
Pathogenesis of Sarcopenia in Chronic Kidney Disease—The Role of Inflammation, Metabolic Dysregulation, Gut Dysbiosis, and microRNA
 , ,
, ,
Abstract
:1. Introduction
2. Mechanisms Linking Chronic Kidney Disease and Sarcopenia
2.1. Inflammation
2.2. Metabolic and Hormonal Dysregulation
2.3. Gut Microbiota Dysbiosis
2.3.1. Metabolites of the Gut Microbiome and Their Impact on CKD and Sarcopenia
2.3.2. Effects of Specific Bacteria on the Development of CKD and Sarcopenia
2.3.3. The Impact of Diet and Drugs
2.4. microRNA
3. Biomarkers
4. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Levey, A.S.; de Jong, P.E.; Coresh, J.; El Nahas, M.; Astor, B.C.; Matsushita, K.; Gansevoort, R.T.; Kasiske, B.L.; Eckardt, K.U. The definition, classification, and prognosis of chronic kidney disease: A KDIGO Controversies Conference report. Kidney Int. 2011, 80, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Group KDIGOKC-MUW. KDIGO 2017 Clinical Practice Guideline Update for the Diagnosis, Evaluation, Prevention, and Treatment of Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD). Kidney Int. Suppl. 2017, 7, 1–59. [Google Scholar] [CrossRef] [PubMed]
- Ammirati, A.L. Chronic Kidney Disease. Rev. Assoc. Med. Bras. 2020, 66 (Suppl. S1), s03–s09. [Google Scholar] [CrossRef] [PubMed]
- Lucas, G.N.C.; Leitão, A.C.C.; Alencar, R.L.; Xavier, R.M.F.; Daher, E.F.; Silva Junior, G.B.D. Pathophysiological aspects of nephropathy caused by non-steroidal anti-inflammatory drugs. J. Bras. Nefrol. 2019, 41, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Collaboration, G.C.K.D. Global, regional, and national burden of chronic kidney disease, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef]
- Turin, T.C.; Tonelli, M.; Manns, B.J.; Ravani, P.; Ahmed, S.B.; Hemmelgarn, B.R. Chronic kidney disease and life expectancy. Nephrol. Dial. Transplant. 2012, 27, 3182–3186. [Google Scholar] [CrossRef] [PubMed]
- Drawz, P.; Rahman, M. Chronic kidney disease. Ann. Intern. Med. 2015, 162, ITC1–ITC16. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.K.; Knicely, D.H.; Grams, M.E. Chronic Kidney Disease Diagnosis and Management: A Review. JAMA 2019, 322, 1294–1304. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, A.; Mattace-Raso, F.; Soler, M.J.; Fouque, D. Ageing meets kidney disease. Nephrol. Dial. Transplant. 2023, 38, 523–526. [Google Scholar] [CrossRef]
- Mohanasundaram, S.; Fernando, E. Uremic Sarcopenia. Indian. J. Nephrol. 2022, 32, 399–405. [Google Scholar] [CrossRef]
- Duarte, M.P.; Almeida, L.S.; Neri, S.G.R.; Oliveira, J.S.; Wilkinson, T.J.; Ribeiro, H.S.; Lima, R.M. Prevalence of sarcopenia in patients with chronic kidney disease: A global systematic review and meta-analysis. J. Cachexia Sarcopenia Muscle 2024, 15, 501–512. [Google Scholar] [CrossRef]
- Barbosa, A.C.C.; Brison, R.S.; Gomes, C.C.; Wilkinson, T.J.; Duarte, M.P.; Gruezo, N.D.; Ribeiro, H.S. Should we consider sarcopenia in pediatric patients with chronic kidney disease? A preliminary cross-sectional analysis. Pediatr. Nephrol. 2024, 39, 539–545. [Google Scholar] [CrossRef]
- Sun, S.; Lee, H.; Yim, H.W.; Won, H.S.; Ko, Y.H. The impact of sarcopenia on health-related quality of life in elderly people: Korean National Health and Nutrition Examination Survey. Korean J. Intern. Med. 2019, 34, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Guo, X.; Xu, X.; Liang, S.; Wang, W.; Zhu, F.; Wang, S.; Wu, J.; Zhang, L.; Sun, X.; et al. Association between sarcopenia and frailty in elderly patients with chronic kidney disease. J. Cachexia Sarcopenia Muscle 2023, 14, 1855–1864. [Google Scholar] [CrossRef]
- Wilkinson, T.J.; Miksza, J.; Yates, T.; Lightfoot, C.J.; Baker, L.A.; Watson, E.L.; Zaccardi, F.; Smith, A.C. Association of sarcopenia with mortality and end-stage renal disease in those with chronic kidney disease: A UK Biobank study. J. Cachexia Sarcopenia Muscle 2021, 12, 586–598. [Google Scholar] [CrossRef]
- Petropoulou, E.; Landini, L.; Athanasiadis, L.; Dialektakou, K.; Honka, M.J.; Rebelos, E. Sarcopenia and chronic illness: From diagnosis to treatment approaches. Recenti Prog. Med. 2021, 112, 720–727. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Cui, L.; Xu, H. Association between systemic inflammation response index and chronic kidney disease: A population-based study. Front. Endocrinol. 2024, 15, 1329256. [Google Scholar] [CrossRef]
- Li, L.; Chen, K.; Wen, C.; Ma, X.; Huang, L. Association between systemic immune-inflammation index and chronic kidney disease: A population-based study. PLoS ONE 2024, 19, e0292646. [Google Scholar] [CrossRef] [PubMed]
- Fox, E.R.; Benjamin, E.J.; Sarpong, D.F.; Nagarajarao, H.; Taylor, J.K.; Steffes, M.W.; Salahudeen, A.K.; Flessner, M.F.; Akylbekova, E.L.; Fox, C.S.; et al. The relation of C-reactive protein to chronic kidney disease in African Americans: The Jackson Heart Study. BMC Nephrol. 2010, 11, 1. [Google Scholar] [CrossRef]
- Adejumo, O.A.; Okaka, E.I.; Okwuonu, C.G.; Iyawe, I.O.; Odujoko, O.O. Serum C-reactive protein levels in pre-dialysis chronic kidney disease patientsin southern Nigeria. Ghana. Med. J. 2016, 50, 31–38. [Google Scholar] [CrossRef]
- Spoto, B.; Leonardis, D.; Parlongo, R.M.; Pizzini, P.; Pisano, A.; Cutrupi, S.; Testa, A.; Tripepi, G.; Zoccali, C.; Mallamaci, F. Plasma cytokines, glomerular filtration rate and adipose tissue cytokines gene expression in chronic kidney disease (CKD) patients. Nutr. Metab. Cardiovasc. Dis. 2012, 22, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Herbelin, A.; Ureña, P.; Nguyen, A.T.; Zingraff, J.; Descamps-Latscha, B. Elevated circulating levels of interleukin-6 in patients with chronic renal failure. Kidney Int. 1991, 39, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Lousa, I.; Belo, L.; Valente, M.J.; Rocha, S.; Preguiça, I.; Rocha-Pereira, P.; Beirão, I.; Mira, F.; Alves, R.; Reis, F.; et al. Inflammatory biomarkers in staging of chronic kidney disease: Elevated TNFR2 levels accompanies renal function decline. Inflamm. Res. 2022, 71, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Frieling, J.T.; van Hamersvelt, H.W.; Wijdenes, J.; Hendriks, T.; Sauerwein, R.W.; van Der Linden, C.J. Circulating concentrations of soluble interleukin 6 receptors gp80 and gp130 in chronic renal failure and effects of renal replacement therapy. Am. J. Nephrol. 1999, 19, 571–575. [Google Scholar] [CrossRef] [PubMed]
