1. Introduction
The periodontal ligament (PDL) is a unique thin connective tissue that covers the root of the tooth between the dental alveolar bone and the tooth cementum. The immediate function of the PDL is to attach the tooth to the alveolar bone through a dense network of fibers that allow withstanding the forces of mastication. This ligament tissue has a complex cellular content, made up of fibroblast-like cells, osteoblasts, osteoclasts, epithelial cell rests of Malassez, cementoblasts and odontoclasts, and exhibits highly structured microstructures, such as networks of blood vessels and sensory nerve endings [
1,
2,
3]. In addition, PDL also contains mesenchymal stem cells (MSCs) able to differentiate into osteoblasts, cementoblasts and fibroblasts, allowing for periodontium regeneration and tissue repair [
4,
5]. Beyond its mechanical and regenerative roles, PDL also exhibits immune regulatory functions, eliciting a regulatory immune response to face acute inflammation triggered by periodontal pathogens [
6,
7,
8,
9]. Particularly, PDL cells have been shown to produce various cytokines and chemokines in response to different inflammatory stimuli, indicating that PDL cells are fibroblast-like cells able to act as immune cells [
10].
While the oral cavity harbors a wide variety of pathogenic viruses, virtually nothing is known about the interplay between PDL and viral infections. Nevertheless, PDL exhibits distinct features that may facilitate viral infections, such as its cellular diversity and a vascular network intimately connected to tissue structures. Particularly, the human herpes simplex virus type-1 (HSV-1) is a common oral virus widely distributed in the human population [
11]. Over 70% of the population shed HSV-1 asymptomatically in the oral cavity at least once a month, with many individuals appearing to shed oral HSV-1 more than 6 times per month [
12]. Oral HSV-1 shedding can eventually result in a mild disease, such as herpes labialis, the vesicular lesions on or near the lips that are commonly known as cold sores [
13]. HSV-1 is also capable of causing much more serious illnesses, including herpes stromal keratitis, herpes encephalitis and disseminated neonatal infections [
14,
15]. Recent evidence also suggests a potential contribution of HSV-1 to Alzheimer’s disease [
16].
Over the past decade, several studies have highlighted the likely role of various human herpesviruses (HHVs), including Epstein–Barr virus (EBV), cytomegalovirus and HSV-1, in the pathogenesis of periodontitis [
17,
18]. Recent meta-analysis and examination survey based on large cohorts of patients revealed that HSV-1 was significantly associated with periodontitis, and notably with severe periodontitis [
19,
20]. While infections of epithelial cells and plasma cells have been proposed to support EBV spread in periodontal lesions [
21,
22,
23] the mechanisms supporting HSV-1 periodontal pathogenesis are still elusive. It is well established that HSV-1 primarily exhibits tropism for epithelial cells and fibroblasts, and infects skin and connective dermal tissues. Subsequently, it infects the termini of neurons and travels in a retrograde manner to the neuronal cell body, where it remains in a latent state until reactivated by different stimuli [
24,
25]. Here, we explore the hypothesis that HSV-1 may infect mesenchymal cells originating from PDL, examining the possibility that the PDL may serve as a target tissue capable of sustaining HSV-1 infection in the periodontium.
