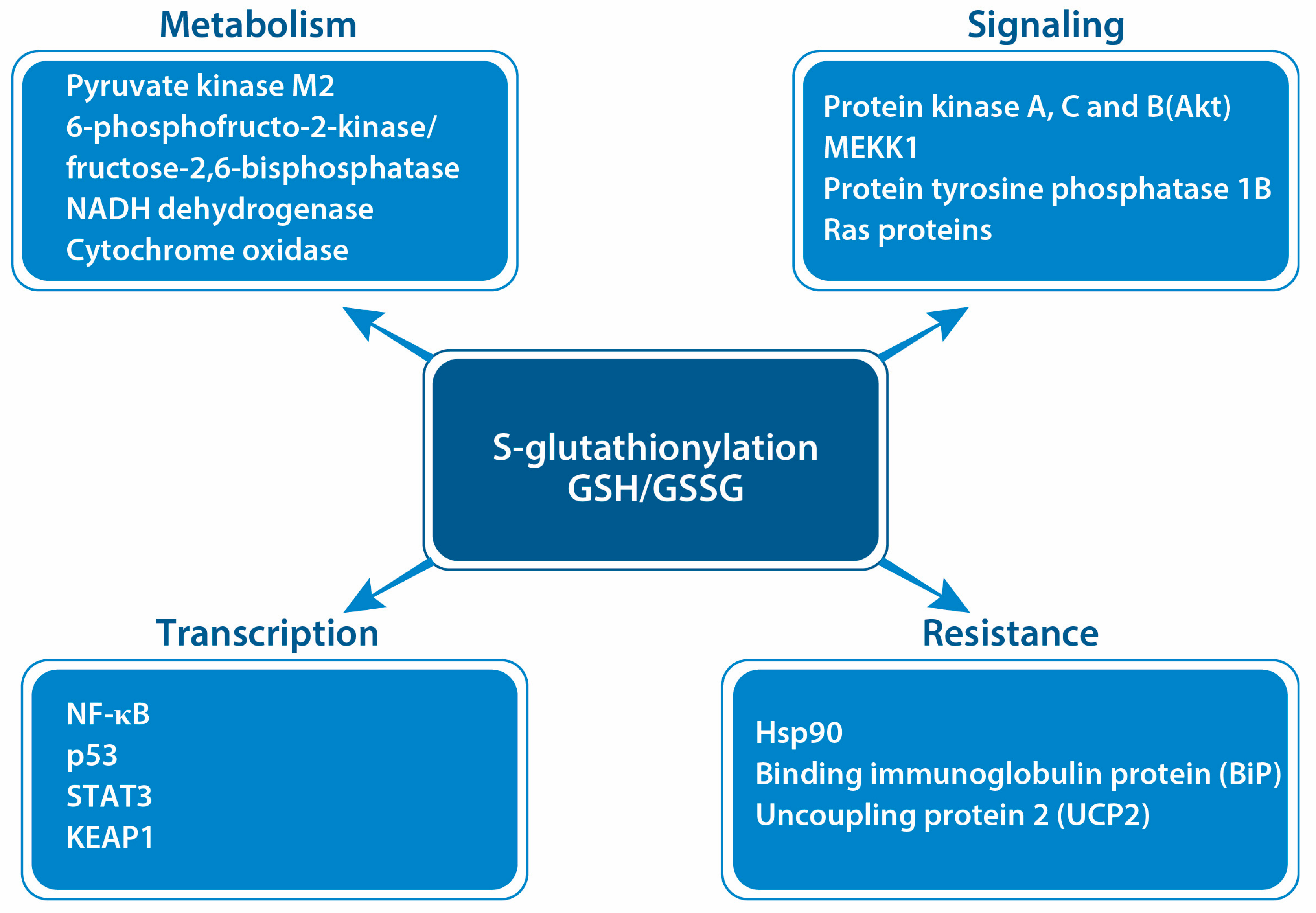
5. Protein S-Glutathionylation in Cancer
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Vázquez-Meza, H.; Vilchis-Landeros, M.M.; Vázquez-Carrada, M.; Uribe-Ramírez, D.; Matuz-Mares, D. Cellular Compartmentalization, Glutathione Transport and Its Relevance in Some Pathologies. Antioxidants 2023, 12, 834. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.-H.; Wang, H.-C.; Chang, C.-J.; Lee, S.-Y. Mitochondrial Glutathione in Cellular Redox Homeostasis and Disease Manifestation. Int. J. Mol. Sci. 2024, 25, 1314. [Google Scholar] [CrossRef] [PubMed]
- Georgiou-Siafis, S.K.; Tsiftsoglou, A.S. The Key Role of GSH in Keeping the Redox Balance in Mammalian Cells: Mechanisms and Significance of GSH in Detoxification via Formation of Conjugates. Antioxidants 2023, 12, 1953. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, L.; Sandhu, J.K.; Harper, M.-E.; Cuperlovic-Culf, M. Role of Glutathione in Cancer: From Mechanisms to Therapies. Biomolecules 2020, 10, 1429. [Google Scholar] [CrossRef] [PubMed]
- Kuehne, A.; Emmert, H.; Soehle, J.; Winnefeld, M.; Fischer, F.; Wenck, H.; Gallinat, S.; Terstegen, L.; Lucius, R.; Hildebrand, J.; et al. Acute Activation of Oxidative Pentose Phosphate Pathway as First-Line Response to Oxidative Stress in Human Skin Cells. Mol. Cell 2015, 59, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Gamcsik, M.P.; Kasibhatla, M.S.; Teeter, S.D.; Colvin, O.M. Glutathione Levels in Human Tumors. Biomarkers 2012, 17, 671–691. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- Wang, Y.; Qi, H.; Liu, Y.; Duan, C.; Liu, X.; Xia, T.; Chen, D.; Piao, H.-L.; Liu, H.-X. The Double-Edged Roles of ROS in Cancer Prevention and Therapy. Theranostics 2021, 11, 4839–4857. [Google Scholar] [CrossRef]
- O’Brien, M.L.; Tew, K.D. Glutathione and Related Enzymes in Multidrug Resistance. Eur. J. Cancer 1996, 32, 967–978. [Google Scholar] [CrossRef]
- Valenti, G.E.; Tasso, B.; Traverso, N.; Domenicotti, C.; Marengo, B. Glutathione in Cancer Progression and Chemoresistance: An Update. Red. Exp. Med. 2023, 2023, e220023. [Google Scholar] [CrossRef]
- Jones, C.M.; Lawrence, A.; Wardman, P.; Burkitt, M.J. Electron Paramagnetic Resonance Spin Trapping Investigation into the Kinetics of Glutathione Oxidation by the Superoxide Radical: Re-Evaluation of the Rate Constant. Free Radic. Biol. Med. 2002, 32, 982–990. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, M.; Lehnig, M.; Korth, H.G.; Sustmann, R.; de Groot, H. Inhibition of peroxynitrite-induced nitration of tyrosine by glutathione in the presence of carbon dioxide through both radical repair and peroxynitrate formation. Chemistry 2001, 7, 3313–3320. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C.; Metodiewa, D. Reactivity of Biologically Important Thiol Compounds with Superoxide and Hydrogen Peroxide. Free Radic. Biol. Med. 1999, 27, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Cassier-Chauvat, C.; Marceau, F.; Farci, S.; Ouchane, S.; Chauvat, F. The Glutathione System: A Journey from Cyanobacteria to Higher Eukaryotes. Antioxidants 2023, 12, 1199. [Google Scholar] [CrossRef]
- Xing, F.; Hu, Q.; Qin, Y.; Xu, J.; Zhang, B.; Yu, X.; Wang, W. The Relationship of Redox with Hallmarks of Cancer: The Importance of Homeostasis and Context. Front. Oncol. 2022, 12, 862743. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.; Pan, X.; Wei, G.; Hua, Y. Research Progress of Glutathione Peroxidase Family (GPX) in Redoxidation. Front. Pharmacol. 2023, 14, 1147414. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.R.; Carvalho, R.V.; Coelho, L.L.; De Souza Gonzaga, B.M.; Da Gloria Bonecini-Almeida, M.; Garzoni, L.R.; Araujo-Jorge, T.C. Current Understanding of Human Polymorphism in Selenoprotein Genes: A Review of Its Significance as a Risk Biomarker. Int. J. Mol. Sci. 2024, 25, 1402. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, H.; Zhou, J.-D.; Shao, Q. Glutathione Peroxidase GPX1 and Its Dichotomous Roles in Cancer. Cancers 2022, 14, 2560. [Google Scholar] [CrossRef]
- Mannervik, B.; Morgenstern, R. Glutathione Transferases. In Comprehensive Toxicology, 4th ed.; Elsevier eBooks: Amsterdam, The Netherlands, 2024; pp. 1–50. [Google Scholar] [CrossRef]
- Mazari, A.M.A.; Zhang, L.; Ye, Z.-W.; Zhang, J.; Tew, K.D.; Townsend, D.M. The Multifaceted Role of Glutathione S-Transferases in Health and Disease. Biomolecules 2023, 13, 688. [Google Scholar] [CrossRef]
- Townsend, D.M.; Shen, H.; Staros, A.L.; Gaté, L.; Tew, K.D. Efficacy of a glutathione S-transferase pi-activated prodrug in platinum-resistant ovarian cancer cells. Mol. Cancer Ther. 2002, 1, 1089–1095. [Google Scholar]
- Lv, N.; Huang, C.; Huang, H.; Dong, Z.; Chen, X.; Lu, C.; Zhang, Y. Overexpression of Glutathione S-Transferases in Human Diseases: Drug Targets and Therapeutic Implications. Antioxidants 2023, 12, 1970. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.M.; Manevich, Y.; He, L.; Hutchens, S.; Pazoles, C.J.; Tew, K.D. Novel Role for Glutathione S-Transferase π. J. Biol. Chem. 2009, 284, 436–445. [Google Scholar] [CrossRef]
- Meister, A.; Anderson, M.E. GLUTATHIONE. Ann. Rev. Biochem. 1983, 52, 711–760. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.E. Assay of the Enzymes of Glutathione Biosynthesis. Anal. Biochem. 2022, 644, 114218. [Google Scholar] [CrossRef]
- Lien, E.C.; Lyssiotis, C.A.; Juvekar, A.; Hu, H.; Asara, J.M.; Cantley, L.C.; Toker, A. Glutathione Biosynthesis Is a Metabolic Vulnerability in PI(3)K/Akt-Driven Breast Cancer. Nat. Cell Biol. 2016, 18, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. Regulation of Glutathione Synthesis. Mol. Aspects Med. 2009, 30, 42–59. [Google Scholar] [CrossRef]
- Lapenna, D. Glutathione and Glutathione-Dependent Enzymes: From Biochemistry to Gerontology and Successful Aging. Ageing Res. Rev. 2023, 92, 102066. [Google Scholar] [CrossRef] [PubMed]
- Ristoff, E.; Larsson, A. Inborn Errors in the Metabolism of Glutathione. Orphanet. J. Rare Dis. 2007, 2, 16. [Google Scholar] [CrossRef]
- Almusafri, F.; Elamin, H.E.; Khalaf, T.; Ali, A.; Ben-Omran, T.; El-Hattab, A.W. Clinical and Molecular Characterization of 6 Children with Glutamate-Cysteine Ligase Deficiency Causing Hemolytic Anemia. Blood Cells Mol. Dis. 2017, 65, 73–77. [Google Scholar] [CrossRef]
- Da, D.; Pan, Z.; Zhang, L.; Dang, Y.; Dang, C.; Huang, Y.; Shi, D.; Li, H. Glutamate-Cysteine Ligase Catalytic and Its Modifier Function as Novel Immunotargets in Gastric Adenocarcinoma. Asian J. Surg. 2023, 46, 143–149. [Google Scholar] [CrossRef]
- Zhang, L.; Tang, M.; Tao, X.; Shao, Q.; Thomas, V.; Shimizu, S.; Kasano, M.; Ishikawa, Y.; Inukai, T.; Nomura, D.K. Covalent Targeting of Glutamate Cysteine Ligase to Inhibit Glutathione Synthesis. ChemBioChem 2023, 24, e202300371. [Google Scholar] [CrossRef] [PubMed]
- Xue, Z.; Nuerrula, Y.; Sitiwaerdi, Y.; Eli, M. Nuclear Factor Erythroid 2-Related Factor 2 Promotes Radioresistance by Regulating Glutamate-Cysteine Ligase Modifier Subunit and Its Unique Immunoinvasive Pattern. Biomol. Biomed. 2024, 24, 545–559. [Google Scholar] [CrossRef] [PubMed]
- Hiyama, N.; Ando, T.; Maemura, K.; Sakatani, T.; Amano, Y.; Watanabe, K.; Kage, H.; Yatomi, Y.; Nagase, T.; Nakajima, J.; et al. Glutamate-Cysteine Ligase Catalytic Subunit Is Associated with Cisplatin Resistance in Lung Adenocarcinoma. Jpn. J. Clin. Oncol. 2018, 48, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Zhang, Z.; Weng, Y.; Zeng, J. Ferroptosis-Related Gene GCLC Is a Novel Prognostic Molecular and Correlates with Immune Infiltrates in Lung Adenocarcinoma. Cells 2022, 11, 3371. [Google Scholar] [CrossRef] [PubMed]
- Dequanter, D.; Van De Velde, M.; Bar, I.; Nuyens, V.; Rousseau, A.F.; Nagy, N.; Vanhamme, L.; Vanhaeverbeek, M.; Brohée, D.; Delrée, P.; et al. Nuclear Localization of Glutamate-Cysteine Ligase Is Associated with Proliferation in Head and Neck Squamous Cell Carcinoma. Oncol. Lett. 2016, 11, 3660–3668. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Hua, K.T.; Li, K.; Kao, H.; Hong, R.; Ko, J.; Hsiao, M.; Kuo, M.-L.; Tan, C.-T. Histone Methyltransferase G9A Drives Chemotherapy Resistance by Regulating the Glutamate–Cysteine Ligase Catalytic Subunit in Head and Neck Squamous Cell Carcinoma. Mol. Cancer Ther. 2017, 16, 1421–1434. [Google Scholar] [CrossRef] [PubMed]
- Bykanova, M.; Solodilova, M.; Azarova, I.; Klyosova, E.; Бушуева, О.Ю.; Polonikova, A.; Churnosov, M.; Polonikov, A. Genetic Variation at the Catalytic Subunit of Glutamate Cysteine Ligase Contributes to the Susceptibility to Sporadic Colorectal Cancer: A Pilot Study. Mol. Biol. Rep. 2022, 49, 6145–6154. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, Z.; Yuan, J.; Zhang, Y.; Jin, X. Altered Glutamate Cysteine Ligase Expression and Activity in Renal Cell Carcinoma. Biomed. Rep. 2014, 2, 831–834. [Google Scholar] [CrossRef] [PubMed]
- Koyani, C.N.; Kitz, K.; Rossmann, C.; Bernhart, E.; Huber, E.; Trummer, C.; Windischhofer, W.; Sattler, W.; Malle, E. Activation of the MAPK/Akt/Nrf2-Egr1/HO-1-GCLc Axis Protects MG-63 Osteosarcoma Cells against 15d-PGJ2-Mediated Cell Death. Biochem. Pharmacol. 2016, 104, 29–41. [Google Scholar] [CrossRef]
- Laoukili, J.; Constantinides, A.; Wassenaar, E.; Elias, S.G.; Raats, D.; Van Schelven, S.J.; Van Wettum, J.; Volckmann, R.; Koster, J.; Huitema, A.D.R.; et al. Peritoneal Metastases from Colorectal Cancer Belong to Consensus Molecular Subtype 4 and Are Sensitised to Oxaliplatin by Inhibiting Reducing Capacity. Br. J. Cancer 2022, 126, 1824–1833. [Google Scholar] [CrossRef]