- Verzola, D.; Bonanni, A.; Sofia, A.; Montecucco, F.; D’Amato, E.; Cademartori, V.; Parodi, E.L.; Viazzi, F.; Venturelli, C.; Brunori, G.; et al. Toll-like receptor 4 signalling mediates inflammation in skeletal muscle of patients with chronic kidney disease. J. Cachexia Sarcopenia Muscle 2017, 8, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, C.S.L.; Thang, L.A.N.; Maier, A.B. Markers of inflammation and their association with muscle strength and mass: A systematic review and meta-analysis. Ageing Res. Rev. 2020, 64, 101185. [Google Scholar] [CrossRef] [PubMed]
- Souza, V.A.; Oliveira, D.; Barbosa, S.R.; Corrêa, J.O.D.A.; Colugnati, F.A.B.; Mansur, H.N.; Fernandes, N.M.D.S.; Bastos, M.G. Sarcopenia in patients with chronic kidney disease not yet on dialysis: Analysis of the prevalence and associated factors. PLoS ONE 2017, 12, e0176230. [Google Scholar] [CrossRef] [PubMed]
- Wåhlin-Larsson, B.; Wilkinson, D.J.; Strandberg, E.; Hosford-Donovan, A.; Atherton, P.J.; Kadi, F. Mechanistic Links Underlying the Impact of C-Reactive Protein on Muscle Mass in Elderly. Cell Physiol. Biochem. 2017, 44, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Bolster, D.R.; Crozier, S.J.; Kimball, S.R.; Jefferson, L.S. AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. J. Biol. Chem. 2002, 277, 23977–23980. [Google Scholar] [CrossRef]
- Zeng, Z.; Liang, J.; Wu, L.; Zhang, H.; Lv, J.; Chen, N. Exercise-Induced Autophagy Suppresses Sarcopenia Through Akt/mTOR and Akt/FoxO3a Signal Pathways and AMPK-Mediated Mitochondrial Quality Control. Front. Physiol. 2020, 11, 583478. [Google Scholar] [CrossRef]
- Huang, Y.; Zhu, X.; Chen, K.; Lang, H.; Zhang, Y.; Hou, P.; Ran, L.; Zhou, M.; Zheng, J.; Yi, L.; et al. Resveratrol prevents sarcopenic obesity by reversing mitochondrial dysfunction and oxidative stress via the PKA/LKB1/AMPK pathway. Aging 2019, 11, 2217–2240. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Zhang, H.; Zeng, Z.; Wu, L.; Zhang, Y.; Guo, Y.; Lv, J.; Wang, C.; Fan, J.; Chen, N. Lifelong Aerobic Exercise Alleviates Sarcopenia by Activating Autophagy and Inhibiting Protein Degradation via the AMPK/PGC-1α Signaling Pathway. Metabolites 2021, 11, 323. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandris, C.; Lauro, R.; Presta, I.; Sesti, G. C-reactive protein induces phosphorylation of insulin receptor substrate-1 on Ser307 and Ser 612 in L6 myocytes, thereby impairing the insulin signalling pathway that promotes glucose transport. Diabetologia 2007, 50, 840–849. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Chen, Y.; Zhang, Y.; Wang, X.; Yang, N.; Zhu, D. Extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase pathway is involved in myostatin-regulated differentiation repression. Cancer Res. 2006, 66, 1320–1326. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.Y.; Hsiao, A.W.; Shiu, H.T.; Wong, N.; Wang, A.Y.; Lee, C.W.; Lee, O.K.; Lee, W.Y. Mesenchymal stem cells alleviate dexamethasone-induced muscle atrophy in mice and the involvement of ERK1/2 signalling pathway. Stem Cell Res. Ther. 2023, 14, 195. [Google Scholar] [CrossRef] [PubMed]
- Hsu, B.G.; Wang, C.H.; Lai, Y.H.; Kuo, C.H.; Lin, Y.L. Association of endothelial dysfunction and peripheral arterial disease with sarcopenia in chronic kidney disease. J. Cachexia Sarcopenia Muscle 2024, 15, 1199–1208. [Google Scholar] [CrossRef] [PubMed]
- Bian, A.L.; Hu, H.Y.; Rong, Y.D.; Wang, J.; Wang, J.X.; Zhou, X.Z. A study on relationship between elderly sarcopenia and inflammatory factors IL-6 and TNF-α. Eur. J. Med. Res. 2017, 22, 25. [Google Scholar] [CrossRef] [PubMed]
- Goodman, M.N. Interleukin-6 induces skeletal muscle protein breakdown in rats. Proc. Soc. Exp. Biol. Med. 1994, 205, 182–185. [Google Scholar] [CrossRef] [PubMed]
- Fujita, J.; Tsujinaka, T.; Yano, M.; Ebisui, C.; Saito, H.; Katsume, A.; Akamatsu, K.; Ohsugi, Y.; Shiozaki, H.; Monden, M. Anti-interleukin-6 receptor antibody prevents muscle atrophy in colon-26 adenocarcinoma-bearing mice with modulation of lysosomal and ATP-ubiquitin-dependent proteolytic pathways. Int. J. Cancer 1996, 68, 637–643. [Google Scholar] [CrossRef]
- Costelli, P.; Bossola, M.; Muscaritoli, M.; Grieco, G.; Bonelli, G.; Bellantone, R.; Doglietto, G.B.; Baccino, F.M.; Rossi Fanelli, F. Anticytokine treatment prevents the increase in the activity of ATP-ubiquitin- and Ca(2+)-dependent proteolytic systems in the muscle of tumour-bearing rats. Cytokine 2002, 19, 1–5. [Google Scholar] [CrossRef]
- Raj, D.S.; Moseley, P.; Dominic, E.A.; Onime, A.; Tzamaloukas, A.H.; Boyd, A.; Shah, V.O.; Glew, R.; Wolfe, R.; Ferrando, A. Interleukin-6 modulates hepatic and muscle protein synthesis during hemodialysis. Kidney Int. 2008, 73, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S. Therapeutic targeting of IL-6 trans-signaling. Cytokine 2021, 144, 155577. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S. Interleukin-6 signalling in health and disease. F1000Research 2020, 9, 1013. [Google Scholar] [CrossRef] [PubMed]
- Zanders, L.; Kny, M.; Hahn, A.; Schmidt, S.; Wundersitz, S.; Todiras, M.; Lahmann, I.; Bandyopadhyay, A.; Wollersheim, T.; Kaderali, L.; et al. Sepsis induces interleukin 6, gp130/JAK2/STAT3, and muscle wasting. J. Cachexia Sarcopenia Muscle 2022, 13, 713–727. [Google Scholar] [CrossRef] [PubMed]
- Bonetto, A.; Aydogdu, T.; Jin, X.; Zhang, Z.; Zhan, R.; Puzis, L.; Koniaris, L.G.; Zimmers, T.A. JAK/STAT3 pathway inhibition blocks skeletal muscle wasting downstream of IL-6 and in experimental cancer cachexia. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E410–E421. [Google Scholar] [CrossRef] [PubMed]
- Bermejo-Álvarez, I.; Pérez-Baos, S.; Gratal, P.; Medina, J.P.; Largo, R.; Herrero-Beaumont, G.; Mediero, A. Effects of Tofacitinib on Muscle Remodeling in Experimental Rheumatoid Sarcopenia. Int. J. Mol. Sci. 2023, 24, 13181. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Li, B.; Zhan, R.Z.; Rao, L.; Bursac, N. Exercise mimetics and JAK inhibition attenuate IFN-γ-induced wasting in engineered human skeletal muscle. Sci. Adv. 2021, 7, eabd9502. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Yamada, E.; Uehara, R.; Okada, S.; Chikuda, H.; Yamada, M. Role of Fyn and the interleukin-6-STAT-3-autophagy axis in sarcopenia. iScience 2023, 26, 107717. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wang, B.; Hassounah, F.; Price, S.R.; Klein, J.; Mohamed, T.M.A.; Wang, Y.; Park, J.; Cai, H.; Zhang, X.; et al. The impact of senescence on muscle wasting in chronic kidney disease. J. Cachexia Sarcopenia Muscle 2023, 14, 126–141. [Google Scholar] [CrossRef]