3. Discussion
In this study, we established a primary cell culture model representative of mesenchymal PDL cells. Although this culture system oversimplifies the PDL tissue complexity, it provides a reliable and suitable model for HSV-1 infection of PDL considering that, apart from neurons, the main cell tropism of HSV-1 are stromal cells. One of the main advantages of this model is its ability to maintain a homogeneous phenotype of PDL-derived mesenchymal cells over the long term in cell culture while preserving their capacity for differentiation. Indeed, more than 99% of cultured PDL cells met all the required criteria to be identified as mesenchymal cells with a fibroblast phenotype, in particular, plastic adhesion, expression of mesenchymal markers (CD105, CD29, vimentin), lack of hematopoietic markers (CD45) and the ability to differentiate into osteoblasts, adipocytes and chondrocytes [
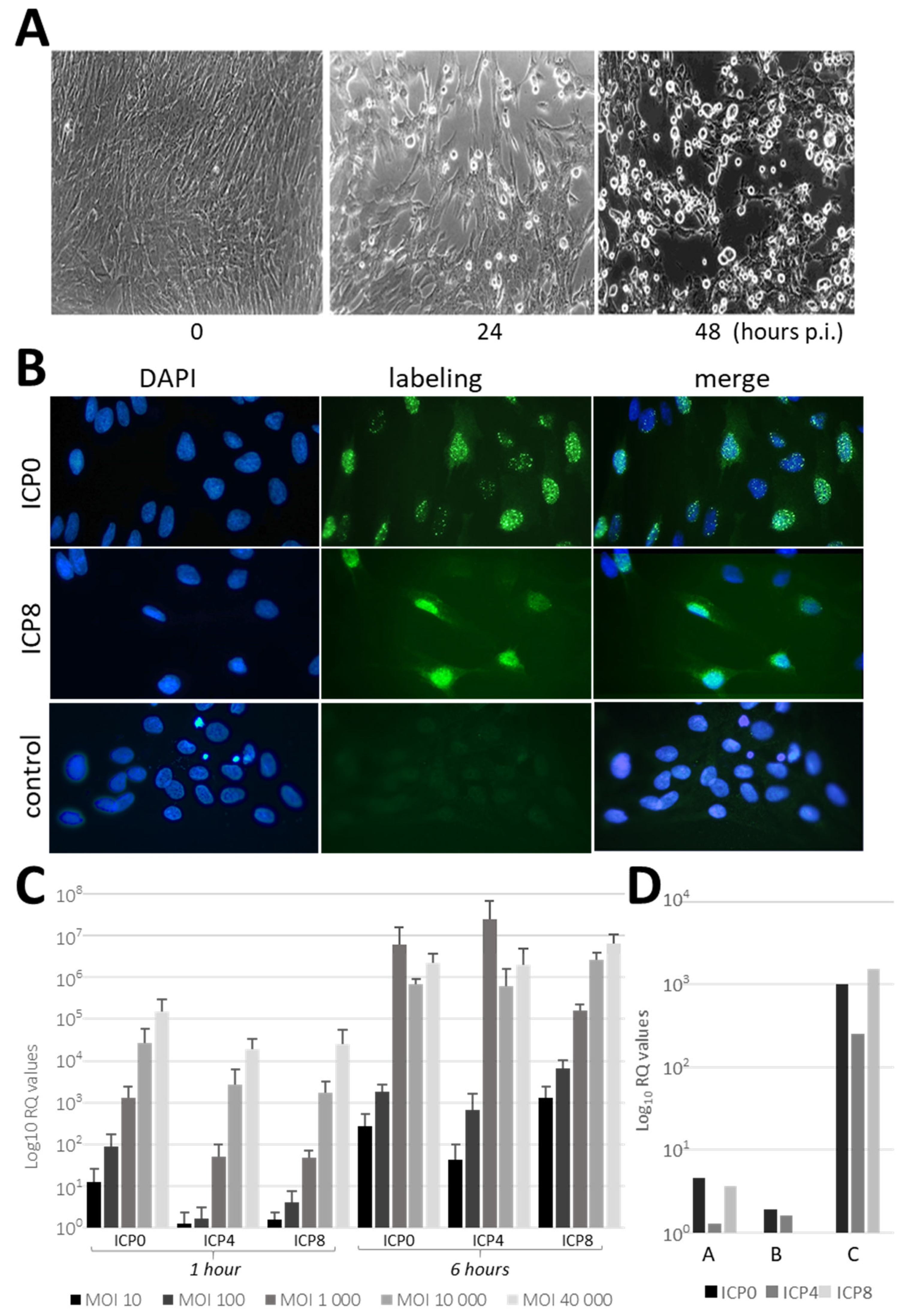
26]. These observations thus clearly established that HSV-1 infects mesenchymal fibroblasts derived from PDL. However, more exhaustive experiments would be needed to determine whether there are minor phenotypic differences within this fibroblast population and whether these subtypes could present differences in sensitivity to HSV-1. Using this system, we brought the first evidence that PDL may support HSV-1 infection. HSV-1 was shown to quickly spread in PDL cell cultures promoting profound morphological changes and cytopathic effects within 24 h. This model proves to be highly effective for supporting HSV-1 infection in vitro, demonstrating the capability to produce large amounts of infectious HSV-1, at levels comparable to or even higher than those produced by Vero cells.