- Sun, J.; Zhou, C.; Ma, Q.; Chen, W.; Atyah, M.; Yin, Y.; Fu, P.; Liu, S.; Hu, B.; Ren, N.; et al. High GCLC Level in Tumor Tissues Is Associated with Poor Prognosis of Hepatocellular Carcinoma after Curative Resection. J. Cancer 2019, 10, 3333–3343. [Google Scholar] [CrossRef] [PubMed]
- Njålsson, R.; Norgren, S. Physiological and Pathological Aspects of GSH Metabolism. Acta Paediatr. 2005, 94, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Bansal, A.; Simon, M.C. Glutathione Metabolism in Cancer Progression and Treatment Resistance. J. Cell Biol. 2018, 217, 2291–2298. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Zhang, Y.; Sedlazeck, F.J.; Creighton, C.J. Germline Structural Variation Globally Impacts the Cancer Transcriptome Including Disease-Relevant Genes. Cell Rep. Med. 2024, 5, 101446. [Google Scholar] [CrossRef] [PubMed]
- Njålsson, R.; Carlsson, K.S.; Winkler, A.; Larsson, A.; Norgren, S. Diagnostics in Patients with Glutathione Synthetase Deficiency but without Mutations in the Exons of the GSS Gene. Hum. Mutat. 2003, 22, 497. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yuan, D.; Liu, Y.; Ma, Y.; Song, J.; Wang, Q.; Yang, Y. Five Chinese Patients with 5-Oxoprolinuria Due to Glutathione Synthetase and 5-Oxoprolinase Deficiencies. Brain Dev. 2015, 37, 952–959. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Cao, Z.; Wang, W.; Zhang, C.; Wang, Y.; Pan, L.; Jia, B.; Zhang, K.; Zhang, W.; Li, W.; et al. Engineered Extracellular Vesicle-Delivered CRISPR/CAS9 for Radiotherapy Sensitization of Glioblastoma. ACS Nano 2023, 17, 16432–16447. [Google Scholar] [CrossRef]
- Ke, H.; Lin, J.; Ye, Y.; Wu, W.J.; Lin, H.; Wei, H.; Huang, M.; Chang, D.W.; Dinney, C.P.; Wu, X. Genetic Variations in Glutathione Pathway Genes Predict Cancer Recurrence in Patients Treated with Transurethral Resection and Bacillus Calmette–Guerin Instillation for Non-Muscle Invasive Bladder Cancer. Ann. Surg. Oncol. 2015, 22, 4104–4110. [Google Scholar] [CrossRef] [PubMed]
- Strohkamp, S.; Gemoll, T.; Humborg, S.; Hartwig, S.; Lehr, S.; Freitag-Wolf, S.; Becker, S.; Franzén, B.; Pries, R.; Wollenberg, B.; et al. Protein Levels of Clusterin and Glutathione Synthetase in Platelets Allow for Early Detection of Colorectal Cancer. Cell Mol. Life Sci. 2017, 75, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Z.; Wu, M.; Zhang, R. Advances and Perspectives of Responsive Probes for Measuring γ-Glutamyl Transpeptidase. ACS Meas. Sci. Au. 2023, 4, 54–75. [Google Scholar] [CrossRef]
- Mitrić, A.; Castellano, I. Targeting Gamma-Glutamyl Transpeptidase: A Pleiotropic Enzyme Involved in Glutathione Metabolism and in the Control of Redox Homeostasis. Free Radic. Biol. Med. 2023, 208, 672–683. [Google Scholar] [CrossRef] [PubMed]
- Takemura, K.; Board, P.G.; Koga, F. A Systematic Review of Serum γ-Glutamyltransferase as a Prognostic Biomarker in Patients with Genitourinary Cancer. Antioxidants 2021, 10, 549. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Li, Y.; Chang, L.; Tian, X. Prognostic and Clinicopathological Significance of Gamma-Glutamyltransferase in Patients with Hepatocellular Carcinoma. Medicine 2019, 98, e15603. [Google Scholar] [CrossRef]
- Takemura, K.; Ito, M.; Nakanishi, Y.; Kataoka, M.; Sakamoto, K.; Suzuki, H.; Tobisu, K.; Koga, F. Serum γ-Glutamyltransferase as a Prognostic Biomarker in Metastatic Castration-Resistant Prostate Cancer Treated with Enzalutamide. Anticancer. Res. 2019, 39, 5773–5780. [Google Scholar] [CrossRef] [PubMed]
- Takemura, K.; Fukushima, H.; Ito, M.; Kataoka, M.; Nakanishi, Y.; Sakamoto, K.; Suzuki, H.; Tobisu, K.; Koga, F. Prognostic Significance of Serum γ-Glutamyltransferase in Patients with Advanced Urothelial Carcinoma. Urol. Oncol. 2019, 37, 108–115. [Google Scholar] [CrossRef]
- King, J.B.; West, M.B.; Cook, P.F.; Hanigan, M.H. A Novel, Species-Specific Class of Uncompetitive Inhibitors of γ-Glutamyl Transpeptidase. J. Biol. Chem. 2009, 284, 9059–9065. [Google Scholar] [CrossRef] [PubMed]
- Lu, E.; Wolfreys, F.; Muppidi, J.R.; Xu, Y.; Cyster, J.G. S-Geranylgeranyl-l-Glutathione Is a Ligand for Human B Cell-Confinement Receptor P2RY8. Nature 2019, 567, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Pascale, R.M.; Simile, M.M.; Calvisi, D.F.; Feo, C.F.; Feo, F. S-Adenosylmethionine: From the Discovery of Its Inhibition of Tumorigenesis to Its Use as a Therapeutic Agent. Cells 2022, 11, 409. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhu, Y.; Awakawa, T.; Abe, I. Unusual Cysteine Modifications in Natural Product Biosynthesis. RSC Chem. Biol. 2024, 5, 293–311. [Google Scholar] [CrossRef]
- Hakimi, A.A.; Reznik, E.; Lee, C.; Creighton, C.J.; Brannon, A.R.; Luna, A.; Aksoy, B.A.; Liu, E.M.; Shen, R.; Lee, W.; et al. An Integrated Metabolic Atlas of Clear Cell Renal Cell Carcinoma. Cancer Cell 2016, 29, 104–116. [Google Scholar] [CrossRef]
- Clasen, J.; Heath, A.K.; Van Puyvelde, H.; Huybrechts, I.; Park, J.Y.; Ferrari, P.; Scélo, G.; Ulvik, A.; Midttun, Ø.; Ueland, P.M.; et al. Biomarkers of the Transsulfuration Pathway and Risk of Renal Cell Carcinoma in the European Prospective Investigation into Cancer and Nutrition (EPIC) Study. Int. J. Cancer 2022, 151, 708–716. [Google Scholar] [CrossRef] [PubMed]
- Fantone, S.; Piani, F.; Olivieri, F.; Rippo, M.R.; Sirico, A.; Di Simone, N.; Marzioni, D.; Tossetta, G. Role of SLC7A11/XCT in Ovarian Cancer. Int. J. Mol. Sci. 2024, 25, 587. [Google Scholar] [CrossRef] [PubMed]
- Jyotsana, N.; Ta, K.T.L.; DelGiorno, K.E. The Role of Cystine/Glutamate Antiporter SLC7A11/XCT in the Pathophysiology of Cancer. Front. Oncol. 2022, 12, 858462. [Google Scholar] [CrossRef] [PubMed]
- Koch, K.; Hartmann, R.; Suwala, A.; Rios, D.H.; Kamp, M.A.; Sabel, M.; Steiger, H.; Willbold, D.; Schmidt-Wolf, I.G.H.; Kahlert, U.D.; et al. Overexpression of Cystine/Glutamate Antiporter xCT Correlates with Nutrient Flexibility and ZEB1 Expression in Highly Clonogenic Glioblastoma Stem-like Cells (GSCs). Cancers 2021, 13, 6001. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Delaidelli, A.; Minaker, S.W.; Zhang, H.; Čolović, M.; Yang, H.; Negri, G.L.; Von Karstedt, S.; Lockwood, W.W.; Schaffer, P.; et al. Cystine/Glutamate Antiporter xCT (SLC7A11) Facilitates Oncogenic RAS Transformation by Preserving Intracellular Redox Balance. Proc. Natl. Acad. Sci. USA 2019, 116, 9433–9442. [Google Scholar] [CrossRef] [PubMed]
- Mesclon, F.; Lambert-Langlais, S.; Carraro, V.; Parry, L.; Hainault, I.; Jousse, C.; Maurin, A.; Bruhat, A.; Fafournoux, P.; Avérous, J. Decreased ATF4 Expression as a Mechanism of Acquired Resistance to Long-Term Amino Acid Limitation in Cancer Cells. Oncotarget 2017, 8, 27440–27453. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Roh, J. SLC7A11 as a Gateway of Metabolic Perturbation and Ferroptosis Vulnerability in Cancer. Antioxidants 2022, 11, 2444. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Hou, Q.; Zhang, X.; Ma, Y.; Yuan, J.; Li, S.; Zhao, X.; Sun, L.; Wang, H.; Zheng, H. Anesthetic Propofol Inhibits Ferroptosis and Aggravates Distant Cancer Metastasis via Nrf2 Upregulation. Free Radic. Biol. Med. 2023, 195, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Kilberg, M.S.; Shan, J.; Su, N. ATF4-Dependent Transcription Mediates Signaling of Amino Acid Limitation. Trends Endocrinol. Metab. 2009, 20, 436–443. [Google Scholar] [CrossRef]
- De La Vega, M.R.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef]
- Iqbal, M.J.; Kabeer, A.; Abbas, Z.; Siddiqui, H.A.; Calina, D.; Sharifi-Rad, J.; Cho, W.C. Interplay of Oxidative Stress, Cellular Communication and Signaling Pathways in Cancer. Cell Commun. Signal. 2024, 22, 7. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.-J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a P53-Mediated Activity during Tumour Suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, Y.; Du, T.; Yang, H.; Lei, L.; Guo, M.; Ding, H.; Zhang, J.; Wang, H.; Chen, X.; et al. ATF3 Promotes Erastin-Induced Ferroptosis by Suppressing System Xc–. Cell Death Differ. 2019, 27, 662–675. [Google Scholar] [CrossRef] [PubMed]
- Bi, R.; Hu, R.; Jiang, L.; Wen, B.; Jiang, Z.; Liu, H.; Mei, J. Butyrate Enhances Erastin-induced Ferroptosis of Lung Cancer Cells via Modulating the ATF3/SLC7A11 pathway. Environ. Toxicol. 2023, 39, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Shin, C.S.; Mishra, P.; Watrous, J.D.; Carelli, V.; D’Aurelio, M.; Jain, M.; Chan, D.C. The Glutamate/Cystine xCT Antiporter Antagonizes Glutamine Metabolism and Reduces Nutrient Flexibility. Nat. Commun. 2017, 8, 15074. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xia, X.; Huang, P. XCT: A Critical Molecule That Links Cancer Metabolism to Redox Signaling. Mol. Ther. 2020, 28, 2358–2366. [Google Scholar] [CrossRef]
- Bonifácio, V.D.B.; Pereira, S.A.; Serpa, J.; Vicente, J.B. Cysteine Metabolic Circuitries: Druggable Targets in Cancer. Br. J. Cancer 2020, 124, 862–879. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Pavlova, N.N.; Thompson, C.B. Cancer Cell Metabolism: The Essential Role of the Nonessential Amino Acid, Glutamine. EMBO J. 2017, 36, 1302–1315. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Venneti, S.; Nagrath, D. Glutaminolysis: A Hallmark of Cancer Metabolism. Annu. Rev. Biomed. Eng. 2017, 19, 163–194. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. C-Myc Suppression of miR-23a/b Enhances Mitochondrial Glutaminase Expression and Glutamine Metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to Clinic: Glutamine Metabolism to Cancer Therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef] [PubMed]