- Baker, L.A.; O’Sullivan, T.F.; Robinson, K.A.; Graham-Brown, M.P.M.; Major, R.W.; Ashford, R.U.; Smith, A.C.; Philp, A.; Watson, E.L. Primary skeletal muscle cells from chronic kidney disease patients retain hallmarks of cachexia in vitro. J. Cachexia Sarcopenia Muscle 2022, 13, 1238–1249. [Google Scholar] [CrossRef]
- Chalupsky, M.; Goodson, D.A.; Gamboa, J.L.; Roshanravan, B. New insights into muscle function in chronic kidney disease and metabolic acidosis. Curr. Opin. Nephrol. Hypertens. 2021, 30, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Visser, W.J.; van de Braak, E.E.M.; de Mik-van Egmond, A.M.E.; van der Burgh, A.C.; de Roos, N.M.; Jans, I.; van der Hoef, I.; Olieman, J.F.; Hoorn, E.J.; Severs, D. Effects of correcting metabolic acidosis on muscle mass and functionality in chronic kidney disease: A systematic review and meta-analysis. J. Cachexia Sarcopenia Muscle 2023, 14, 2498–2508. [Google Scholar] [CrossRef] [PubMed]
- Raphael, K.L. Metabolic Acidosis in CKD: Core Curriculum 2019. Am. J. Kidney Dis. 2019, 74, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Raphael, K.L. Metabolic Acidosis and Subclinical Metabolic Acidosis in CKD. J. Am. Soc. Nephrol. 2018, 29, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Mitch, W.E.; Medina, R.; Grieber, S.; May, R.C.; England, B.K.; Price, S.R.; Bailey, J.L.; Goldberg, A.L. Metabolic acidosis stimulates muscle protein degradation by activating the adenosine triphosphate-dependent pathway involving ubiquitin and proteasomes. J. Clin. Investig. 1994, 93, 2127–2133. [Google Scholar] [CrossRef] [PubMed]
- Ballmer, P.E.; McNurlan, M.A.; Hulter, H.N.; Anderson, S.E.; Garlick, P.J.; Krapf, R. Chronic metabolic acidosis decreases albumin synthesis and induces negative nitrogen balance in humans. J. Clin. Investig. 1995, 95, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Reaich, D.; Channon, S.M.; Scrimgeour, C.M.; Goodship, T.H. Ammonium chloride-induced acidosis increases protein breakdown and amino acid oxidation in humans. Am. J. Physiol. 1992, 263, E735–E739. [Google Scholar] [CrossRef]
- Verove, C.; Maisonneuve, N.; El Azouzi, A.; Boldron, A.; Azar, R. Effect of the correction of metabolic acidosis on nutritional status in elderly patients with chronic renal failure. J. Ren. Nutr. 2002, 12, 224–228. [Google Scholar] [CrossRef]
- Movilli, E.; Zani, R.; Carli, O.; Sangalli, L.; Pola, A.; Camerini, C.; Cancarini, G.C.; Scolari, F.; Feller, P.; Maiorca, R. Correction of metabolic acidosis increases serum albumin concentrations and decreases kinetically evaluated protein intake in haemodialysis patients: A prospective study. Nephrol. Dial. Transplant. 1998, 13, 1719–1722. [Google Scholar] [CrossRef]
- Patschan, D.; Patschan, S.; Ritter, O. Chronic Metabolic Acidosis in Chronic Kidney Disease. Kidney Blood Press. Res. 2020, 45, 812–822. [Google Scholar] [CrossRef]
- Watanabe, H.; Enoki, Y.; Maruyama, T. Sarcopenia in Chronic Kidney Disease: Factors, Mechanisms, and Therapeutic Interventions. Biol. Pharm. Bull. 2019, 42, 1437–1445. [Google Scholar] [CrossRef] [PubMed]
- Rysz, J.; Franczyk, B.; Ławiński, J.; Olszewski, R.; Ciałkowska-Rysz, A.; Gluba-Brzózka, A. The Impact of CKD on Uremic Toxins and Gut Microbiota. Toxins 2021, 13, 252. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, G.G.C.; Dellê, H.; Brito, R.B.O.; Cardoso, V.O.; Fernandes, K.P.S.; Mesquita-Ferrari, R.A.; Cunha, R.S.; Stinghen, A.E.M.; Dalboni, M.A.; Barreto, F.C. Indoxyl Sulfate Contributes to Uremic Sarcopenia by Inducing Apoptosis in Myoblasts. Arch. Med. Res. 2020, 51, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Enoki, Y.; Watanabe, H.; Arake, R.; Sugimoto, R.; Imafuku, T.; Tominaga, Y.; Ishima, Y.; Kotani, S.; Nakajima, M.; Tanaka, M.; et al. Indoxyl sulfate potentiates skeletal muscle atrophy by inducing the oxidative stress-mediated expression of myostatin and atrogin-1. Sci. Rep. 2016, 6, 32084. [Google Scholar] [CrossRef]
- Jheng, J.R.; Chen, Y.S.; Ao, U.I.; Chan, D.C.; Huang, J.W.; Hung, K.Y.; Tarng, D.C.; Chiang, C.K. The double-edged sword of endoplasmic reticulum stress in uremic sarcopenia through myogenesis perturbation. J. Cachexia Sarcopenia Muscle 2018, 9, 570–584. [Google Scholar] [CrossRef]
- Nigam, S.K.; Bush, K.T. Uraemic syndrome of chronic kidney disease: Altered remote sensing and signalling. Nat. Rev. Nephrol. 2019, 15, 301–316. [Google Scholar] [CrossRef]
- Stockler-Pinto, M.B.; Soulage, C.O.; Borges, N.A.; Cardozo, L.; Dolenga, C.J.; Nakao, L.S.; Pecoits-Filho, R.; Fouque, D.; Mafra, D. From bench to the hemodialysis clinic: Protein-bound uremic toxins modulate NF-kappaB/Nrf2 expression. Int. Urol. Nephrol. 2018, 50, 347–354. [Google Scholar] [CrossRef]
- Gomez-Garcia, E.F.; Del Campo, F.M.; Cortes-Sanabria, L.; Mendoza-Carrera, F.; Avesani, C.M.; Stenvinkel, P.; Lindholm, B.; Cueto-Manzano, A.M. Transcription factor NRF2 as potential therapeutic target for preventing muscle wasting in aging chronic kidney disease patients. J. Nephrol. 2022, 35, 2215–2225. [Google Scholar] [CrossRef]
- Waller, S. Parathyroid hormone and growth in chronic kidney disease. Pediatr. Nephrol. 2011, 26, 195–204. [Google Scholar] [CrossRef]
- Kir, S.; Komaba, H.; Garcia, A.P.; Economopoulos, K.P.; Liu, W.; Lanske, B.; Hodin, R.A.; Spiegelman, B.M. PTH/PTHrP Receptor Mediates Cachexia in Models of Kidney Failure and Cancer. Cell Metab. 2016, 23, 315–323. [Google Scholar] [CrossRef]
- Mak, R.H.; Cheung, W.W.; Roberts, C.T. The growth hormone-insulin-like growth factor-I axis in chronic kidney disease. Growth Horm. IGF Res. 2008, 18, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Gungor, O.; Ulu, S.; Hasbal, N.B.; Anker, S.D.; Kalantar-Zadeh, K. Effects of hormonal changes on sarcopenia in chronic kidney disease: Where are we now and what can we do? J. Cachexia Sarcopenia Muscle 2021, 12, 1380–1392. [Google Scholar] [CrossRef] [PubMed]
- Palmer, S.C.; McGregor, D.O.; Macaskill, P.; Craig, J.C.; Elder, G.J.; Strippoli, G.F. Meta-analysis: Vitamin D compounds in chronic kidney disease. Ann. Intern. Med. 2007, 147, 840–853. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.H.; Chen, J.J.; Wu, C.Y.; Lin, T.Y.; Hung, S.C.; Yang, H.Y. Immunosenescence, gut dysbiosis, and chronic kidney disease: Interplay and implications for clinical management. Biomed. J. 2023, 47, 100638. [Google Scholar] [CrossRef] [PubMed]