Furthermore, HSV-1 was also able to infect PDL fragments revealing viral permissiveness in the whole tissue. Previous studies have shown that HSV-1 invasion in oral mucosa can be restricted by mechanical barriers [
27]; however, we did not observe any restriction and delay regarding HSV-1 infection in cultured PDL cells and PDL fragments, since immediate-early (ICP0 and ICP4) and early viral transcripts (ICP8) were expressed as soon as 1 h after viral exposure. However, we assume that our experiments may have some limitations, considering microlesions caused by scraping and PDL detachment could have favored the viral entry and diffusion into the tissue. In whole tissue, the level of HSV-1 infection greatly varied between samples, which may reflect the differences in tissue organization and cell content among PDL samples collected by dissection from different individuals. Additionally, the presence of different factors may regulate HSV-1 infection, as suggested by previous studies demonstrating that salivary factors can modulate the infection of AG09319 gingival fibroblasts [
28].
Overall, while our study benefited from using simplified and reliable in vitro models of PDL, this approach also presents a significant limitation. Although in vitro models offer controlled experimental conditions suitable for mechanistic studies, they lack the complexity of in vivo infections where interactions with immune cells are crucial. The absence of immune cells in our experimental setup restricts our understanding of how HSV-1 interacts with the host immune system during periodontal infections. To address this limitation, future investigations could explore more sophisticated models that better simulate the oral environment. Several potential in vivo models could be considered for studying HSV-1 infection in PDL cells and its interactions within the oral environment. Organotypic culture models of human oral tissues provide a more realistic three-dimensional environment to examine HSV-1 entry, replication and spread. Mouse models provide a controlled setting to explore HSV-1 pathogenesis and immune responses in the oral cavity. Humanized mouse models, involving engrafting mice with human immune cells or tissues, enhance translational relevance by allowing the study of HSV-1 infection in the presence of human immune components. Finally, clinical studies involving HSV-1-infected individuals with periodontal diseases will provide valuable clinical data on viral load, persistence and immune responses directly in affected tissues. Our ongoing research includes one such clinical study to further explore these aspects.
Innate immunity is crucial for an effective host defense against pathogenic microorganisms in periodontal tissues. PDL cells have been shown to synthesize immunomodulatory cytokines that are believed to influence the local response to infections [
6,
7,
8,
9]. We thus analyzed the innate immune response of the PDL exposed to HSV-1 and concluded that viral infection promoted an acute pro-inflammatory immune response in both cultured PDL cells and PDL fragments with a significant increase in TNF-α and IL-1β and induction of innate antiviral cytokines such as the type I interferons (IFN-α and IFN-β) and type III interferon (IFN-λ). The level of IFN induction in response to HSV-1 appeared lower in HSV-1-infected PDL fragments than in cultured PDL cells. However, this difference may be explained by the duration of viral exposure, which was adjusted to each experimental system (i.e., 4 h versus 24 h, respectively). Moreover, the innate response of PDL cells was shown to be over-stimulated when cells were infected with the virus in the presence of bacterial-derived products such as PEG. Although future studies will be needed to confirm synergistic effects through in-depth quantitative methods, the initial results presented here show that the combination of HSV-1 and PEG strongly enhances the inflammatory response of PDL cells. It has been proposed that the exacerbation of periodontal pathogenesis may involve synergistic interactions between periodontal bacterial dysbiosis and viral infections. Our results thus highlight the possibility that PDL may contribute to acute periodontal inflammation when exposed to different pathogen-associated molecular patterns from both bacteria and viruses.