- Locasale, J.W.; Grassian, A.; Melman, T.; Lyssiotis, C.A.; Mattaini, K.; Bass, A.J.; Heffron, G.J.; Metallo, C.M.; Muranen, T.; Sharfi, H.; et al. Phosphoglycerate Dehydrogenase Diverts Glycolytic Flux and Contributes to Oncogenesis. Nat. Genet. 2011, 43, 869–874. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Nilsson, R.; Sharma, S.; Madhusudhan, N.; Kitami, T.; Souza, A.; Kafri, R.; Kirschner, M.W.; Clish, C.B.; Mootha, V.K. Metabolite Profiling Identifies a Key Role for Glycine in Rapid Cancer Cell Proliferation. Science 2012, 336, 1040–1044. [Google Scholar] [CrossRef] [PubMed]
- Ghanem, N.; El-Baba, C.; Araji, K.; El-Khoury, R.; Usta, J.; Darwiche, N. The Pentose Phosphate Pathway in Cancer: Regulation and Therapeutic Opportunities. Chemotherapy 2021, 66, 179–191. [Google Scholar] [CrossRef]
- Li, B.; Qiu, B.; Lee, D.S.M.; Walton, Z.E.; Ochocki, J.D.; Mathew, L.K.; Mancuso, A.; Gade, T.P.F.; Keith, B.; Nissim, I.; et al. Fructose-1,6-Bisphosphatase Opposes Renal Carcinoma Progression. Nature 2014, 513, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Chen, D.; Wu, Y.; Zhou, H.; Diao, W.; Liu, G.; Li, Q. A Feedback Loop of PPP and PI3K/AKT Signal Pathway Drives Regorafenib-Resistance in HCC. Cancer Metab. 2023, 11, 27. [Google Scholar] [CrossRef] [PubMed]
- Jaganjac, M.; Milkovic, L.; Sunjic, S.B.; Zarkovic, N. The NRF2, Thioredoxin, and Glutathione System in Tumorigenesis and Anticancer Therapies. Antioxidants 2020, 9, 1151. [Google Scholar] [CrossRef] [PubMed]
- Romero, R.; Sayin, V.I.; Davidson, S.M.; Bauer, M.R.; Singh, S.X.; LeBoeuf, S.E.; Karakousi, T.R.; Ellis, D.C.; Bhutkar, A.; Sánchez-Rivera, F.J.; et al. Keap1 Loss Promotes Kras-Driven Lung Cancer and Results in Dependence on Glutaminolysis. Nat. Med. 2017, 23, 1362–1368. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Liang, L.; Matsumoto, M.; Iwata, K.; Umemura, A.; He, F. Reactive Oxygen Species and NRF2 Signaling, Friends or Foes in Cancer? Biomolecules 2023, 13, 353. [Google Scholar] [CrossRef]
- Vernier, M.; Dufour, C.R.; McGuirk, S.; Scholtes, C.; Li, X.; Bourmeau, G.; Kuasne, H.; Park, M.; St-Pierre, J.; Audet-Walsh, E.; et al. Estrogen-Related Receptors Are Targetable ROS Sensors. Genes Dev. 2020, 34, 544–559. [Google Scholar] [CrossRef]
- Cuperlovic-Culf, M.; Cormier, K.; Touaibia, M.; Reyjal, J.; Robichaud, S.; Belbraouet, M.; Turcotte, S. 1H NMR Metabolomics Analysis of Renal Cell Carcinoma Cells: Effect of VHL Inactivation on Metabolism. Int. J. Cancer 2016, 138, 2439–2449. [Google Scholar] [CrossRef] [PubMed]
- Flohé, L.; Toppo, S.; Orian, L. The Glutathione Peroxidase Family: Discoveries and Mechanism. Free Radic. Biol. Med. 2022, 187, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R.; Maiorino, M. Glutathione Peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, N.V.; Nogueira, C.W.; Nogara, P.A.; De, A.F.; Aschner, M. Organoselenium Compounds as Mimics of Selenoproteins and Thiol Modifier Agents. Metallomics 2017, 9, 1703–1734. [Google Scholar] [CrossRef] [PubMed]
- Vašková, J.; Kočan, L.; Vaŝko, L.; Perjési, P. Glutathione-Related Enzymes and Proteins: A Review. Molecules 2023, 28, 1447. [Google Scholar] [CrossRef] [PubMed]
- Deponte, M. Glutathione Catalysis and the Reaction Mechanisms of Glutathione-Dependent Enzymes. Biochim. Biophys. Acta 2013, 1830, 3217–3266. [Google Scholar] [CrossRef] [PubMed]
- Flohé, L.; Toppo, S.; Cozza, G.; Ursini, F. A Comparison of Thiol Peroxidase Mechanisms. Antioxid. Redox Signal. 2011, 15, 763–780. [Google Scholar] [CrossRef] [PubMed]
- Toppo, S.; Flohé, L.; Ursini, F.; Vanin, S.; Maiorino, M. Catalytic Mechanisms and Specificities of Glutathione Peroxidases: Variations of a Basic Scheme. Biochim. Biophys. Acta 2009, 1790, 1486–1500. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, Y.; Yan, L.; Zhu, C.; Zou, Z. GPX4, Ferroptosis, and Diseases. Biomed. Pharmacother. 2024, 174, 116512. [Google Scholar] [CrossRef]
- Kipp, A.P. Selenium-dependent glutathione peroxidases during tumor development. Adv. Cancer Res. 2017, 136, 109–138. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R.; Flohé, L. Regulatory Phenomena in the Glutathione Peroxidase Superfamily. Antioxid. Redox Signal. 2020, 33, 498–516. [Google Scholar] [CrossRef] [PubMed]
- Miwa, T.; Adachi, T.; Ito, Y.; Hirano, K.; Sugiura, M. Purification and Properties of Glutathione Peroxidase from Human Liver. Chem. Pharm. Bull. 1983, 31, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Handy, D.E.; Loscalzo, J. The Role of Glutathione Peroxidase-1 in Health and Disease. Free Radic. Biol. Med. 2022, 188, 146–161. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Qiu, H.; Xu, J.; Mo, J.; Liu, Y.; Gui, Y.; Huang, G.; Zhang, S.; Yao, H.; Huang, X.; et al. Expression and Prognostic Potential of GPX1 in Human Cancers Based on Data Mining. Ann. Transl. Med. 2020, 8, 124. [Google Scholar] [CrossRef] [PubMed]
- Gouazé, V.; Mirault, M.-É.; Carpentier, S.; Salvayre, R.; Levade, T.; Andrieu-Abadie, N. Glutathione Peroxidase-1 Overexpression Prevents Ceramide Production and Partially Inhibits Apoptosis in Doxorubicin-Treated Human Breast Carcinoma Cells. Mol. Pharmacol. 2001, 60, 488–496. [Google Scholar] [PubMed]
- Gan, X.; Chen, B.; Shen, Z.; Liu, Y.; Li, H.; Xie, X.; Xu, X.; Li, H.; Huang, Z.; Chen, J. High GPX1 expression promotes esophageal squamous cell carcinoma invasion, migration, proliferation and cisplatin-resistance but can be reduced by vitamin D. Int. J. Clin. Exp. Med. 2014, 7, 2530–2540. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Liu, Y.; Huang, Z.; Li, H.; Gan, X.; Shen, Z. 1,25-Dihydroxyvitamin D3 Alleviates Salivary Adenoid Cystic Carcinoma Progression by Suppressing GPX1 Expression through the NF-κB Pathway. Int. J. Oncol. 2016, 48, 1271–1279. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hinkhouse, M.M.; Sun, W.; Weydert, C.J.; Ritchie, J.M.; Oberley, L.W.; Cullen, J.J. Redox Regulation of Pancreatic Cancer Cell Growth: Role of Glutathione Peroxidase in the Suppression of the Malignant Phenotype. Hum. Gene Ther. 2004, 15, 239–250. [Google Scholar] [CrossRef]
- Meng, Q.; Shi, S.; Liang, C.; Liang, D.; Hua, J.; Zhang, B.; Xu, J.; Lei, Y. Abrogation of Glutathione Peroxidase-1 Drives EMT and Chemoresistance in Pancreatic Cancer by Activating ROS-Mediated Akt/GSK3β/Snail Signaling. Oncogene 2018, 37, 5843–5857. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Choi, A.; Jun, Y.; Kim, N.; Yook, J.I.; Kim, S.Y.; Lee, S.; Kang, S.W. Glutathione Peroxidase-1 Regulates Adhesion and Metastasis of Triple-Negative Breast Cancer Cells via FAK Signaling. Redox Biol. 2020, 29, 101391. [Google Scholar] [CrossRef]
- Lee, S.-M.; Lee, E.K.; Kang, D.H.; Lee, J.; Hong, S.H.; Jeong, W.; Kang, S.W. Glutathione Peroxidase-1 Regulates ASK1-Dependent Apoptosis via Interaction with TRAF2 in RIPK3-Negative Cancer Cells. Exp. Mol. Med. 2021, 53, 1080–1091. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Xu, T.; Li, S.; Ruan, H. GPX1, a Biomarker for the Diagnosis and Prognosis of Kidney Cancer, Promotes the Progression of Kidney Cancer. Aging 2019, 11, 12165–12176. [Google Scholar] [CrossRef]
- Yang, W.; Shen, Y.; Wei, J.; Liu, F. MicroRNA-153/Nrf-2/GPx1 Pathway Regulates Radiosensitivity and Stemness of Glioma Stem Cells via Reactive Oxygen Species. Oncotarget 2015, 6, 22006–22027. [Google Scholar] [CrossRef]
- Chen, B.; Shen, Z.; Wu, D.; Xie, X.; Xu, X.; Lv, L.; Dai, H.; Chen, J.; Gan, X. Glutathione Peroxidase 1 Promotes NSCLC Resistance to Cisplatin via ROS-Induced Activation of PI3K/AKT Pathway. BioMed Res. Int. 2019, 2019, 7640547. [Google Scholar] [CrossRef] [PubMed]
- Min, S.Y.; Kim, H.S.; Jung, E.J.; Jung, E.J.; Jee, C.D.; Kim, W.H. Prognostic significance of glutathione peroxidase 1 (GPX1) downregulation and correlation with aberrant promoter methylation in human gastric cancer. Anticancer Res. 2012, 32, 3169–3175. [Google Scholar] [PubMed]
- Meng, Q.; Xu, J.; Liang, C.; Liu, J.; Hua, J.; Zhang, Y.; Ni, Q.; Shi, S.; Lei, Y. GPx1 Is Involved in the Induction of Protective Autophagy in Pancreatic Cancer Cells in Response to Glucose Deprivation. Cell Death Dis. 2018, 9, 1187. [Google Scholar] [CrossRef] [PubMed]
- Esworthy, R.S.; Doroshow, J.H.; Chu, F. The Beginning of GPX2 and 30 Years Later. Free Radic. Biol. Med. 2022, 188, 419–433. [Google Scholar] [CrossRef] [PubMed]
- Hashinokuchi, A.; Matsubara, T.; Ono, Y.; Shunichi, S.; Matsudo, K.; Nagano, T.; Kinoshita, F.; Akamine, T.; Kohno, M.; Takenaka, T.; et al. Clinical and Prognostic Significance of Glutathione Peroxidase 2 in Lung Adenocarcinoma. Ann. Surg. Oncol. 2024, 31, 4822–4829. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Xu, Q.; Jing, X.; Chi, X.; Zhang, Z.; Meng, X.; Liu, X.; Yan, J.; Li, X.; Shao, S. GPX2 Promotes EMT and Metastasis in Non-small Cell Lung Cancer by Activating PI3K/AKT/mTOR/Snail Signaling Axis. FASEB Bioadv. 2023, 5, 233–250. [Google Scholar] [CrossRef]
- Naiki, T.; Naiki-Ito, A.; Iida, K.; Etani, T.; Kato, H.; Suzuki, S.; Yamashita, Y.; Kawai, N.; Yasui, T.; Takahashi, S. GPX2 Promotes Development of Bladder Cancer with Squamous Cell Differentiation through the Control of Apoptosis. Oncotarget 2018, 9, 15847–15859. [Google Scholar] [CrossRef]