- Stavropoulou, E.; Kantartzi, K.; Tsigalou, C.; Aftzoglou, K.; Voidarou, C.; Konstantinidis, T.; Chifiriuc, M.C.; Thodis, E.; Bezirtzoglou, E. Microbiome, Immunosenescence, and Chronic Kidney Disease. Front. Med. 2021, 8, 661203. [Google Scholar] [CrossRef] [PubMed]
- Ramezani, A.; Raj, D.S. The gut microbiome, kidney disease, and targeted interventions. J. Am. Soc. Nephrol. 2014, 25, 657–670. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The human microbiome project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Zhang, J.; Li, W.; Li, Y.; Jia, L.; Liu, Z.; Fu, W.; Zhang, A. Yi-Shen-Hua-Shi regulates intestinal microbiota dysbiosis and protects against proteinuria in patients with chronic kidney disease: A randomized controlled study. Pharm. Biol. 2024, 62, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Price, J.; Abu-Ali, G.; Huttenhower, C. The healthy human microbiome. Genome Med. 2016, 8, 51. [Google Scholar] [CrossRef]
- Wilck, N.; Matus, M.G.; Kearney, S.M.; Olesen, S.W.; Forslund, K.; Bartolomaeus, H.; Haase, S.; Mähler, A.; Balogh, A.; Markó, L.; et al. Salt-responsive gut commensal modulates T. Nature 2017, 551, 585–589. [Google Scholar] [CrossRef]
- Hobby, G.P.; Karaduta, O.; Dusio, G.F.; Singh, M.; Zybailov, B.L.; Arthur, J.M. Chronic kidney disease and the gut microbiome. Am. J. Physiol. Renal Physiol. 2019, 316, F1211–F1217. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Piceno, Y.M.; DeSantis, T.Z.; Pahl, M.; Andersen, G.L.; Vaziri, N.D. Expansion of urease- and uricase-containing, indole- and p-cresol-forming and contraction of short-chain fatty acid-producing intestinal microbiota in ESRD. Am. J. Nephrol. 2014, 39, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Wong, J.; Pahl, M.; Piceno, Y.M.; Yuan, J.; DeSantis, T.Z.; Ni, Z.; Nguyen, T.H.; Andersen, G.L. Chronic kidney disease alters intestinal microbial flora. Kidney Int. 2013, 83, 308–315. [Google Scholar] [CrossRef]
- Vaziri, N.D.; Zhao, Y.Y.; Pahl, M.V. Altered intestinal microbial flora and impaired epithelial barrier structure and function in CKD: The nature, mechanisms, consequences and potential treatment. Nephrol. Dial. Transplant. 2016, 31, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Eckert, M.; Meek, S.E.; Ball, K.L. A novel repressor domain is required for maximal growth inhibition by the IRF-1 tumor suppressor. J. Biol. Chem. 2006, 281, 23092–23102. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, J.C.; Dinatale, B.C.; Murray, I.A.; Flaveny, C.A.; Liu, Q.; Laurenzana, E.M.; Lin, J.M.; Strom, S.C.; Omiecinski, C.J.; Amin, S.; et al. The uremic toxin 3-indoxyl sulfate is a potent endogenous agonist for the human aryl hydrocarbon receptor. Biochemistry 2010, 49, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Sato, E.; Mori, T.; Mishima, E.; Suzuki, A.; Sugawara, S.; Kurasawa, N.; Saigusa, D.; Miura, D.; Morikawa-Ichinose, T.; Saito, R.; et al. Metabolic alterations by indoxyl sulfate in skeletal muscle induce uremic sarcopenia in chronic kidney disease. Sci. Rep. 2016, 6, 36618. [Google Scholar] [CrossRef] [PubMed]
- Fahal, I.H. Uraemic sarcopenia: Aetiology and implications. Nephrol. Dial. Transplant. 2014, 29, 1655–1665. [Google Scholar] [CrossRef]
- Xu, Y.; Mao, T.; Wang, Y.; Qi, X.; Zhao, W.; Chen, H.; Zhang, C.; Li, X. Effect of Gut Microbiota-Mediated Tryptophan Metabolism on Inflammaging in Frailty and Sarcopenia. J. Gerontol. A Biol. Sci. Med. Sci. 2024, 79, glae044. [Google Scholar] [CrossRef]
- Hui, Y.; Zhao, J.; Yu, Z.; Wang, Y.; Qin, Y.; Zhang, Y.; Xing, Y.; Han, M.; Wang, A.; Guo, S.; et al. The Role of Tryptophan Metabolism in the Occurrence and Progression of Acute and Chronic Kidney Diseases. Mol. Nutr. Food Res. 2023, 67, e2300218. [Google Scholar] [CrossRef]
- Mor, A.; Kalaska, B.; Pawlak, D. Kynurenine Pathway in Chronic Kidney Disease: What’s Old, What’s New, and What’s Next? Int. J. Tryptophan Res. 2020, 13, 1178646920954882. [Google Scholar] [CrossRef] [PubMed]
- Ballesteros, J.; Rivas, D.; Duque, G. The Role of the Kynurenine Pathway in the Pathophysiology of Frailty, Sarcopenia, and Osteoporosis. Nutrients 2023, 15, 3132. [Google Scholar] [CrossRef] [PubMed]
- Chatzipetrou, V.; Bégin, M.J.; Hars, M.; Trombetti, A. Sarcopenia in Chronic Kidney Disease: A Scoping Review of Prevalence, Risk Factors, Association with Outcomes, and Treatment. Calcif. Tissue Int. 2022, 110, 1–31. [Google Scholar] [CrossRef] [PubMed]
- van Krimpen, S.J.; Jansen, F.A.C.; Ottenheim, V.L.; Belzer, C.; van der Ende, M.; van Norren, K. The Effects of Pro-, Pre-, and Synbiotics on Muscle Wasting, a Systematic Review-Gut Permeability as Potential Treatment Target. Nutrients 2021, 13, 1115. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Johnson, D.W.; Morrison, M.; Pascoe, E.M.; Coombes, J.S.; Forbes, J.M.; Szeto, C.C.; McWhinney, B.C.; Ungerer, J.P.; Campbell, K.L. Synbiotics Easing Renal Failure by Improving Gut Microbiology (SYNERGY): A Randomized Trial. Clin. J. Am. Soc. Nephrol. 2016, 11, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, K.; Tsuchiya, A.; Nakano, O.; Kuroki, Y.; Oka, K.; Minemura, A.; Matsumoto, A.; Takahashi, M.; Kadota, Y.; Tochio, T.; et al. Increase in muscle mass associated with the prebiotic effects of 1-kestose in super-elderly patients with sarcopenia. Biosci. Microbiota Food Health 2021, 40, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Takkavatakarn, K.; Wuttiputinun, T.; Phannajit, J.; Praditpornsilpa, K.; Eiam-Ong, S.; Susantitaphong, P. Protein-bound uremic toxin lowering strategies in chronic kidney disease: A systematic review and meta-analysis. J. Nephrol. 2021, 34, 1805–1817. [Google Scholar] [CrossRef] [PubMed]
- Sato, E.; Saigusa, D.; Mishima, E.; Uchida, T.; Miura, D.; Morikawa-Ichinose, T.; Kisu, K.; Sekimoto, A.; Saito, R.; Oe, Y.; et al. Impact of the Oral Adsorbent AST-120 on Organ-Specific Accumulation of Uremic Toxins: LC-MS/MS and MS Imaging Techniques. Toxins 2017, 10, 19. [Google Scholar] [CrossRef] [PubMed]
- Cha, R.H.; Kang, S.H.; Han, M.Y.; An, W.S.; Kim, S.H.; Kim, J.C. Effects of AST-120 on muscle health and quality of life in chronic kidney disease patients: Results of RECOVERY study. J. Cachexia Sarcopenia Muscle 2022, 13, 397–408. [Google Scholar] [CrossRef]
- Lee, S.M.; Han, M.Y.; Kim, S.H.; Cha, R.H.; Kang, S.H.; Kim, J.C.; An, W.S. Indoxyl Sulfate Might Play a Role in Sarcopenia, While Myostatin Is an Indicator of Muscle Mass in Patients with Chronic Kidney Disease: Analysis from the RECOVERY Study. Toxins 2022, 14, 660. [Google Scholar] [CrossRef]