In conclusion, the present study provides evidence to support that PDL may represent a reservoir for HSV-1 spread not only in the periodontium but also beyond in the oral cavity. HSV-1 shedding is common in the oral cavity, and its abundance in inflammatory periodontal sites has been reported. Furthermore, PDL is densely innervated and vascularized tissue, with HSV-1 infections typically occurring via the neural route and possibly also via the hematogenous route [
29]. These factors collectively suggest a high probability of HSV-1 spreading in PDL in vivo, although further investigations are needed for confirmation. However, the presence of HSV-1 in PDL may promote severe immune dysfunction and significant alterations in PDL organization due to its cytopathogenic effect. This assumption opens the door to further investigations into the infection of periodontal tissues by HHVs and highlights the possible need for antiviral therapy. This therapy should be based on the administration of anti-HHV drugs to patients suffering from periodontal diseases, notably periodontitis.
4. Materials and Methods
4.1. Preparation of PDL Fragments and PDL Single-Cell Suspensions
PDLs were collected from healthy individuals undergoing surgery for wisdom teeth removal. In total, PDLs from 9 healthy donors were used in this study, 6 for flow cytometry analysis and PDL cell seeding, and 3 for ex vivo HSV-1 infection of PDL fragments (donors A, B and C). This study is classified as non-interventional research involving acts devoid of risks for the patients (category 3 in the context of the French ‘Jardé Law’). Informed consent was obtained from all subjects involved in the study to inform him/her of his/her right to oppose the use of his/her specimens and data for research purposes (authorized biomedical collection N°DC-2022–5040, French Research Ministry).
Extracted teeth were immersed in phosphate-buffered saline (PBS) containing antibiotics (100 U/mL penicillin and 100 μg/mL streptomycin) and antifungals (2.5 µg/mL amphotericin B and 0.5 µg/mL caspofungin) and kept at 4 °C for less than 24 h. After 3 successive washings in PBS, rare pieces of gingival tissue still attached to the tooth were carefully removed by dissection. PDL tissues were then collected through scalpel-scraping of the mid-third of the root surface, washed with PBS and chopped into small pieces of tissue a few millimeters in size (PDL fragments). A single-cell suspension of PDL cells was obtained after digestion of PDL fragments with 3 mg/mL type I collagenase and 4 mg/mL dispase II (Life Science, Sunnyvale, CA, USA) for 30 to 40 min at 37 °C with vigorous shaking every 10 min. The digested PDL fragments were then passed through a 70 μm cell strainer (BD Falcon, Franklin Lakes, NJ, USA) and centrifuged at 400× g for 5 min. The cells were resuspended in α-MEM supplemented with 0.292 mg/mL L-Glutamine and 20% heat-inactivated fetal calf serum (FCS), and then counted using a Malassez-counting chamber.