- Brzozowa-Zasada, M.; Ianaro, A.; Piecuch, A.; Michalski, M.; Matysiak, N.; Stęplewska, K. Immunohistochemical Expression of Glutathione Peroxidase-2 (GPX-2) and Its Clinical Relevance in Colon Adenocarcinoma Patients. Int. J. Mol. Sci. 2023, 24, 14650. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Zhu, X.; Shen, Y.; He, Q.; Qin, Y.; Shao, Y.; Yuan, L.; Ye, H. GPX2 Predicts Recurrence-Free Survival and Triggers the Wnt/β-Catenin/EMT Pathway in Prostate Cancer. PeerJ 2022, 10, e14263. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Liang, H.; Galbo, P.M.; Dharmaratne, M.; Kulkarni, A.; Fard, A.T.; Aoun, M.L.; Martínez–López, N.; Suyama, K.; Benard, O.; et al. Redox Signaling by Glutathione Peroxidase 2 Links Vascular Modulation to Metabolic Plasticity of Breast Cancer. Proc. Natl. Acad. Sci. USA 2022, 119, e2107266119. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R.; Kipp, A. Glutathione Peroxidases in Different Stages of Carcinogenesis. Biochim. Biophys. Acta 2009, 1790, 1555–1568. [Google Scholar] [CrossRef] [PubMed]
- Geng, D.; Zhou, Y.; Wang, M. Advances in the Role of GPX3 in Ovarian Cancer (Review). Int. J. Oncol. 2024, 64, 31. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Xie, Q.; Liu, X.; Wan, C.; Wu, W.; Fang, K.; Yao, Y.; Cheng, P.; Deng, D.; Liu, Z. Identification the Prognostic Value of Glutathione Peroxidases Expression Levels in Acute Myeloid Leukemia. Ann. Transl. Med. 2020, 8, 678. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Bai, W.; Huang, F.; Tang, J.; Lin, X. Downregulation of microRNA-196a Inhibits Stem Cell Self-Renewal Ability and Stemness in Non-Small-Cell Lung Cancer through Upregulating GPX3 Expression. Int. J. Biochem. Cell Biol. 2019, 115, 105571. [Google Scholar] [CrossRef] [PubMed]
- Worley, B.L.; Kim, Y.S.; Mardini, J.; Zaman, R.; Leon, K.E.; Vallur, P.G.; Nduwumwami, A.J.; Warrick, J.I.; Timmins, P.F.; Kesterson, J.P.; et al. GPx3 Supports Ovarian Cancer Progression by Manipulating the Extracellular Redox Environment. Redox Biol. 2019, 25, 101051. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Chen, N.; Wang, X.; Li, P.; Liu, L.; Zheng, R.; Liu, W.; Jiang, K.; Zhao, J. Prognostic Value and Immunological Roles of GPX3 in Gastric Cancer. Int. J. Med. Sci. 2023, 20, 1399–1416. [Google Scholar] [CrossRef]
- Yi, Z.; Jiang, L.; Zhao, L.; Zhou, M.; Ni, Y.; Yang, Y.; Yang, H.; Yang, L.; Zhang, Q.; Kuang, Y.; et al. Glutathione Peroxidase 3 (GPX3) Suppresses the Growth of Melanoma Cells through Reactive Oxygen Species (ROS)-dependent Stabilization of Hypoxia-inducible Factor 1-α and 2-α. J. Cell Biochem. 2019, 120, 19124–19136. [Google Scholar] [CrossRef]
- Lee, S.-H.; Golinska, M.A.; Griffiths, J.R. HIF-1-Independent Mechanisms Regulating Metabolic Adaptation in Hypoxic Cancer Cells. Cells 2021, 10, 2371. [Google Scholar] [CrossRef]
- Hu, Q.; Chen, J.; Yang, W.B.; Xu, M.; Zhou, J.; Tan, J.; Huang, T. GPX3 Expression Was Down-Regulated but Positively Correlated with Poor Outcome in Human Cancers. Front. Oncol. 2023, 13, 990551. [Google Scholar] [CrossRef]
- Savić, Ž.; Ćorić, V.; Vidović, S.; Vidović, V.; Bećarević, J.; Milovač, I.; Reljic, Z.; Mirjanić-Azarić, B.; Škrbić, R.; Gajanin, R.; et al. GPX3 Rs8177412 Polymorphism Modifies Risk of Upper Urothelial Tumors in Patients with Balkan Endemic Nephropathy. Medicina 2023, 59, 1421. [Google Scholar] [CrossRef] [PubMed]
- Noci, S.; Dugo, M.; Bertola, F.; Melotti, F.; Vannelli, A.; Dragani, T.A.; Galvan, A. A Subset of Genetic Susceptibility Variants for Colorectal Cancer Also Has Prognostic Value. Pharmacogenomics J. 2015, 16, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhao, W.; Gu, D.; Du, M.; Gong, W.; Tan, Y.; Wang, M.; Wen, J.; Zhai, Y.; Xu, Z. Association of Antioxidative Enzymes Polymorphisms with Efficacy of Platin and Fluorouracil-Based Adjuvant Therapy in Gastric Cancer. Cell Physiol. Biochem. 2018, 48, 2247–2257. [Google Scholar] [CrossRef]
- Wang, J.; Yang, I.; Wu, D.; Huang, S.-W.; Wu, J.; Juo, S.H. Functional Glutathione Peroxidase 3 Polymorphisms Associated with Increased Risk of Taiwanese Patients with Gastric Cancer. Clin. Chim. Acta 2010, 411, 1432–1436. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, Y.; Kuang, S.; Zhang, Y.; Qin, H.; Xie, J.-F. GPX3-Mediated Oxidative Stress Affects Pyrimidine Metabolism Levels in Stomach Adenocarcinoma via the AMPK/MTOR Pathway. Int. J. Clin. Pract. 2024, 2024, 1–18. [Google Scholar] [CrossRef]
- Wang, Z.; Zhu, J.; Liu, Y.; Wang, Z.; Cao, X.; Gu, Y. Tumor-Polarized GPX3 + AT2 Lung Epithelial Cells Promote Premetastatic Niche Formation. Proc. Natl. Acad. Sci. USA 2022, 119, e2201899119. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, L.; Gao, X.; Zhu, D. GPX3 Represses Pancreatic Cancer Cell Proliferation, Migration and Invasion, and Improves Their Chemo-sensitivity by Regulating the JNK/c-Jun Signaling Pathway. Exp. Ther. Med. 2024, 27, 118. [Google Scholar] [CrossRef]
- Cai, M.; Sikong, Y.; Wang, Q.; Zhu, S.; Pang, F.; Cui, X.-D. Gpx3 Prevents Migration and Invasion in Gastric Cancer by Targeting NFкB/Wnt5a/JNK Signaling. Int. J. Clin. Exp. Pathol. 2019, 12, 1194–1203. [Google Scholar]
- Mosca, L.; Ilari, A.; Fazi, F.; Assaraf, Y.G.; Colotti, G. Taxanes in Cancer Treatment: Activity, Chemoresistance and Its Overcoming. Drug Resist. Updat. 2021, 54, 100742. [Google Scholar] [CrossRef] [PubMed]
- Kelner, M.J.; Montoya, M.A. Structural Organization of the Human Selenium-Dependent Phospholipid Hydroperoxide Glutathione Peroxidase Gene (GPX4): Chromosomal Localization to 19p13.3. Biochem. Biophys. Res. Commun. 1998, 249, 53–55. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Kang, R.; Klionsky, D.J.; Tang, D. GPX4 in Cell Death, Autophagy, and Disease. Autophagy 2023, 19, 2621–2638. [Google Scholar] [CrossRef] [PubMed]
- Puglisi, R.; Tramer, F.; Panfili, E.; Micali, F.; Sandri, G.; Boitani, C. Differential Splicing of the Phospholipid Hydroperoxide Glutathione Peroxidase Gene in Diploid and Haploid Male Germ Cells in the RAT1. Biol. Reprod. 2003, 68, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Shi, Z.; Sun, Y.; Ning, H.; Xu, G.; Zhang, L. Prospects for Anti-Tumor Mechanism and Potential Clinical Application Based on Glutathione Peroxidase 4 Mediated Ferroptosis. Int. J. Mol. Sci. 2023, 24, 1607. [Google Scholar] [CrossRef] [PubMed]
- Forcina, G.C.; Dixon, S.J. GPX4 at the Crossroads of Lipid Homeostasis and Ferroptosis. Proteomics 2019, 19, e1800311. [Google Scholar] [CrossRef] [PubMed]
- Ursini, F.; Maiorino, M. Lipid Peroxidation and Ferroptosis: The Role of GSH and GPx4. Free Radic. Biol. Med. 2020, 152, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Weaver, K.; Skouta, R. The Selenoprotein Glutathione Peroxidase 4: From Molecular Mechanisms to Novel Therapeutic Opportunities. Biomedicines 2022, 10, 891. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, X.; Jin, S.; Chen, Y.; Guo, R. Ferroptosis in Cancer Therapy: A Novel Approach to Reversing Drug Resistance. Mol. Cancer 2022, 21, 47. [Google Scholar] [CrossRef]
- Feng, H.; Stockwell, B.R. Unsolved Mysteries: How Does Lipid Peroxidation Cause Ferroptosis? PLoS Biol. 2018, 16, e2006203. [Google Scholar] [CrossRef]
- Hassannia, B.; Vandenabeele, P.; Vanden Berghe, T. Targeting Ferroptosis to Iron out Cancer. Cancer Cell 2019, 35, 830–849. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zhang, Y.; Lin, S.; Liu, Y.; Li, W. Correction for: Suppressing the KIF20A/NUAK1/Nrf2/GPX4 Signaling Pathway Induces Ferroptosis and Enhances the Sensitivity of Colorectal Cancer to Oxaliplatin. Aging 2021, 13, 19077. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Chen, K.; Zhang, J.; Zhang, X. Inhibition of GPX4 or mTOR Overcomes Resistance to Lapatinib via Promoting Ferroptosis in NSCLC Cells. Biochem. Biophys. Res. Commun. 2021, 567, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, S.; Chen, Q.; Xia, T.-L.; Luo, D.-H.; Li, L.; Liu, S.-L.; Guo, S.-S.; Liu, L.; Du, C.; et al. EBV Infection-Induced GPX4 Promotes Chemoresistance and Tumor Progression in Nasopharyngeal Carcinoma. Cell Death Differ. 2022, 29, 1513–1527. [Google Scholar] [CrossRef]
- Battaglia, A.M.; Chirillo, R.; Aversa, I.; Sacco, A.; Costanzo, F.; Biamonte, F. Ferroptosis and Cancer: Mitochondria Meet the “Iron Maiden” Cell Death. Cells 2020, 9, 1505. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Song, J.; Yung, B.C.; Zhou, Z.; Wu, A.; Chen, X. Emerging Strategies of Cancer Therapy Based on Ferroptosis. Adv. Mater. 2018, 30, e1704007. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Xu, B.; Han, Q.; Zhou, H.; Xia, Y.; Gong, C.; Dai, X.; Li, Z.; Wu, G. Ferroptosis: A Novel Anti-Tumor Action for Cisplatin. Cancer Res. Treat. 2018, 50, 445–460. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.H.; Shimoni, Y.; Yang, W.S.; Subramaniam, P.S.; Iyer, A.; Nicoletti, P.; Martínez, M.R.; López, G.; Mattioli, M.; Realubit, R.; et al. Elucidating Compound Mechanism of Action by Network Perturbation Analysis. Cell 2015, 162, 441–451. [Google Scholar] [CrossRef]
- Costa, I.; Barbosa, D.J.; Benfeito, S.; Silva, V.; Chavarria, D.; Borges, F.; Fernando, R.; Silva, R. Molecular Mechanisms of Ferroptosis and Their Involvement in Brain Diseases. Pharmacol. Ther. 2023, 244, 108373. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, Y.; Zhang, D.; Meng, J.; Che, N.; Zhao, X.; Liu, T. PTGER3 Knockdown Inhibits the Vulnerability of Triple-negative Breast Cancer to Ferroptosis. Cancer Sci. 2024, 115, 2067–2081. [Google Scholar] [CrossRef]
- Yuan, M.; Chen, L.; Wang, C.; Miao, Y.; Song, C.; Shi, J.; Wang, L. AURKA Knockdown Inhibits Esophageal Squamous Cell Carcinoma Progression through Ferroptosis. Heliyon 2024, 10, e28365. [Google Scholar] [CrossRef]