- Jerez-Morales, A.; Merino, J.S.; Diaz-Castillo, S.T.; Smith, C.T.; Fuentealba, J.; Bernasconi, H.; Echeverria, G.; Garcia-Cancino, A. The Administration of the Synbiotic Lactobacillus bulgaricus 6c3 Strain, Inulin and Fructooligosaccharide Decreases the Concentrations of Indoxyl Sulfate and Kidney Damage in a Rat Model. Toxins 2021, 13, 192. [Google Scholar] [CrossRef] [PubMed]
- Ertuglu, L.; Yildiz, A.; Gamboa, J.; Ikizler, T.A. Skeletal muscle energetics in patients with moderate to advanced kidney disease. Kidney Res. Clin. Pract. 2022, 41, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Lau, W.L.; Savoj, J.; Nakata, M.B.; Vaziri, N.D. Altered microbiome in chronic kidney disease: Systemic effects of gut-derived uremic toxins. Clin. Sci. 2018, 132, 509–522. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, M.; Ueno, M.; Itoh, Y.; Suda, W.; Hattori, M. Uremic Toxin-Producing Gut Microbiota in Rats with Chronic Kidney Disease. Nephron 2017, 135, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, K.; Wakino, S.; Irie, J.; Miyamoto, J.; Matsui, A.; Tajima, T.; Itoh, T.; Oshima, Y.; Yoshifuji, A.; Kimura, I.; et al. Contribution of uremic dysbiosis to insulin resistance and sarcopenia. Nephrol. Dial. Transplant. 2020, 35, 1501–1517. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Pahl, M.V.; Vaziri, N.D.; Blake, D.R. Effect of hemodialysis and diet on the exhaled breath methanol concentration in patients with ESRD. J. Ren. Nutr. 2012, 22, 357–364. [Google Scholar] [CrossRef]
- Salvi, P.S.; Cowles, R.A. Butyrate and the Intestinal Epithelium: Modulation of Proliferation and Inflammation in Homeostasis and Disease. Cells 2021, 10, 1775. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Wang, Q.; Lv, Y.; Xia, F.; Chen, X.; Mao, Y.; Wang, X.; Ding, G.; Yu, J. Serum metabolome and gut microbiome alterations are associated with low handgrip strength in older adults. Aging 2024, 16, 2638–2656. [Google Scholar] [CrossRef] [PubMed]
- Kuppusamy, P.; Soundharrajan, I.; Kim, D.H.; Hwang, I.; Choi, K.C. 4-hydroxy-3-methoxy cinnamic acid accelerate myoblasts differentiation on C2C12 mouse skeletal muscle cells via AKT and ERK 1/2 activation. Phytomedicine 2019, 60, 152873. [Google Scholar] [CrossRef]
- Sugimura, Y.; Yang, Y.; Kanda, A.; Mawatari, A.; Tamada, Y.; Mikami, T.; Nakaji, S.; Ihara, K. Association between Gut Microbiota and Muscle Strength in Japanese General Population of the Iwaki Health Promotion Project. Microorganisms 2024, 12, 622. [Google Scholar] [CrossRef]
- Picca, A.; Ponziani, F.R.; Calvani, R.; Marini, F.; Biancolillo, A.; Coelho-Junior, H.J.; Gervasoni, J.; Primiano, A.; Putignani, L.; Del Chierico, F.; et al. Gut Microbial, Inflammatory and Metabolic Signatures in Older People with Physical Frailty and Sarcopenia: Results from the BIOSPHERE Study. Nutrients 2019, 12, 65. [Google Scholar] [CrossRef] [PubMed]
- Donohoe, D.R.; Garge, N.; Zhang, X.; Sun, W.; O’Connell, T.M.; Bunger, M.K.; Bultman, S.J. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab. 2011, 13, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhou, J.; Wang, S.; Xiong, J.; Chen, Y.; Liu, Y.; Xiao, T.; Li, Y.; He, T.; Bi, X.; et al. Indoxyl sulfate induces intestinal barrier injury through IRF1-DRP1 axis-mediated mitophagy impairment. Theranostics 2020, 10, 7384–7400. [Google Scholar] [CrossRef] [PubMed]
- Cigarran Guldris, S.; González Parra, E.; Cases Amenós, A. Gut microbiota in chronic kidney disease. Nefrologia 2017, 37, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Xu, X.; Deji, Y.; Gao, S.; Wu, C.; Song, Q.; Shi, Z.; Xiang, X.; Zang, J.; Su, J. Bifidobacterium as a Potential Biomarker of Sarcopenia in Elderly Women. Nutrients 2023, 15, 1266. [Google Scholar] [CrossRef] [PubMed]
- Lohia, S.; Vlahou, A.; Zoidakis, J. Microbiome in Chronic Kidney Disease (CKD): An Omics Perspective. Toxins 2022, 14, 176. [Google Scholar] [CrossRef] [PubMed]
- Fielding, R.A.; Reeves, A.R.; Jasuja, R.; Liu, C.; Barrett, B.B.; Lustgarten, M.S. Muscle strength is increased in mice that are colonized with microbiota from high-functioning older adults. Exp. Gerontol. 2019, 127, 110722. [Google Scholar] [CrossRef]
- Bjørkhaug, S.T.; Aanes, H.; Neupane, S.P.; Bramness, J.G.; Malvik, S.; Henriksen, C.; Skar, V.; Medhus, A.W.; Valeur, J. Characterization of gut microbiota composition and functions in patients with chronic alcohol overconsumption. Gut Microbes 2019, 10, 663–675. [Google Scholar] [CrossRef] [PubMed]
- Manickam, R.; Oh, H.Y.P.; Tan, C.K.; Paramalingam, E.; Wahli, W. Metronidazole Causes Skeletal Muscle Atrophy and Modulates Muscle Chronometabolism. Int. J. Mol. Sci. 2018, 19, 2418. [Google Scholar] [CrossRef]
- Ren, X.; Hao, S.; Yang, C.; Yuan, L.; Zhou, X.; Zhao, H.; Yao, J. Alterations of intestinal microbiota in liver cirrhosis with muscle wasting. Nutrition 2021, 83, 111081. [Google Scholar] [CrossRef]
- Ticinesi, A.; Mancabelli, L.; Tagliaferri, S.; Nouvenne, A.; Milani, C.; Del Rio, D.; Lauretani, F.; Maggio, M.G.; Ventura, M.; Meschi, T. The Gut-Muscle Axis in Older Subjects with Low Muscle Mass and Performance: A Proof of Concept Study Exploring Fecal Microbiota Composition and Function with Shotgun Metagenomics Sequencing. Int. J. Mol. Sci. 2020, 21, 8496. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Fan, Y.; Li, A.; Shen, Q.; Wu, J.; Ren, L.; Lu, H.; Ding, S.; Ren, H.; Liu, C.; et al. Alterations of the Human Gut Microbiome in Chronic Kidney Disease. Adv. Sci. 2020, 7, 2001936. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, H.; Chen, L.; Liang, S.S.; Shi, K.; Meng, W.; Xue, J.; He, Q.; Jiang, H. Effect of probiotics on the intestinal microbiota of hemodialysis patients: A randomized trial. Eur. J. Nutr. 2020, 59, 3755–3766. [Google Scholar] [CrossRef]
- Kim, J.E.; Kim, H.E.; Park, J.I.; Cho, H.; Kwak, M.J.; Kim, B.Y.; Yang, S.H.; Lee, J.P.; Kim, D.K.; Joo, K.W.; et al. The Association between Gut Microbiota and Uremia of Chronic Kidney Disease. Microorganisms 2020, 8, 907. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.K.; Su, S.C.; Chang, L.C.; Yang, K.J.; Lee, C.C.; Hsu, H.J.; Chen, Y.T.; Sun, C.Y.; Wu, I.W. Oral Absorbent AST-120 Is Associated with Compositional and Functional Adaptations of Gut Microbiota and Modification of Serum Short and Medium-Chain Fatty Acids in Advanced CKD Patients. Biomedicines 2022, 10, 2234. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Jose, A.; Alonzo-Palma, N.; Malik, T.; Shankaranarayanan, D.; Regunathan-Shenk, R.; Raj, D.S. Butyrate producing microbiota are reduced in chronic kidney diseases. Sci. Rep. 2021, 11, 23530. [Google Scholar] [CrossRef]