4.2. PDL Cell Culture and Differentiation
PDL single-cell suspensions were plated into 6-well plates (Falcon, Dutscher, Bernolsheim, France) containing α-MEM supplemented with 20% heat-inactivated FCS, 100 U/mL penicillin, 100 μg/mL streptomycin, 2.5 µg/mL amphotericin B and 0.5 µg/mL caspofungin and then incubated at 37 °C with 5% CO2 in a humidified atmosphere. Cells were typically plated at a density of 3 × 105 cells per well and dissociated with 0.25% Trypsin/0.02% EDTA upon reaching 80–90% confluency. The cells were subcultured in α-MEM supplemented with 10% heat-inactivated FCS, 100 U/mL penicillin and 100 μg/mL streptomycin, defined as a complete culture medium (CCM). Cells from passages P1 to P7 were used for experiments. For osteogenic and adipogenic cell differentiation, PDL cells at 80% confluency were incubated for 4 weeks, either in osteogenic medium (CCM supplemented with 50 µg/mL L-ascorbic acid 2-phosphate, 5 mM sodium β-glycerophosphate, 100 nM dexamethasone) or in adipogenic medium (CCM supplemented with 10 µg/mL insulin (Gibco, Grand Island, NY, USA), 100 nM dexamethasone, 1 µM rosiglitazone). Osteogenesis was demonstrated by alizarin red S staining of calcium deposits, while adipogenesis was demonstrated using oil red O staining of lipids. For chondrogenic differentiation, PDL cells (4 × 105 cells in 20 µL of CCM) were placed into 12-well plates and incubated at 37 °C with 5% CO2 for 3 h attachment period to create micromass cultures. After gently adding additional CCM, the micromass cultures were rested for an additional 24 h. Differentiation was carried out by adding chondrogenic medium (CCM with 50 µg/mL L-ascorbic acid 2-phosphate, 100 nM dexamethasone, 1× insulin-transferrin-selenium premix and 10 ng/mL TGF-β3; Peprotech, Cranbury, NJ, USA). Micromasses were harvested after 3 weeks of differentiation, rinsed twice with PBS and fixed with 4% formaldehyde solution for 24 h. Micromasses were embedded in paraffin and sectioned as 5 µm thick slices. The paraffin sections were then deparaffinized in xylene, rehydrated and stained with alcian blue to visualize proteoglycans. The specificity of the detection procedures used to visualize differentiated cells was verified using non-differentiated cells as controls.
4.3. Flow Cytometry Analysis
Flow cytometry analysis of single-cell suspensions of PDL cells and cultured PDL cells was performed with fluorochrome-conjugated mouse monoclonal antibodies for the detection of cell surface markers (BD Biosciences, Franklin Lakes, NJ, USA). Hematopoietic cells were identified as cells expressing CD45. Mesenchymal cells were identified as stromal cells expressing the mesenchymal cell markers endoglin (CD105) and CD29 [
26]. The gating strategy of flow cytometry analysis was as follows: the region of interest (ROI) was placed on a size/structure dot-plot (FSC/SSC) to eliminate debris and residues from dissociation. Within this ROI, doublets were excluded by size and then by structure. The viability of this population was then assessed by excluding 7-AAD-positive cells. From the pool of viable cells, CD45 cells were then discriminated. Finally, the expression of CD105 and CD29 markers was assessed on CD45
neg cells. Analysis was performed using a BD FACS Canto II, and results were analyzed with FACSDiva software v. 6.1.3 (BD Biosciences, Franklin Lakes, NJ, USA).
4.4. HSV-1 Infection Assays
The HSV-1 isolate used in this study was a clinical strain collected from the oral cavity of a healthy individual. It was initially propagated and characterized on Vero cells, a highly permissive kidney epithelial cell lineage from the African green monkey. HSV-1 viral stock was then produced in α-MEM medium using PDL cells as amplifying cells. The viral titer of the stock was determined by testing serial viral dilutions to define the 50% tissue culture infectious dose (TCID50) that promoted a cytopathic effect on PDL cells [
30]. The multiplicity of infection (MOI) was calculated using the Reed and Muench method.
HSV-1 infection of PDL cells: In total, 6 different batches of PDL cells (from different donors) were used for HSV-1 infection and analyzed with IF staining and RT-PCRs. Of note, the cultured cells were initially characterized by IF vimentin staining, confirming their fibroblastic nature (
Figure S1), which provided the basis for their use in subsequent infection experiments. Sub-confluent monolayers of PDL cells, cultured either in plastic wells (for RNA extraction) or on treated-glass slides (for IF staining), were infected with HSV-1 at different MOIs. At 1 h, 6 h, or 24 h p.i., PDL cells were rinsed twice with α-MEM medium before RNA extraction or IF staining. HSV-1 infections were also performed in the presence of 1 and 10 µg/mL of peptidoglycan (PEG) from
Micrococcus luteus (Sigma-Aldrich, St. Louis, MO, USA).
HSV-1 infection of PDL fragments: PDL fragments were infected by direct incubation in a cell-free virus suspension (4 × 107 TCID50/mL) for 1 h at 37 °C. Subsequently, PDL fragments were washed twice by centrifugation in α-MEM medium and incubated for an additional 3 h at 37 °C in 1 mL of α-MEM medium before the tissues were lysed for RNA extraction (RNeasy Mini kit®, Qiagen, Hilden, Germany).