- Gomaa, A.R.; Peng, D.; Chen, Z.; Soutto, M.; Abouelezz, K.F.M.; Corvalán, A.; El-Rifai, W. Epigenetic Regulation of AURKA by miR-4715-3p in Upper Gastrointestinal Cancers. Sci. Rep. 2019, 9, 16970. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; He, Y.; Lan, J.; Luo, S.; Sun, B.; Xiao, C.; Yu, W.; Zeng, Z.; Lei, S. FOXP3 Promote the Progression of Glioblastoma via Inhibiting Ferroptosis Mediated by Linc00857/miR-1290/GPX4 Axis. Cell Death Dis. 2024, 15, 239. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.B.; Robson, A.; Houghton, B.C.; Jepson, C.A.; Ford, W.C.L.; Frayne, J. Epididymal Specific, Selenium-Independent GPX5 Protects Cells from Oxidative Stress-Induced Lipid Peroxidation and DNA Mutation. Hum. Reprod. 2013, 28, 2332–2342. [Google Scholar] [CrossRef]
- Tan, S.; Liu, Q.; Yang, J.; Cai, J.; Yu, M.; Yu-Bin, J. Macranthoidin B (MB) Promotes Oxidative Stress-Induced Inhibiting of HEPA1-6 Cell Proliferation via Selenoprotein. Biol. Trace Elem. Res. 2022, 201, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Rusolo, F.; Capone, F.; Pasquale, R.; Angiolillo, A.; Colonna, G.; Castello, G.; Costantini, M.; Costantini, S. Comparison of the Seleno-Transcriptome Expression between Human Non-Cancerous Mammary Epithelial Cells and Two Human Breast Cancer Cell Lines. Oncol. Lett. 2017, 13, 2411–2417. [Google Scholar] [CrossRef] [PubMed]
- Mariotti, M.; Ridge, P.G.; Zhang, Y.; Lobanov, A.; Pringle, T.H.; Guigó, R.; Hatfield, D.L.; Gladyshev, V.N. Composition and Evolution of the Vertebrate and Mammalian Selenoproteomes. PLoS ONE 2012, 7, e33066. [Google Scholar] [CrossRef] [PubMed]
- Maiorino, M.; Bosello-Travain, V.; Cozza, G.; Miotto, G.; Roveri, A.; Toppo, S.; Zaccarin, M.; Ursini, F. Understanding Mammalian Glutathione Peroxidase 7 in the Light of Its Homologs. Free Radic. Biol. Med. 2015, 83, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.D.; Saaranen, M.J.; Karala, A.; Lappi, A.; Wang, L.; Raykhel, I.; Alanen, H.I.; Salo, K.E.H.; Wang, C.-C.; Ruddock, L.W. Two Endoplasmic Reticulum PDI Peroxidases Increase the Efficiency of the Use of Peroxide during Disulfide Bond Formation. J. Mol. Biol. 2011, 406, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Buday, K.; Conrad, M. Emerging Roles for Non-Selenium Containing ER-Resident Glutathione Peroxidases in Cell Signaling and Disease. Biol. Chem. 2020, 402, 271–287. [Google Scholar] [CrossRef]
- Bosello-Travain, V.; Conrad, M.; Cozza, G.; Negro, A.; Quartesan, S.; Rossetto, M.; Roveri, A.; Toppo, S.; Ursini, F.; Zaccarin, M.; et al. Protein Disulfide Isomerase and Glutathione Are Alternative Substrates in the One Cys Catalytic Cycle of Glutathione Peroxidase 7. Biochim. Biophys. Acta 2013, 1830, 3846–3857. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, L.; Niu, Y.; Sitia, R.; Wang, C.C. Glutathione Peroxidase 7 Utilizes Hydrogen Peroxide Generated by ERO1A to Promote Oxidative Protein Folding. Antioxid. Redox Signal. 2014, 20, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Guerriero, E.; Capone, F.; Accardo, M.; Sorice, A.; Costantini, M.; Colonna, G.; Castello, G.; Costantini, S. GPX4 and GPX7 Over-Expression in Human Hepatocellular Carcinoma Tissues. Eur. J. Histochem. 2015, 59, 2540. [Google Scholar] [CrossRef]
- Chen, Z.; Hu, T.; Zhu, S.; Mukaisho, K.; El-Rifai, W.; Peng, D.F. Glutathione Peroxidase 7 Suppresses Cancer Cell Growth and Is Hypermethylated in Gastric Cancer. Oncotarget 2017, 8, 54345–54356. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Chen, X.; Liu, Z.; Zhang, R.; Zhang, C.; Yang, Q.; Yao, P.; Jiang, Q.; Wu, J.; Zhao, S. The Increasing Expression of GPX7 Related to the Malignant Clinical Features Leading to Poor Prognosis of Glioma Patients. Chin. Neurosurg. J. 2021, 7, 21. [Google Scholar] [CrossRef]
- Zhao, Q.; Zhang, L.; Wang, Y.; Sun, Y.; Wang, T.; Cao, J.; Qi, M.; Du, X.; Xia, Z.; Zhang, R.; et al. A Bioinformatic Analysis: The Overexpression and Prognostic Potential of GPX7 in Lower-Grade Glioma. Int. J. Gen. Med. 2022, 15, 4321–4337. [Google Scholar] [CrossRef]
- Ramming, T.; Hansen, H.G.; Nagata, K.; Ellgaard, L.; Appenzeller-Herzog, C. GPx8 Peroxidase Prevents Leakage of H2O2 from the Endoplasmic Reticulum. Free Radic. Biol. Med. 2014, 70, 106–116. [Google Scholar] [CrossRef]
- Yoboue, E.D.; Rimessi, A.; Anelli, T.; Pinton, P.; Sitia, R. Regulation of Calcium Fluxes by GPX8, a Type-II Transmembrane Peroxidase Enriched at the Mitochondria-Associated Endoplasmic Reticulum Membrane. Antioxid. Redox Signal. 2017, 27, 583–595. [Google Scholar] [CrossRef]
- Khatib, A.; Solaimuthu, B.; Yosef, M.B.; Rmaileh, A.A.; Tanna, M.; Oren, G.; Frisch, M.S.; Axelrod, J.H.; Lichtenstein, M.; Shaul, Y.D. The Glutathione Peroxidase 8 (GPX8)/IL-6/STAT3 Axis Is Essential in Maintaining an Aggressive Breast Cancer Phenotype. Proc. Natl. Acad. Sci. USA 2020, 117, 21420–21431. [Google Scholar] [CrossRef]
- Yang, Z.; Qin, Y.; Sun, X.; Xiong, K.; Zhu, X.; Wang, Y.; Ren, Q.-Y.; Wu, G.; Shi-Min, W.; Cao, X.; et al. GPX8 as a Novel Prognostic Factor and Potential Therapeutic Target in Primary Glioma. J. Immunol. Res. 2022, 2022, 8025055. [Google Scholar] [CrossRef]
- Zhang, X.; Xu, H.; Zhang, Y.; Sun, C.-Y.; Li, Z.; Hu, C.; Zhao, D.; Guo, C. Immunohistochemistry and Bioinformatics Identify GPX8 as a Potential Prognostic Biomarker and Target in Human Gastric Cancer. Front. Oncol. 2022, 12, 878546. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.M.; Nguyen, T.H.; Kim, H.S.; Dao, T.T.P.; Moon, Y.; Seo, M.; Kang, S.-O.; Mai, V.-H.; An, Y.; Jung, C.; et al. GPX8 Regulates Clear Cell Renal Cell Carcinoma Tumorigenesis through Promoting Lipogenesis by NNMT. J. Exp. Clin. Cancer Res. 2023, 42, 42. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Han, T.; Yan, D.; Liang, C.; Gao, L.; Liu, Y.; Zhou, J.; Guo, J.; Ge, D.; Wu, J.; et al. GPX8+ Cancer-Associated Fibroblast, as a Cancer-Promoting Factor in Lung Adenocarcinoma, Is Related to the Immunosuppressive Microenvironment. BMC Med. Genom. 2024, 17, 77. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Flanagan, J.U.; Jowsey, I.R. Glutathione transferases. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 51–88. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ye, Z.-W.; Morgenstern, R.; Townsend, D.M.; Tew, K.D. Microsomal Glutathione Transferase 1 in Cancer and the Regulation of Ferroptosis. Adv. Cancer Res. 2023, 160, 107–132. [Google Scholar] [CrossRef] [PubMed]
- Morel, F.; Aninat, C. The Glutathione Transferase Kappa Family. Drug Metab. Rev. 2011, 43, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, H.J.; Babbitt, P.C. Glutathione Transferases Are Structural and Functional Outliers in the Thioredoxin Fold. Biochemistry 2009, 48, 11108–11116. [Google Scholar] [CrossRef] [PubMed]
- Board, P.G.; Coggan, M.; Chelvanayagam, G.; Easteal, S.; Jermiin, L.S.; Schulte, G.K.; Danley, D.E.; Hoth, L.R.; Griffor, M.C.; Kamath, A.V.; et al. Identification, Characterization, and Crystal Structure of the Omega Class Glutathione Transferases. J. Biol. Chem. 2000, 275, 24798–24806. [Google Scholar] [CrossRef] [PubMed]
- Dourado, D.; Fernandes, P.; Ramos, M. Mammalian Cytosolic Glutathione Transferases. Curr. Protein Pept. Sci. 2008, 9, 325–337. [Google Scholar] [CrossRef]
- Dourado, D.F.a.R.; Fernandes, P.A.; Mannervik, B.; Ramos, M.J. Glutathione Transferase: New Model for Glutathione Activation. Chemistry 2008, 14, 9591–9598. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.R.; Reindl, K.M. Glutathione S-Transferases in Cancer. Antioxidants 2021, 10, 701. [Google Scholar] [CrossRef] [PubMed]
- Ketterer, B. Detoxication Reactions of Glutathione and Glutathione Transferases. Xenobiotica 1986, 16, 957–973. [Google Scholar] [CrossRef]
- Singhal, S.S.; Singh, S.P.; Singhal, P.; Horne, D.; Singhal, J.; Awasthi, S. Antioxidant Role of Glutathione S-Transferases: 4-Hydroxynonenal, a Key Molecule in Stress-Mediated Signaling. Toxicol. Appl. Pharmacol. 2015, 289, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Grussy, K.; Łaska, M.; Moczurad, W.; Król-Kulikowska, M.; Ściskalska, M. The Importance of Polymorphisms in the Genes Encoding Glutathione S-Transferase Isoenzymes in Development of Selected Cancers and Cardiovascular Diseases. Mol. Biol. Rep. 2023, 50, 9649–9661. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.R.; Chen, J.C.; Li, M.D.; Li, T.; Tan, Y.; Zhang, M. A meta-analysis of glutathione S-transferase M1 and T1 genetic polymorphism in relation to susceptibility to nasopharyngeal carcinoma. Int. J. Clin. Exp. Med. 2015, 8, 10626–10632. [Google Scholar] [PubMed]
- Lalosevic, M.S.; Coric, V.; Pekmezovic, T.; Simic, T.; Markovic, A.P.; Ercegovac, M.P. GSTM1 and GSTP1 Polymorphisms Affect Outcome in Colorectal Adenocarcinoma. Medicina 2024, 60, 553. [Google Scholar] [CrossRef] [PubMed]
- Lalosevic, M.L.j.S.; Coric, V.M.; Pekmezovic, T.D.; Simic, T.P.; Ercegovac, M.S.P.; Markovic, A.R.P.; Krivokapic, Z.V. Deletion and Single Nucleotide Polymorphisms in Common Glutathione-S Transferases Contribute to Colorectal Cancer Development. Pathol. Oncol. Res. 2019, 25, 1579–1587. [Google Scholar] [CrossRef] [PubMed]
- Elderdery, A.Y.; Idris, H.M.E.; Tebien, E.M.; Diab, N.A.; Hamza, S.M.A.; Suliman, B.A.; Alhamidi, A.H.; Omer, N.E.; Mills, J. Impact of GSTT1 and GSTM1 Polymorphisms in the Susceptibility to Philadelphia Negative Chronic Myeloid Leukaemia. Curr. Cancer Drug Targets 2023, 23, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Board, P.G.; Menon, D. Structure, Function and Disease Relevance of Omega-Class Glutathione Transferases. Arch Toxicol. 2016, 90, 1049–1067. [Google Scholar] [CrossRef]