- Hsu, C.K.; Su, S.C.; Chang, L.C.; Shao, S.C.; Yang, K.J.; Chen, C.Y.; Chen, Y.T.; Wu, I.W. Effects of Low Protein Diet on Modulating Gut Microbiota in Patients with Chronic Kidney Disease: A Systematic Review and Meta-analysis of International Studies. Int. J. Med. Sci. 2021, 18, 3839–3850. [Google Scholar] [CrossRef]
- Chen, H.Y.; Sun, C.Y.; Lee, C.C.; Wu, I.W.; Chen, Y.C.; Lin, Y.H.; Fang, W.C.; Pan, H.C. Ketoanalogue supplements reduce mortality in patients with pre-dialysis advanced diabetic kidney disease: A nationwide population-based study. Clin. Nutr. 2021, 40, 4149–4160. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Esteve, E.; Tremaroli, V.; Khan, M.T.; Caesar, R.; Mannerås-Holm, L.; Ståhlman, M.; Olsson, L.M.; Serino, M.; Planas-Fèlix, M.; et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat. Med. 2017, 23, 850–858. [Google Scholar] [CrossRef]
- Vich Vila, A.; Collij, V.; Sanna, S.; Sinha, T.; Imhann, F.; Bourgonje, A.R.; Mujagic, Z.; Jonkers, D.M.A.E.; Masclee, A.A.M.; Fu, J.; et al. Impact of commonly used drugs on the composition and metabolic function of the gut microbiota. Nat. Commun. 2020, 11, 362. [Google Scholar] [CrossRef]
- Maier, L.; Pruteanu, M.; Kuhn, M.; Zeller, G.; Telzerow, A.; Anderson, E.E.; Brochado, A.R.; Fernandez, K.C.; Dose, H.; Mori, H.; et al. Extensive impact of non-antibiotic drugs on human gut bacteria. Nature 2018, 555, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Aya, V.; Flórez, A.; Perez, L.; Ramírez, J.D. Association between physical activity and changes in intestinal microbiota composition: A systematic review. PLoS ONE 2021, 16, e0247039. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Liu, S.; Chen, L.; Shen, J.; Niu, Y.; Wang, T.; Zhang, W.; Fu, L. Effect of exercise and butyrate supplementation on microbiota composition and lipid metabolism. J. Endocrinol. 2019, 243, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Zeng, Y.; Tu, Q.; Jiao, Y.; Yao, S.; Chen, Y.; Sun, L.; Xia, Q.; Luo, Y.; Yuan, L.; et al. Butyrate alleviates renal fibrosis in CKD by regulating NLRP3-mediated pyroptosis via the STING/NF-κB/p65 pathway. Int. Immunopharmacol. 2023, 124, 111010. [Google Scholar] [CrossRef] [PubMed]
- Lan, Z.; Chen, A.; Li, L.; Ye, Y.; Liang, Q.; Dong, Q.; Wang, S.; Fu, M.; Li, Y.; Liu, X.; et al. Downregulation of HDAC9 by the ketone metabolite β-hydroxybutyrate suppresses vascular calcification. J. Pathol. 2022, 258, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Chang, C.I.; Hou, C.Y.; Chang-Chien, G.P.; Lin, S.; Hsu, C.N. Dietary Resveratrol Butyrate Monoester Supplement Improves Hypertension and Kidney Dysfunction in a Young Rat Chronic Kidney Disease Model. Nutrients 2023, 15, 635. [Google Scholar] [CrossRef] [PubMed]
- Li, H.B.; Xu, M.L.; Xu, X.D.; Tang, Y.Y.; Jiang, H.L.; Li, L.; Xia, W.J.; Cui, N.; Bai, J.; Dai, Z.M.; et al. Attenuates CKD via Butyrate-Renal GPR43 Axis. Circ. Res. 2022, 131, e120–e134. [Google Scholar] [CrossRef] [PubMed]
- Mach, N.; Fuster-Botella, D. Endurance exercise and gut microbiota: A review. J. Sport. Health Sci. 2017, 6, 179–197. [Google Scholar] [CrossRef] [PubMed]
- Lambert, K.; Rinninella, E.; Biruete, A.; Sumida, K.; Stanford, J.; Raoul, P.; Mele, M.C.; Wang, A.Y.; Mafra, D. Targeting the Gut Microbiota in Kidney Disease: The Future in Renal Nutrition and Metabolism. J. Ren. Nutr. 2023, 33, S30–S39. [Google Scholar] [CrossRef]
- Garmaa, G.; Bunduc, S.; Kói, T.; Hegyi, P.; Csupor, D.; Ganbat, D.; Dembrovszky, F.; Meznerics, F.A.; Nasirzadeh, A.; Barbagallo, C.; et al. A Systematic Review and Meta-Analysis of microRNA Profiling Studies in Chronic Kidney Diseases. Non-Coding RNA 2024, 10, 30. [Google Scholar] [CrossRef]
- Scullion, K.M.; Vliegenthart, A.D.B.; Rivoli, L.; Oosthuyzen, W.; Farrah, T.E.; Czopek, A.; Webb, D.J.; Hunter, R.W.; Bailey, M.A.; Dhaun, N.; et al. Circulating argonaute-bound microRNA-126 reports vascular dysfunction and treatment response in acute and chronic kidney disease. iScience 2021, 24, 101937. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.H.; Hu, Z.; Klein, J.D.; Zhang, L.; Fang, F.; Mitch, W.E. Decreased miR-29 suppresses myogenesis in CKD. J. Am. Soc. Nephrol. 2011, 22, 2068–2076. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhang, A.; Wang, H.; Klein, J.D.; Tan, L.; Wang, Z.M.; Du, J.; Naqvi, N.; Liu, B.C.; Wang, X.H. Limits Muscle Wasting and Cardiac Fibrosis through Exosome-Mediated microRNA Transfer in Chronic Kidney Disease. Theranostics 2019, 9, 1864–1877. [Google Scholar] [CrossRef]
- Bodine, S.C.; Stitt, T.N.; Gonzalez, M.; Kline, W.O.; Stover, G.L.; Bauerlein, R.; Zlotchenko, E.; Scrimgeour, A.; Lawrence, J.C.; Glass, D.J.; et al. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat. Cell Biol. 2001, 3, 1014–1019. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Wang, H.; Wang, B.; Yuan, Y.; Klein, J.D.; Wang, X.H. Exogenous miR-26a suppresses muscle wasting and renal fibrosis in obstructive kidney disease. FASEB J. 2019, 33, 13590–13601. [Google Scholar] [CrossRef] [PubMed]
- Dey, B.K.; Gagan, J.; Yan, Z.; Dutta, A. miR-26a is required for skeletal muscle differentiation and regeneration in mice. Genes. Dev. 2012, 26, 2180–2191. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.F.; Tellam, R.L. MicroRNA-26a targets the histone methyltransferase Enhancer of Zeste homolog 2 during myogenesis. J. Biol. Chem. 2008, 283, 9836–9843. [Google Scholar] [CrossRef]
- Caretti, G.; Di Padova, M.; Micales, B.; Lyons, G.E.; Sartorelli, V. The Polycomb Ezh2 methyltransferase regulates muscle gene expression and skeletal muscle differentiation. Genes. Dev. 2004, 18, 2627–2638. [Google Scholar] [CrossRef]
- Juan, A.H.; Derfoul, A.; Feng, X.; Ryall, J.G.; Dell’Orso, S.; Pasut, A.; Zare, H.; Simone, J.M.; Rudnicki, M.A.; Sartorelli, V. Polycomb EZH2 controls self-renewal and safeguards the transcriptional identity of skeletal muscle stem cells. Genes. Dev. 2011, 25, 789–794. [Google Scholar] [CrossRef]
- Yong, H.; Wu, G.; Chen, J.; Liu, X.; Bai, Y.; Tang, N.; Liu, L.; Wei, J. lncRNA MALAT1 Accelerates Skeletal Muscle Cell Apoptosis and Inflammatory Response in Sepsis by Decreasing BRCA1 Expression by Recruiting EZH2. Mol. Ther. Nucleic Acids 2020, 21, 1120–1121. [Google Scholar] [CrossRef]