4.5. Immunofluorescent Staining
PDL cells were seeded in CCM on type I collagen-coated coverslips at 50% confluency for 2 days. Adherent PDL cells were infected with HSV-1 at MOI 100 for 1 h and 24 h, then rinsed twice with 1× PBS at room temperature and fixed in 3.7% paraformaldehyde/PBS for 15 min. The cells were then quenched in 50 mM NH4Cl/PBS for 30 min and permeabilized for 4 min in 0.1% Triton X-100/PBS. After two 5 min washes in PBS, cells were incubated for 60 min in a 1% bovine serum albumin (BSA)/PBS solution to block non-specific antibody binding. The incubation with mouse primary antibodies (diluted 1:200 in 1% BSA/PBS) was carried out overnight at 4 °C. Mouse primary antibodies were HSV-1 ICP0 (11060), ICP4 (H943) and ICP8 (10A3) from Santa Cruz Biotechnology, human vimentin (Clone V9) from Dako/Agilent (Glostrup, Denmark) and mouse IgG1 Isotype Control from ThermoFischer Scientific (Waltham, MA, USA). After three 3 min washes in PBS, cells were incubated with a fluorescent secondary antibody (diluted 1:1000 in 1% BSA/PBS) and co-stained with 2 µg/mL DAPI. The secondary antibody was an Alexa Fluor® 488-conjugated donkey anti-mouse IgG H&L from Abcam (Cambridge, UK). After a 30 min incubation in the dark at room temperature, the coverslips were washed three times for 3 min in PBS, once with distilled water and mounted onto microscope slides using Fluoromount Aqueous Mounting Medium (Sigma Aldrich, L’lsle-d’Abeau Chesnes, France). Image acquisition was performed using a Zeiss microscope (Oberkochen, Germany). Unless otherwise indicated, all reagents used for differentiation and staining were from Sigma Aldrich (L’lsle-d’Abeau Chesnes, France).
4.6. RNA Extraction and Reverse-Transcription PCR (RT-PCR) Analysis
PDL cells grown on plastic wells were lysed for RNA extraction according to the manufacturer’s recommendations (RNeasy Mini kit®, Qiagen). RNA extraction from PDL fragments was performed using a GentleMACS M Tube (Miltenyi, Bergisch Gladbach, Germany) with the RNA_01 program (for fresh tissue) and Qiagen RNeasy Mini kit® (tissues protocol) according to the manufacturers’ instructions. RNA quantification was performed using a microvolume spectrophotometer (SimpliNano™, Biochrom, Holliston MA, USA).
Viral and human transcripts were detected by RT-PCR using Power SYBR
® Green PCR Master Mix (Applied Biosystems™, Carlsbad, CA, USA). PCR experiments were performed using QuantStudio™ 5 (Applied Biosystems™) in a 20 µL final volume using 20 ng cDNA from PDL cells and 5.6 ng cDNA from PDL fragments (equivalent RNA). Amplification conditions were as follows: 95 °C, 10 min; (95 °C, 15 s; 60 °C, 1 min) cycled 40 times. Each sample was run in triplicates using specific primer sets (
Table 1) for the immediate-early (ICP0 and ICP4) and early (ICP8) HSV-1 viral transcripts, pro-inflammatory interleukin-1 beta (IL-1β), tumor necrosis factor alpha (TNF-α), type 1 interferons alpha-1 and beta-1 (IFN-α and IFN-β) and type III interferon lambda 1 (IFN-λ). Relative gene expression levels were calculated using the 2
−ΔΔCT method, with the GAPDH gene as the reference gene. Uninfected PDL cells were used as reference samples to normalize PDL cell gene expression, and uninfected PDL fragments from the same donors served as reference controls for comparison with HSV-1 exposed fragments.


 ,
, 


{kind=link}
{kind=link}
{kind=link}