- Petrovic, M.; Simic, T.; Djukic, T.; Radic, T.; Savic-Radojevic, A.; Zekovic, M.; Durutovic, O.; Janicic, A.; Milojevic, B.; Kajmakovic, B.; et al. The Polymorphisms in GSTO Genes (GSTO1 Rs4925, GSTO2 Rs156697, and GSTO2 Rs2297235) Affect the Risk for Testicular Germ Cell Tumor Development: A Pilot Study. Life 2023, 13, 1269. [Google Scholar] [CrossRef]
- Xu, Y.-T.; Wang, J.; Yin, R.; Qiu, M.-T.; Xu, L.; Wang, J.; Xu, L. Genetic Polymorphisms in Glutathione S-Transferase Omega (GSTO) and Cancer Risk: A Meta-Analysis of 20 Studies. Sci. Rep. 2014, 4, 6578. [Google Scholar] [CrossRef] [PubMed]
- Simic, P.; Coric, V.; Pljesa, I.; Savic-Radojevic, A.; Zecevic, N.; Kocic, J.; Simic, T.; Pazin, V.; Pljesa-Ercegovac, M. The Role of Glutathione Transferase Omega-Class Variant Alleles in Individual Susceptibility to Ovarian Cancer. Int. J. Mol. Sci. 2024, 25, 4986. [Google Scholar] [CrossRef] [PubMed]
- Piacentini, S.; Monaci, P.M.; Polimanti, R.; Manfellotto, D.; Fuciarelli, M. GSTO2*N142D Gene Polymorphism Associated with Hypothyroidism in Italian Patients. Mol. Biol. Rep. 2012, 40, 1967–1971. [Google Scholar] [CrossRef] [PubMed]
- Tiongco, R.E.; Cayanan, N.D.; Catacata, M.; Dominguez, M.J. Ile105Val Polymorphism in the GSTP1 Gene Is Associated with Susceptibility to Acute Myeloid Leukemia: An Updated Systematic Review and Meta-Analysis. Biomarkers 2024, 29, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Feroz, Z.; Tiwari, S.; Vijayaraghavalu, S.; Kumar, M. GSTs Genetic Polymorphism, Gene–Environment Interaction and Association with Gallbladder Cancer Risk in North Indian Population: A Case-Controlled Study. J. Cancer Res. Ther. 2023, 19, 1908–1914. [Google Scholar] [CrossRef]
- Kuang, M.; Xu, W.; Cao, C.X.; Shen, L.L.; Chang, J.; Zhang, X.L.; Chen, J.F.; Tang, C.J. Glutathione S-Transferase P1 Rs1695 A>G Polymorphism and Breast Cancer Risk: Evidence from a Meta-Analysis. Genet. Mol. Res. 2016, 15, 10–4238. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Cho, Y.-A.; Kim, D.-C.; Lee, K.-E. Association between Genetic Polymorphism of GSTP1 and Toxicities in Patients Receiving Platinum-Based Chemotherapy: A Systematic Review and Meta-Analysis. Pharmaceuticals 2022, 15, 439. [Google Scholar] [CrossRef]
- Jiao, H.; Song, A.; Cheng, L.; Zhou, D.; Luan, J.; Lin, H.; Zhang, Z. Association between GSTP1 I105V Polymorphisms and Responses to GSTP1 Inhibitor Treatment: In Silico and in Vitro Insights. J. Biomol. Struct. Dyn. 2024, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kudhair, B.K.; Alabid, N.N.; Taheri-Kafrani, A.; Lafta, I.J. Correlation of GSTP1 Gene Variants of Male Iraqi Waterpipe (Hookah) Tobacco Smokers and the Risk of Lung Cancer. Mol. Biol. Rep. 2020, 47, 2677–2684. [Google Scholar] [CrossRef]
- Wang, Z.; Qu, K.; Niu, W.; Lin, T.; Xu, X.; Huang, Z.; Liu, S.; Liu, S.; Chang, H.; Liu, Y.; et al. Glutathione S-Transferase P1 Gene Rs4147581 Polymorphism Predicts Overall Survival of Patients with Hepatocellular Carcinoma: Evidence from an Enlarged Study. Tumor Biol. 2015, 37, 943–952. [Google Scholar] [CrossRef]
- Santric, V.; Djokic, M.; Suvakov, S.; Pljesa-Ercegovac, M.; Nikitovic, M.; Radic, T.; Acimovic, M.; Stankovic, V.; Bumbasirevic, U.; Milojevic, B.; et al. GSTP1 Rs1138272 Polymorphism Affects Prostate Cancer Risk. Medicina 2020, 56, 128. [Google Scholar] [CrossRef]
- Cui, J.; Li, G.; Yin, J.; Li, L.; Tan, Y.; Wei, H.; Liu, B.; Deng, L.; Tang, J.; Chen, Y.; et al. GSTP1 and Cancer: Expression, Methylation, Polymorphisms and Signaling (Review). Int. J. Oncol. 2020, 56, 867–878. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.M.; Tew, K.D.; He, L.; King, J.B.; Hanigan, M.H. Role of Glutathione S-Transferase Pi in Cisplatin-Induced Nephrotoxicity. Biomed. Pharmacother. 2009, 63, 79–85. [Google Scholar] [CrossRef]
- Jenderny, S.; Lin, H.; Garrett, T.; Tew, K.D.; Townsend, D.M. Protective Effects of a Glutathione Disulfide Mimetic (NOV-002) against Cisplatin Induced Kidney Toxicity. Biomed. Pharmacother. 2010, 64, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Tew, K.D.; Monks, A.; Barone, L.; Rosser, D.; Akerman, G.; Montali, J.A.; Wheatley, J.B.; Schmidt, D.E., Jr. Glutathione-associated enzymes in the human cell lines of the National Cancer Institute Drug Screening Program. Mol. Pharmacol. 1996, 50, 149–159. [Google Scholar]
- Mousseau, M.; Chauvin, C.; Nissou, M.F.; Chaffanet, M.; Plantaz, D.; Pasquier, B.; Schaerer, R.; Benabid, A. A study of the expression of four chemoresistance-related genes in human primary and metastatic brain tumours. Eur. J. Cancer 1993, 29, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liang, S.; Lian, X.; Liu, L.; Zhao, S.; Xuan, Q.; Guo, L.; Liu, H.; Yang, Y.; Dong, T.; et al. Identification of Proteins Responsible for Adriamycin Resistance in Breast Cancer Cells Using Proteomics Analysis. Sci. Rep. 2015, 5, 9301. [Google Scholar] [CrossRef]
- Dong, X.; Yang, Y.; Zhou, Y.; Bi, X.; Zhao, N.; Zhang, Z.; Li, L.; Hang, Q.; Zhang, R.; Chen, D.; et al. Glutathione S-Transferases P1 Protects Breast Cancer Cell from Adriamycin-Induced Cell Death through Promoting Autophagy. Cell Death Differ. 2019, 26, 2086–2099. [Google Scholar] [CrossRef] [PubMed]
- Gurioli, G.; Martignano, F.; Salvi, S.; Costantini, M.; Gunelli, R.; Casadio, V. GSTP1 Methylation in Cancer: A Liquid Biopsy Biomarker? Clin. Chem. Lab. Med. 2018, 56, 702–717. [Google Scholar] [CrossRef]
- Ye, J.; Wu, M.; He, L.; Chen, P.; Liu, H.; Yang, H. Glutathione-S-Transferase p1 Gene Promoter Methylation in Cell-Free DNA as a Diagnostic and Prognostic Tool for Prostate Cancer: A Systematic Review and Meta-Analysis. Int. J. Endocrinol. 2023, 2023, 7279243. [Google Scholar] [CrossRef]
- Danos, P.; Giannoni-Luza, S.; Carrasco, A.G.M.; Acosta, O.; Guevara-Fujita, M.L.; Concha, J.M.C.; Miller, H.G.; Oblitas, J.P.; Cartagena, A.A.; Araujo, J.M.; et al. Promoter Hypermethylation of RARB and GSTP1 Genes in Plasma Cell-free DNA as Breast Cancer Biomarkers in Peruvian Women. Mol. Genet. Genom. Med. 2023, 11, e2260. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Arifoglu, P.; Ronai, Z.; Tew, K.D. Glutathione S-Transferase P1–1 (GSTP1–1) Inhibits c-Jun N-Terminal Kinase (JNK1) Signaling through Interaction with the C Terminus. J. Biol. Chem. 2001, 276, 20999–21003. [Google Scholar] [CrossRef] [PubMed]
- Tew, K.D.; Townsend, D.M. Glutathione-S-Transferases as Determinants of Cell Survival and Death. Antioxid. Redox Signal. 2012, 17, 1728–1737. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Fan, Y.; Xue, B.; Luo, L.; Shen, J.; Zhang, S.; Jiang, Y.; Yin, Z. Human Glutathione S-Transferase P1-1 Interacts with TRAF2 and Regulates TRAF2–ASK1 Signals. Oncogene 2006, 25, 5787–5800. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.; Zhao, S.; Li, J.; Liu, K.; Ma, X.; Zhang, Z.; Wang, R.; Tian, J.; Liu, F.; Song, Y.; et al. NUMA1 Modulates Apoptosis of Esophageal Squamous Cell Carcinoma Cells through Regulating ASK1-JNK Signaling Pathway. Cell Mol. Life Sci. 2023, 80, 211. [Google Scholar] [CrossRef] [PubMed]
- Dorion, S.; Lambert, H.; Landry, J. Activation of the P38 Signaling Pathway by Heat Shock Involves the Dissociation of Glutathione S-Transferase Mu from Ask1. J. Biol. Chem. 2002, 277, 30792–30797. [Google Scholar] [CrossRef]
- Romero, L.; Andrews, K.; Ng, L.; O’Rourke, K.; Maslen, A.; Kirby, G. Human GSTA1-1 Reduces c-Jun N-Terminal Kinase Signalling and Apoptosis in Caco-2 Cells. Biochem. J. 2006, 400, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Sui, X.; Zhang, C.; Wei, K.; Bao, Y.; Xiong, J.; Zhou, Z.; Chen, Z.; Wang, C.; Zhu, H.; et al. Glutathione S-Transferase A1 Suppresses Tumor Progression and Indicates Better Prognosis of Human Primary Hepatocellular Carcinoma. J. Cancer 2020, 11, 83–91. [Google Scholar] [CrossRef]
- Saisawang, C.; Wongsantichon, J.; Robinson, R.C.; Ketterman, A.J. Glutathione Transferase Omega 1-1 (GSTO1-1) Modulates Akt and MEK1/2 Signaling in Human Neuroblastoma Cell SH-SY5Y. Proteins 2019, 87, 588–595. [Google Scholar] [CrossRef]
- Robin, S.K.D.; Ansari, M.; Uppugunduri, C.R.S. Spectrophotometric Screening for Potential Inhibitors of Cytosolic Glutathione S-Transferases. J. Vis. Exp. 2020, 164, e61347. [Google Scholar] [CrossRef]
- Allocati, N.; Masulli, M.; Di Ilio, C.; Federici, L. Glutathione Transferases: Substrates, Inihibitors and pro-Drugs in Cancer and Neurodegenerative Diseases. Oncogenesis 2018, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, S.; Srivastava, S.K.; Ahmad, F.; Ahmad, H.; Ansari, G.A.S. Interactions of Glutathione S-Transferase-π with Ethacrynic Acid and Its Glutathione Conjugate. Biochim. Biophys. Acta 1993, 1164, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Mulder, G.J.; Ouwerkerk-Mahadevan, S. Modulation of Glutathione Conjugation in Vivo: How to Decrease Glutathione Conjugation in Vivo or in Intact Cellular Systems in Vitro. Chem. Biol. Interact. 1997, 105, 17–34. [Google Scholar] [CrossRef] [PubMed]
- Turella, P.; Cerella, C.; Filomeni, G.; Bullo, A.; De Maria, F.; Ghibelli, L.; Ciriolo, M.R.; Cianfriglia, M.; Mattei, M.; Federici, G.; et al. Proapoptotic Activity of New Glutathione S-Transferase Inhibitors. Cancer Res. 2005, 65, 3751–3761. [Google Scholar] [CrossRef] [PubMed]