- Yu, T.; Wang, P.; Wu, Y.; Zhong, J.; Chen, Q.; Wang, D.; Chen, H.; Hu, S.; Wu, Q. MiR-26a Reduces Inflammatory Responses via Inhibition of PGE2 Production by Targeting COX-2. Inflammation 2022, 45, 1484–1495. [Google Scholar] [CrossRef] [PubMed]
- Sirago, G.; Toniolo, L.; Crea, E.; Giacomello, E. A short-term treatment with resveratrol improves the inflammatory conditions of Middle-aged mice skeletal muscles. Int. J. Food Sci. Nutr. 2022, 73, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Fu, X.; Xie, J.; Pan, H.; Han, W.; Huang, W. miR-26a attenuates colitis and colitis-associated cancer by targeting the multiple intestinal inflammatory pathways. Mol. Ther. Nucleic Acids 2021, 24, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Ji, J.; Zhao, T.; Wang, E.; Zhang, A. Exosome-encapsulated miR-26a attenuates aldosterone-induced tubulointerstitial fibrosis by inhibiting the CTGF/SMAD3 signaling pathway. Int. J. Mol. Med. 2023, 51, 11. [Google Scholar] [CrossRef] [PubMed]
- Petrosino, J.M.; Leask, A.; Accornero, F. Genetic manipulation of CCN2/CTGF unveils cell-specific ECM-remodeling effects in injured skeletal muscle. FASEB J. 2019, 33, 2047–2057. [Google Scholar] [CrossRef] [PubMed]
- Ding, N.; Wang, W.; Teng, J.; Zeng, Y.; Zhang, Q.; Dong, L.; Tang, H. miR-26a-5p Regulates Adipocyte Differentiation via Directly Targeting ACSL3 in Adipocytes. Adipocyte 2023, 12, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, B.; Zhang, A.; Hassounah, F.; Seow, Y.; Wood, M.; Ma, F.; Klein, J.D.; Price, S.R.; Wang, X.H. Exosome-Mediated miR-29 Transfer Reduces Muscle Atrophy and Kidney Fibrosis in Mice. Mol. Ther. 2019, 27, 571–583. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wang, J.; He, W.; Zhao, Y.; Zhang, A.; Liu, Y.; Hassounah, F.; Ma, F.; Klein, J.D.; Wang, X.H.; et al. Exogenous miR-29a Attenuates Muscle Atrophy and Kidney Fibrosis in Unilateral Ureteral Obstruction Mice. Hum. Gene Ther. 2020, 31, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Li, L.; Ge, M.; Gu, L.; Zhang, K.; Su, Y.; Zhang, Y.; Lan, M.; Yu, Y.; Wang, T.; et al. MiR-29ab1 Cluster Resists Muscle Atrophy Through Inhibiting MuRF1. DNA Cell Biol. 2021, 40, 1167–1176. [Google Scholar] [CrossRef]
- Chen, F.; Zhou, J.; Li, Y.; Zhao, Y.; Yuan, J.; Cao, Y.; Wang, L.; Zhang, Z.; Zhang, B.; Wang, C.C.; et al. YY1 regulates skeletal muscle regeneration through controlling metabolic reprogramming of satellite cells. EMBO J. 2019, 38, e99727. [Google Scholar] [CrossRef]
- Walowitz, J.L.; Bradley, M.E.; Chen, S.; Lee, T. Proteolytic regulation of the zinc finger transcription factor YY1, a repressor of muscle-restricted gene expression. J. Biol. Chem. 1998, 273, 6656–6661. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.M.; Lee, S.H.; Yum, J.; Yeo, C.Y.; Lee, K.Y. Smurf2 regulates the degradation of YY1. Biochim. Biophys. Acta 2014, 1843, 2005–2011. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chan, M.C.; Yu, Y.; Bei, Y.; Chen, P.; Zhou, Q.; Cheng, L.; Chen, L.; Ziegler, O.; Rowe, G.C.; et al. miR-29b contributes to multiple types of muscle atrophy. Nat. Commun. 2017, 8, 15201. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, T.; Sha, Z.; Tang, H.; Hua, X.; Wang, L.; Wang, Z.; Gao, Z.; Sluijter, J.P.G.; Rowe, G.C.; et al. Angiotensin II-induced muscle atrophy via PPARγ suppression is mediated by miR-29b. Mol. Ther. Nucleic Acids 2021, 23, 743–756. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Li, L.; Ge, M.; Gu, L.; Wang, M.; Zhang, K.; Su, Y.; Zhang, Y.; Lan, M.; Yu, Y.; et al. Overexpression of miR-29 Leads to Myopathy that Resemble Pathology of Ullrich Congenital Muscular Dystrophy. Cells 2019, 8, 459. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhou, L.; Jiang, P.; Lu, L.; Chen, X.; Lan, H.; Guttridge, D.C.; Sun, H.; Wang, H. Loss of miR-29 in myoblasts contributes to dystrophic muscle pathogenesis. Mol. Ther. 2012, 20, 1222–1233. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Song, Y.; Fu, Y.; Li, P. MiR-29b mimics promotes cell apoptosis of smooth muscle cells via targeting on MMP-2. Cytotechnology 2018, 70, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Bar-Shai, M.; Carmeli, E.; Coleman, R.; Rozen, N.; Perek, S.; Fuchs, D.; Reznick, A.Z. The effect of hindlimb immobilization on acid phosphatase, metalloproteinases and nuclear factor-kappaB in muscles of young and old rats. Mech. Ageing Dev. 2005, 126, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Koshizuka, K.; Kikkawa, N.; Hanazawa, T.; Yamada, Y.; Okato, A.; Arai, T.; Katada, K.; Okamoto, Y.; Seki, N. Inhibition of integrin beta1-mediated oncogenic signalling by the antitumor microRNA-29 family in head and neck squamous cell carcinoma. Oncotarget 2018, 9, 3663–3676. [Google Scholar] [CrossRef]
- Rozo, M.; Li, L.; Fan, C.M. Targeting beta1-integrin signaling enhances regeneration in aged and dystrophic muscle in mice. Nat. Med. 2016, 22, 889–896. [Google Scholar] [CrossRef]
- Robinson, C.J.; Thiagarajan, L.; Maynard, R.; Aruketty, M.; Herrera, J.; Dingle, L.; Reid, A.; Wong, J.; Cao, H.; Dooley, J.; et al. Release of miR-29 Target Laminin C2 Improves Skin Repair. Am. J. Pathol. 2024, 194, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Duhachek-Muggy, S.; Zolkiewska, A. ADAM12-L is a direct target of the miR-29 and miR-200 families in breast cancer. BMC Cancer 2015, 15, 93. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Li, R.; Workeneh, B.; Dong, Y.; Wang, X.; Hu, Z. Transcription factor FoxO1, the dominant mediator of muscle wasting in chronic kidney disease, is inhibited by microRNA-486. Kidney Int. 2012, 82, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Fu, X.; Chen, X.; Xu, S.; Yu, J. Silencing of miR-486 alleviates LPS-stimulated inflammatory response of macrophages through targeting SIRT1. RSC Adv. 2019, 9, 17057–17064. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Liu, D.; Jiang, S.; Li, H.; Chen, L.; Wu, Y.; Essien, A.E.; Opoku, M.; Naranmandakh, S.; Liu, S.; et al. SIRT1 signaling pathways in sarcopenia: Novel mechanisms and potential therapeutic targets. Biomed. Pharmacother. 2024, 177, 116917. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cai, Y.; Huang, H.; Chen, X.; Chen, X.; Chen, X.; Mai, H.; Li, X.; Zhao, J.; Yang, J.; et al. miR-486-3p Influences the Neurotoxicity of a-Synuclein by Targeting the SIRT2 Gene and the Polymorphisms at Target Sites Contributing to Parkinson’s Disease. Cell Physiol. Biochem. 2018, 51, 2732–2745. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhang, C.; Zhang, A.; Cai, H.; Price, S.R.; Wang, X.H. MicroRNA-23a and MicroRNA-27a Mimic Exercise by Ameliorating CKD-Induced Muscle Atrophy. J. Am. Soc. Nephrol. 2017, 28, 2631–2640. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Hu, L.; Cheng, J.; Klein, J.D.; Hassounah, F.; Cai, H.; Li, M.; Wang, H.; Wang, X.H. Acupuncture plus low-frequency electrical stimulation (Acu-LFES) attenuates denervation-induced muscle atrophy. J. Appl. Physiol. 2016, 120, 426–436. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Klein, J.D.; Hassounah, F.; Cai, H.; Zhang, C.; Xu, P.; Wang, X.H. Low-frequency electrical stimulation attenuates muscle atrophy in CKD—A potential treatment strategy. J. Am. Soc. Nephrol. 2015, 26, 626–635. [Google Scholar] [CrossRef]