- Turella, P.; Filomeni, G.; Dupuis, M.L.; Ciriolo, M.R.; Molinari, A.; De Maria, F.; Tombesi, M.; Cianfriglia, M.; Federici, G.; Ricci, G.; et al. A Strong Glutathione S-Transferase Inhibitor Overcomes the P-Glycoprotein-Mediated Resistance in Tumor Cells. J. Biol. Chem. 2006, 281, 23725–23732. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Mei, G.; Rosato, N.; Nicolai, E.; Federici, L.; Palumbo, C.; Pastore, A.; Serra, M.; Caccuri, A.M. The Fine-Tuning of TRAF2–GSTP1-1 Interaction: Effect of Ligand Binding and in Situ Detection of the Complex. Cell Death Dis. 2014, 5, e1015. [Google Scholar] [CrossRef] [PubMed]
- Sha, H.; Zou, R.; Lu, Y.; Gan, Y.; Ma, R.; Feng, J.; Chen, D. NBDHEX Re-sensitizes Adriamycin-resistant Breast Cancer by Inhibiting Glutathione S-transferase Pi. Cancer Med. 2022, 12, 5833–5845. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, D.; Sutton, G.R. Ezatiostat Hydrochloride for the Treatment of Myelodysplastic Syndromes. Expert. Opin. Investig. Drugs 2015, 24, 725–733. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, M.L.; Vulevic, B.; Freer, S.; Boyd, J.; Shen, H.; Tew, K.D. Glutathione peptidomimetic drug modulator of multidrug resistance-associated protein. J. Pharmacol. Exp. Ther. 1999, 291, 1348–1355. [Google Scholar]
- Zhang, J.; Ye, Z.-W.; Janssen-Heininger, Y.; Townsend, D.M.; Tew, K.D. Development of Telintra as an Inhibitor of Glutathione S-Transferase P. Handb. Exp. Pharmacol. 2021, 264, 71–91. [Google Scholar] [CrossRef]
- Lyttle, M.H.; Satyam, A.; Hocker, M.D.; Bauer, K.E.; Caldwell, C.G.; Hui, H.C.; Morgan, A.S.; Mergia, A.; Kauvar, L.M. Glutathione-S-Transferase Activates Novel Alkylating Agents. J. Med. Chem. 1994, 37, 1501–1507. [Google Scholar] [CrossRef] [PubMed]
- Dourado, D.F.a.R.; Fernandes, P.A.; Ramos, M.J.; Mannervik, B. Mechanism of Glutathione Transferase P1-1-Catalyzed Activation of the Prodrug Canfosfamide (TLK286, TELCYTA). Biochemistry 2013, 52, 8069–8078. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh, J.J.; Gershenson, D.M.; Choi, H.; Lewis, L.; Patel, K.; Brown, G.L.; Garcia, A.; Spriggs, D.R. Multi-Institutional Phase 2 Study of TLK286 (TELCYTATM, a Glutathione S-Transferase P1-1 Activated Glutathione Analog Prodrug) in Patients with Platinum and Paclitaxel Refractory or Resistant Ovarian Cancer. Int. J. Gynecol. Cancer. 2005, 15, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Vergote, I.; Finkler, N.; Del Campo, J.; Lohr, A.; Hunter, J.; Matei, D.; Kavanagh, J.; Vermorken, J.B.; Meng, L.; Jones, M.; et al. Phase 3 Randomised Study of Canfosfamide (Telcyta®, TLK286) versus Pegylated Liposomal Doxorubicin or Topotecan as Third-Line Therapy in Patients with Platinum-Refractory or -Resistant Ovarian Cancer. Eur. J. Cancer 2009, 45, 2324–2332. [Google Scholar] [CrossRef] [PubMed]
- Tew, K.D. TLK-286: A Novel glutathioneS-Transferase-Activated Prodrug. Expert. Expert. Opin. Investig. Drugs 2005, 14, 1047–1054. [Google Scholar] [CrossRef]
- Ismail, A.; Govindarajan, S.; Mannervik, B. Human GST P1-1 Redesigned for Enhanced Catalytic Activity with the Anticancer Prodrug Telcyta and Improved Thermostability. Cancers 2024, 16, 762. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ye, Z.; Singh, S.; Townsend, D.M.; Tew, K.D. An Evolving Understanding of the S-Glutathionylation Cycle in Pathways of Redox Regulation. Free Radic Biol. Med. 2018, 120, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Chai, Y.C.; Mieyal, J.J. Glutathione and Glutaredoxin—Key Players in Cellular Redox Homeostasis and Signaling. Antioxidants 2023, 12, 1553. [Google Scholar] [CrossRef] [PubMed]
- Oppong, D.; Schiff, W.M.; Shivamadhu, M.C.; Ahn, Y. Chemistry and Biology of Enzymes in Protein Glutathionylation. Curr. Opin. Chem. Biol. 2023, 75, 102326. [Google Scholar] [CrossRef]
- Bechtel, T.J.; Weerapana, E. From Structure to Redox: The Diverse Functional Roles of Disulfides and Implications in Disease. Proteomics 2017, 17, 10. [Google Scholar] [CrossRef]
- Ghezzi, P. Regulation of Protein Function by Glutathionylation. Free Radic. Res. 2005, 39, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Janssen-Heininger, Y.; Nolin, J.D.; Hoffman, S.M.; Van Der Velden, J.; Tully, J.E.; Lahue, K.G.; Abdalla, S.; Chapman, D.G.; Reynaert, N.L.; Van Der Vliet, A.; et al. Emerging Mechanisms of Glutathione-dependent Chemistry in Biology and Disease. J. Cell. Biochem. 2013, 114, 1962–1968. [Google Scholar] [CrossRef]
- Atkins, W.M.; Wang, R.W.; Bird, A.W.; Newton, D.J.; Lu, A.Y.H. The Catalytic Mechanism of Glutathione S-Transferase (GST). Spectroscopic Determination of the pKa of Tyr-9 in Rat Alpha 1-1 GST. J. Biol. Chem. 1993, 268, 19188–19191. [Google Scholar] [CrossRef]
- Arnér, E.S.J.; Holmgren, A. Physiological Functions of Thioredoxin and Thioredoxin Reductase. Eur. J. Biochem. 2000, 267, 6102–6109. [Google Scholar] [CrossRef]
- Findlay, V.J.; Townsend, D.M.; Morris, T.E.; Fraser, J.P.; He, L.; Tew, K.D. A Novel Role for Human Sulfiredoxin in the Reversal of Glutathionylation. Cancer Res. 2006, 66, 6800–6806. [Google Scholar] [CrossRef] [PubMed]
- Gallogly, M.; Mieyal, J.J. Mechanisms of Reversible Protein Glutathionylation in Redox Signaling and Oxidative Stress. Curr. Opin. Pharmacol. 2007, 7, 381–391. [Google Scholar] [CrossRef]
- Ogata, F.T.; Branco, V.; Vale, F.F.; Coppo, L. Glutaredoxin: Discovery, Redox Defense and Much More. Redox Biol. 2021, 43, 101975. [Google Scholar] [CrossRef] [PubMed]
- Beer, S.M.; Taylor, E.; Brown, S.; Dahm, C.C.; Costa, N.J.; Runswick, M.J.; Murphy, M.P. Glutaredoxin 2 Catalyzes the Reversible Oxidation and Glutathionylation of Mitochondrial Membrane Thiol Proteins. J. Biol. Chem. 2004, 279, 47939–47951. [Google Scholar] [CrossRef]
- Ukuwela, A.A.; Bush, A.I.; Wedd, A.G.; Xiao, Z. Reduction Potentials of Protein Disulfides and Catalysis of Glutathionylation and Deglutathionylation by Glutaredoxin Enzymes. Biochem. J. 2017, 474, 3799–3815. [Google Scholar] [CrossRef]
- Pal, D.; Rai, A.; Checker, R.; Patwardhan, R.S.; Singh, B.; Sharma, D.; Sandur, S.K. Role of Protein S-Glutathionylation in Cancer Progression and Development of Resistance to Anti-Cancer Drugs. Arch. Biochem. Biophys. 2021, 704, 108890. [Google Scholar] [CrossRef]
- Peltoniemi, M.; Karala, A.; Jurvansuu, J.; Kinnula, V.L.; Ruddock, L.W. Insights into Deglutathionylation Reactions. J. Biol. Chem. 2006, 281, 33107–33114. [Google Scholar] [CrossRef]
- Brzozowa-Zasada, M.; Piecuch, A.; Bajdak-Rusinek, K.; Gołąbek, K.; Michalski, M.; Matysiak, N.; Czuba, Z. A Prognostic Activity of Glutaredoxin 1 Protein (GRX1) in Colon Cancer. Int. J. Mol. Sci. 2024, 25, 1007. [Google Scholar] [CrossRef] [PubMed]
- Cha, M.; Kim, I.-H. Preferential Overexpression of Glutaredoxin3 in Human Colon and Lung Carcinoma. Cancer Epidemiol. 2009, 33, 281–287. [Google Scholar] [CrossRef]
- Li, B.; Chen, M.; Lu, M.; Jiang, X.-X.; Pan, M.; Mao, J.-W. Glutaredoxin 3 Promotes Migration and Invasion via the Notch Signalling Pathway in Oral Squamous Cell Carcinoma. Free Radic. Res. 2018, 52, 390–401. [Google Scholar] [CrossRef]
- He, F.; Wei, L.; Luo, W.; Liao, Z.; Li, B.; Zhou, X.; Xiao, X.; You, J.; Chen, Y.; Zheng, S.; et al. Glutaredoxin 3 Promotes Nasopharyngeal Carcinoma Growth and Metastasis via EGFR/Akt Pathway and Independent of ROS. Oncotarget 2016, 7, 37000–37012. [Google Scholar] [CrossRef]
- Park, J.W.; Mieyal, J.J.; Rhee, S.G.; Chock, P.B. Deglutathionylation of 2-Cys Peroxiredoxin Is Specifically Catalyzed by Sulfiredoxin. J. Biol. Chem. 2009, 284, 23364–23374. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.; Jiang, H.; Wu, L.; Chawsheen, H.A.; Wei, Q. The Sulfiredoxin–Peroxiredoxin (Srx–Prx) Axis in Cell Signal Transduction and Cancer Development. Cancer Lett. 2015, 366, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Menon, D.; Board, P.G. A Role for Glutathione Transferase Omega 1 (GSTO1-1) in the Glutathionylation Cycle. J. Biol. Chem. 2013, 288, 25769–25779. [Google Scholar] [CrossRef]
- Hughes, M.; Hooftman, A.; Angiari, S.; Tummala, P.; Zasłona, Z.; Runtsch, M.C.; McGettrick, A.F.; Sutton, C.E.; Diskin, C.; Rooke, M.; et al. Glutathione Transferase Omega-1 Regulates NLRP3 Inflammasome Activation through NEK7 Deglutathionylation. Cell Rep. 2019, 29, 151–161. [Google Scholar] [CrossRef]
- Tew, K.D.; Manevich, Y.; Grek, C.L.; Xiong, Y.; Uys, J.D.; Townsend, D.M. The Role of Glutathione S-Transferase P in Signaling Pathways and S-Glutathionylation in Cancer. Free Radic. Biol. Med. 2011, 51, 299–313. [Google Scholar] [CrossRef]
- Webb, B.A.; Chimenti, M.S.; Jacobson, M.P.; Barber, D.L. Dysregulated pH: A Perfect Storm for Cancer Progression. Nat. Rev. Cancer 2011, 11, 671–677. [Google Scholar] [CrossRef]
- Mailloux, R.J. Protein S-Glutathionylation Reactions as a Global Inhibitor of Cell Metabolism for the Desensitization of Hydrogen Peroxide Signals. Redox Biol. 2020, 32, 101472. [Google Scholar] [CrossRef] [PubMed]
- Van Der Velden, J.; Kinsey, M.; Chia, S.B.; Lahue, K.G.; Qian, X.; Janssen-Heininger, Y. GSTP1-Catalyzed PKM2 S-Glutathionylation Regulates Glycolysis in Non-Small Cell Lung Cancer and Is Attenuated with a Clinically Relevant Inhibitor of Glutathione-S-Transferase P. Free Radic Biol. Med. 2017, 112, 104–105. [Google Scholar] [CrossRef]
- Adachi, T.; Pimentel, D.R.; Heibeck, T.H.; Hou, X.; Lee, Y.J.; Jiang, B.; Ido, Y.; Cohen, R.A. S-Glutathiolation of RAS Mediates Redox-Sensitive Signaling by Angiotensin II in Vascular Smooth Muscle Cells. J. Biol. Chem. 2004, 279, 29857–29862. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Dong, X.; Zheng, S.; Sun, J.; Ye, J.; Chen, J.; Fang, Y.; Zhao, B.; Yin, Z.; Cao, P.; et al. GSTpi Regulates VE-Cadherin Stabilization through Promoting S-Glutathionylation of Src. Redox Biol. 2020, 30, 101416. [Google Scholar] [CrossRef]
- Abdelsaid, M.; El-Remessy, A.B. S-Glutathionylation of LMW-PTP Regulates VEGF-Mediated FAK Activation and Endothelial Cell Migration. J. Cell Sci. 2012, 125, 4751–4760. [Google Scholar] [CrossRef] [PubMed]
- Sakai, J.; Li, J.; Subramanian, K.K.; Mondal, S.; Bajrami, B.; Hattori, H.; Jia, Y.; Dickinson, B.C.; Zhong, J.; Ye, K.; et al. Reactive Oxygen Species-Induced Actin Glutathionylation Controls Actin Dynamics in Neutrophils. Immunity 2012, 37, 1037–1049. [Google Scholar] [CrossRef] [PubMed]
- Velu, C.S.; Niture, S.K.; Doneanu, C.E.; Pattabiraman, N.; Srivenugopal, K.S. Human P53 Is Inhibited by Glutathionylation of Cysteines Present in the Proximal DNA-Binding Domain during Oxidative Stress. Biochemistry 2007, 46, 7765–7780. [Google Scholar] [CrossRef] [PubMed]
- Seo, M.; Lee, Y.H. PFKFB3 Regulates Oxidative Stress Homeostasis via Its S-Glutathionylation in Cancer. J. Mol. Biol. 2014, 426, 830–842. [Google Scholar] [CrossRef]
- Huang, Z.; Pinto, J.T.; Deng, H.; Richie, J.P. Inhibition of Caspase-3 Activity and Activation by Protein Glutathionylation. Biochem. Pharmacol. 2008, 75, 2234–2244. [Google Scholar] [CrossRef]
- Humphries, K.M.; Juliano, C.E.; Taylor, S.S. Regulation of CAMP-Dependent Protein Kinase Activity by Glutathionylation. J. Biol. Chem. 2002, 277, 43505–43511. [Google Scholar] [CrossRef] [PubMed]
- Kawano, T.; Inokuchi, J.; Eto, M.; Murata, M.; Kang, J.H. Protein Kinase C (PKC) Isozymes as Diagnostic and Prognostic Biomarkers and Therapeutic Targets for Cancer. Cancers 2022, 14, 5425. [Google Scholar] [CrossRef] [PubMed]
- Benavides, F.; Blando, J.; Pérez, C.F.; Garg, R.; Conti, C.J.; DiGiovanni, J.; Kazanietz, M.G. Transgenic Overexpression of PKCε in the Mouse Prostate Induces Preneoplastic Lesions. Cell Cycle 2011, 10, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Gutiérrez-Uzquiza, Á.; Garg, R.; Barrio-Real, L.; Abera, M.B.; López-Haber, C.; Rosemblit, C.; Lu, H.; Abba, M.C.; Kazanietz, M.G. Transcriptional Regulation of Oncogenic Protein Kinase CΕ (PKCΕ) by STAT1 and SP1 Proteins. J. Biol. Chem. 2014, 289, 19823–19838. [Google Scholar] [CrossRef] [PubMed]
- Ward, N.E.; Stewart, J.R.; Ioannides, C.G.; O’Brian, C.A. Oxidant-Induced S-Glutathiolation Inactivates Protein Kinase C-A (PKC-A): A Potential Mechanism of PKC Isozyme Regulation. Biochemistry 2000, 39, 10319–10329. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.; Clayton, L. Regulation of Protein Phosphatase 2A by Hydrogen Peroxide and Glutathionylation. Biochem. Biophys. Res. Commun. 2002, 293, 610–616. [Google Scholar] [CrossRef]
- Liu, T.; Wang, X.; Zhang, L. [The Correlation between the up-Regulation of Hsp90 and Drug Resistance to Cisplatin in Lung Cancer Cell Line]. Zhongguo Fei Ai Za Zhi 2011, 14, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Tang, J.; Sun, W.; Wang, H. PGK1 Facilities Cisplatin Chemoresistance by Triggering HSP90/ERK Pathway Mediated DNA Repair and Methylation in Endometrial Endometrioid Adenocarcinoma. Mol. Med. 2019, 25, 11. [Google Scholar] [CrossRef]
- Shih, Y.-Y.; Lin, H.; Jan, H.; Chen, Y.; Ong, L.-L.; Yu, A.L.; Lin, C. S-Glutathionylation of Hsp90 Enhances Its Degradation and Correlates with Favorable Prognosis of Breast Cancer. Redox Biol. 2022, 57, 102501. [Google Scholar] [CrossRef]
- Zhang, J.; Ye, Z.; Chen, W.; Culpepper, J.; Jiang, H.; Ball, L.E.; Mehrotra, S.; Blumental-Perry, A.; Tew, K.D.; Townsend, D.M. Altered Redox Regulation and S-Glutathionylation of BiP Contribute to Bortezomib Resistance in Multiple Myeloma. Free Radic. Biol. Med. 2020, 160, 755–767. [Google Scholar] [CrossRef]
- Pfefferle, A.; Mailloux, R.J.; Adjeitey, C.N.K.; Harper, M. Glutathionylation of UCP2 Sensitizes Drug Resistant Leukemia Cells to Chemotherapeutics. Biochim. Biophys. Acta. 2013, 1833, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ludden, C.; Cullen, A.J.; Tew, K.D.; De Barros, A.L.B.; Townsend, D.M. Nuclear Factor Kappa B Expression in Non-Small Cell Lung Cancer. Biomed. Pharmacother. 2023, 167, 115459. [Google Scholar] [CrossRef] [PubMed]
- Butturini, E.; De Prati, A.C.; Boriero, D.; Mariotto, S. Natural Sesquiterpene Lactones Enhance Chemosensitivity of Tumor Cells through Redox Regulation of STAT3 Signaling. Oxid. Med. Cell Longev. 2019, 2019, 4568964. [Google Scholar] [CrossRef]
- Butturini, E.; De Prati, A.C.; Chiavegato, G.; Rigo, A.; Cavalieri, E.; Darra, E.; Mariotto, S. Mild Oxidative Stress Induces S-Glutathionylation of STAT3 and Enhances Chemosensitivity of Tumoural Cells to Chemotherapeutic Drugs. Free Radic. Biol. Med. 2013, 65, 1322–1330. [Google Scholar] [CrossRef] [PubMed]
- Robertson, H.; Dinkova-Kostova, A.T.; Hayes, J.D. NRF2 and the Ambiguous Consequences of Its Activation during Initiation and the Subsequent Stages of Tumourigenesis. Cancers 2020, 12, 3609. [Google Scholar] [CrossRef] [PubMed]
- Hecht, F.; Zocchi, M.; Alimohammadi, F.; Harris, I.S. Regulation of Antioxidants in Cancer. Mol. Cell. 2024, 84, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Holland, R.J.; Hawkins, A.; Eggler, A.L.; Mesecar, A.D.; Fabris, D.; Fishbein, J.C. Prospective Type 1 and Type 2 Disulfides of KEAP1 Protein. Chem. Res. Toxicol. 2008, 21, 2051–2060. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Tew, K.D. Reductive Stress in Cancer. Adv. Cancer Res. 2021, 152, 383–413. [Google Scholar] [CrossRef] [PubMed]
- Krakowiak, A.; Pietrasik, S. New Insights into Oxidative and Reductive Stress Responses and Their Relation to the Anticancer Activity of Selenium-Containing Compounds as Hydrogen Selenide Donors. Biology 2023, 12, 875. [Google Scholar] [CrossRef]
- Xiao, W.; Loscalzo, J. Metabolic Responses to Reductive Stress. Antioxid. Redox Signal. 2020, 32, 1330–1347. [Google Scholar] [CrossRef]
- Kôrge, P.; Calmettes, G.; Weiss, J.N. Increased Reactive Oxygen Species Production during Reductive Stress: The Roles of Mitochondrial Glutathione and Thioredoxin Reductases. Biochim. Biophys. Acta Bioenerg. 2015, 1847, 514–525. [Google Scholar] [CrossRef] [PubMed]
- Chun, K.; Kim, D.-H.; Surh, Y. Role of Reductive versus Oxidative Stress in Tumor Progression and Anticancer Drug Resistance. Cells 2021, 10, 758. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Human Cell Line | Expression (Tumor vs. Normal) | Roles in Cancer | Target Action | Reference | |
|---|---|---|---|---|---|
| Breast cancer | T47D | ---- | Tumor promoter | GPX1 overexpression inhibits doxorubicin-induced apoptosis | [106] |
| MDA-MB-231, MDA-MB-468, Hs578T, BT-549 | Up (mRNA, protein) | Tumor promoter | GPX1 expression promotes migration and invasion | [111] | |
| MDA-MB-231 | ---- | Tumor promoter | GPx1 silencing increases TNF-α-induced apoptosis | [112] | |
| Kidney cancer | A-498, ACHN, 786-O, CAKI-1 | Up (protein) | Tumor promoter | GPX1 knockdown inhibits proliferation and clonogenic capacity | [113] |
| Glioma | Glioma stem cells U87, SU-2 | Up (mRNA, protein) | Tumor promoter | Increased GPX1 expression decreases ROS levels and increases radioresistance | [114] |
| Lung cancer | A549, H1975, H460, H1650, GLC-82, H1993, H2170, Spc-a1, H1299 | Up (protein, in cisplatin-resistant cell lines) | Tumor promoter | GPX1 overexpression inhibits ROS accumulation and leads to cisplatin resistance | [115] |
| Gastric cancer | SNU-1, -5, -16, -216,-484,- 601, -620, -638, -668, -719 cells | Down (mRNA, protein) | Tumor suppressor | Decreased GPX1 expression predicts aggressiveness, lymphatic invasion, and poor survival | [116] |
| Pancreatic cancer | MiaPaCa-2, SW1990, PANC-1 | ---- | Tumor suppressor | GPX1 overexpression sensitizes cells to starvation-induced cell death via the activation of caspase-dependent apoptosis | [117] |
| CST Class | GST Polymorphism | Cancer Type | Related Mechanism | References |
|---|---|---|---|---|
| GSTM | GSTM1-null | Nasopharyngeal cancer Colorectal cancer | Affects the risk of developing nasopharyngeal carcinoma in the Chinese population Associated with the risk of developing colorectal cancer and shorter survival in colorectal cancer patients | [196,197,198] |
| GSTT | GSTT1-null | Colorectal cancer Leukemia | Affects the risk of developing colorectal cancer Affects the risk of developing Philadelphia-negative chronic myeloid leukemia (Ph-ve CML) | [198,199] |
| GSTO | GSTO2*A/G*G/G and GSTO2*A/G*G/G | Testicular cancer | Associated with an increased risk of testicular germ-cell cancer | [200,201,202] |
| GSTP | GSTP1 Ile105Val GSTP1 rs1695 A>G GSTP1-rs1695 GSTP1 rs4147581 GSTP1*Val rs1695 + GSTP1*Val rs1138272 | Leukemia Breast cancer Lung cancer Liver cancer Prostate cancer | Affects the risk of developing acute myeloid leukemia Increases incidence of breast cancer in Asian women Affects the risk of developing lung cancer Prognostic marker for hepatocellular carcinoma Affects the risk of developing prostate cancer | [203,204,205,206,207,208,209,210,211,212] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalinina, E. Glutathione-Dependent Pathways in Cancer Cells. Int. J. Mol. Sci. 2024, 25, 8423. https://doi.org/10.3390/ijms25158423
Kalinina E. Glutathione-Dependent Pathways in Cancer Cells. International Journal of Molecular Sciences. 2024; 25(15):8423. https://doi.org/10.3390/ijms25158423
Chicago/Turabian StyleKalinina, Elena. 2024. "Glutathione-Dependent Pathways in Cancer Cells" International Journal of Molecular Sciences 25, no. 15: 8423. https://doi.org/10.3390/ijms25158423
APA StyleKalinina, E. (2024). Glutathione-Dependent Pathways in Cancer Cells. International Journal of Molecular Sciences, 25(15), 8423. https://doi.org/10.3390/ijms25158423