- Klein, J.D.; Wang, X.H. Electrically stimulated acupuncture increases renal blood flow through exosome-carried miR-181. Am. J. Physiol. Renal Physiol. 2018, 315, F1542–F1549. [Google Scholar] [CrossRef]
- McCarthy, J.J. The MyomiR network in skeletal muscle plasticity. Exerc. Sport. Sci. Rev. 2011, 39, 150–154. [Google Scholar] [CrossRef] [PubMed]
- An, J.N.; Kim, J.K.; Lee, H.S.; Kim, S.G.; Kim, H.J.; Song, Y.R. Serum cystatin C to creatinine ratio is associated with sarcopenia in non-dialysis-dependent chronic kidney disease. Kidney Res. Clin. Pract. 2022, 41, 580–590. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.L.; Wang, C.H.; Chang, I.C.; Hsu, B.G. A Novel Application of Serum Creatinine and Cystatin C to Predict Sarcopenia in Advanced CKD. Front. Nutr. 2022, 9, 828880. [Google Scholar] [CrossRef] [PubMed]
- Buehring, B.; Binkley, N. Myostatin--the holy grail for muscle, bone, and fat? Curr. Osteoporos. Rep. 2013, 11, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Barrett, D.; Bilic, S.; Chyung, Y.; Cote, S.M.; Iarrobino, R.; Kacena, K.; Kalra, A.; Long, K.; Nomikos, G.; Place, A.; et al. A Randomized Phase 1 Safety, Pharmacokinetic and Pharmacodynamic Study of the Novel Myostatin Inhibitor Apitegromab (SRK-015): A Potential Treatment for Spinal Muscular Atrophy. Adv. Ther. 2021, 38, 3203–3222. [Google Scholar] [CrossRef] [PubMed]
- Jin, Q.; Qiao, C.; Li, J.; Xiao, B.; Li, J.; Xiao, X. A GDF11/myostatin inhibitor, GDF11 propeptide-Fc, increases skeletal muscle mass and improves muscle strength in dystrophic mdx mice. Skelet. Muscle 2019, 9, 16. [Google Scholar] [CrossRef] [PubMed]
- Yasar, E.; Tek, N.A.; Tekbudak, M.Y.; Yurtdas, G.; Gulbahar, O.; Uyar, G.O.; Ural, Z.; Celik, O.M.; Erten, Y. The Relationship Between Myostatin, Inflammatory Markers, and Sarcopenia in Patients With Chronic Kidney Disease. J. Ren. Nutr. 2022, 32, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Zeng, Q.; Xu, Q.; Zhou, H.; Tan, R.; Zhong, X.; Liu, Y.; Li, Y.; Liu, Y. The non-linear associations between blood manganese level and sarcopenia in patients undergoing maintenance hemodialysis: A multicenter cross-sectional study. J. Trace Elem. Med. Biol. 2024, 84, 127465. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Mok, B.W.; Deng, S.; Liu, H.; Wang, P.; Song, W.; Chen, P.; Huang, X.; Zheng, M.; Lau, S.Y.; et al. Mammalian cells use the autophagy process to restrict avian influenza virus replication. Cell Rep. 2021, 35, 109213. [Google Scholar] [CrossRef]
- Tavares, F.S.; de Luca Corrêa, H.; Wilund, K.R.; Deus, L.A.; de Araújo, T.B.; Tzanno-Martins, C.; da Motta Vilalva Mestrinho, V.M.; Dos Santos, R.L.; Reis, A.L.; Souza, F.H.; et al. Exploring the impact of short daily haemodialysis on muscle strength and bone health in end-stage kidney disease patients. J. Cachexia Sarcopenia Muscle 2024, 15, 718–725. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, Q.; Chen, Z.; Wang, Y.; Gamboa, J.L.; Ikizler, T.A.; Garibotto, G.; Mitch, W.E. Mechanisms Regulating Muscle Protein Synthesis in CKD. J. Am. Soc. Nephrol. 2020, 31, 2573–2587. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Jiang, X.; Zhong, Q.; Zhang, Y.; Zhang, H.; Liu, Z.; Wang, X. Possible sarcopenia and risk of chronic kidney disease: A four-year follow-up study and Mendelian randomization analysis. Endocr. Res. 2024, 49, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Nakano, Y.; Mandai, S.; Naito, S.; Fujiki, T.; Mori, Y.; Ando, F.; Mori, T.; Susa, K.; Iimori, S.; Sohara, E.; et al. Effect of osteosarcopenia on longitudinal mortality risk and chronic kidney disease progression in older adults. Bone 2024, 179, 116975. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Pathogen/Family | Effect of CKD/Association with Kidney Function | Potential Link with Sarcopenia | References |
|---|---|---|---|
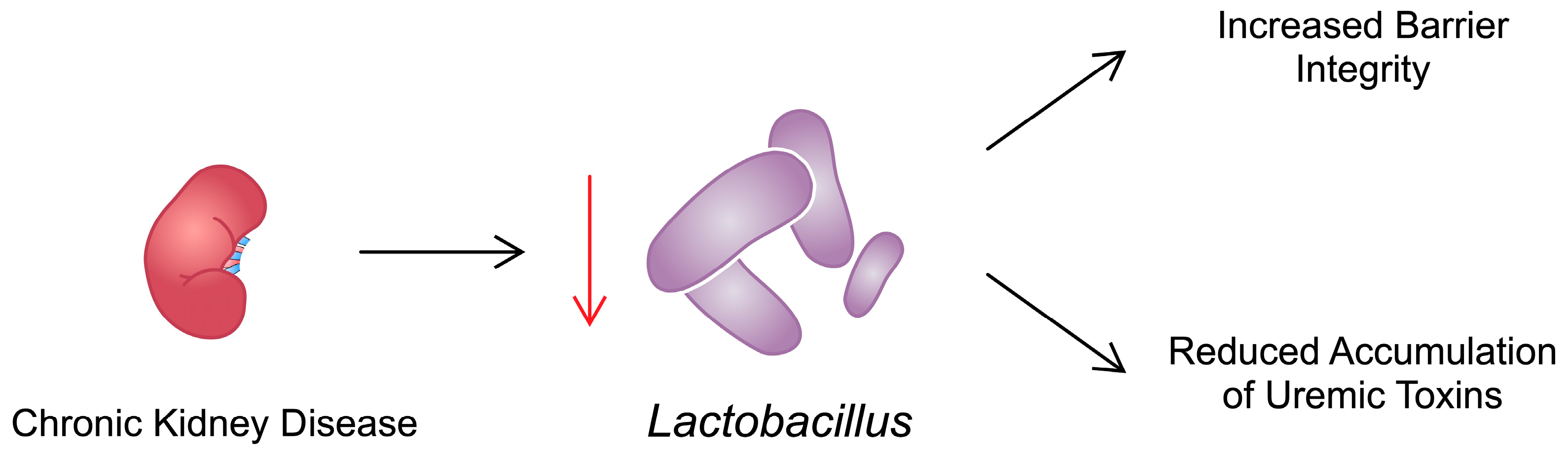
| Lactobacillaceae | Reduction in the number of families with enzymes that form short-chain fatty acids (butyrate) in the intestine in CKD | Lactobacillus pluralis and Lactobacillus spp. supplementation have a positive effect on muscle atrophy | [83,84,94,95,98,113,114] |
| Prevotellaceae | Reduction in the number of families with enzymes that form short-chain fatty acids (butyrate) in the intestine in CKD | Reduced Prevotellaceae abundance in Chinese women Reduced Prevotellaceae abundance in older people with low muscle strength | [83,84,115,116,117] |
| Bifidobacterium | Decrease in CKD | In a rat model, Bifidobacterium supplementation had a positive effect on muscle atrophy Increase in people with physical weakness and sarcopenia | [94,95,96,111,114,116] |
| Proteobacteria | Increase in CKD | Indirect association in an alcohol-abusing group with lower muscle mass based on handgrip strength: increase in Proteobacteria Mice treated with metronidazole showed an increase in Proteobacteria abundance and a decrease in lower limb muscle mass | [114,118,119] |
| Escherichia coli | Increase in E. coli population and enhanced production of indole. | Enhanced E. coli growth shown in patients with cirrhosis and muscular atrophy | [113,114,120] |
| Allobaculum | Increase in CKD | Increased Allobaculum abundance may contribute to a sarcopenic state | [105] |
| Lactonifactor | Decrease in CKD | Decreased Lactonifactor abundance may contribute to a sarcopenic state | [105] |
| Alistipes | Increase (production of indole, a precursor of IS, from tryptophan) | Decreased Alistipes shahii abundance in sarcopenia | [82,104,116,121] |
| Eggerthella | Increase in CKD Eggerthella lenta | Increased Eggerthella abundance | [110,111,116] |
| Blautia | Decrease in CKD | Increased Blautia abundance | [110,122] |
| Faecalibacterium | Decrease in CKD | Decreased Faecalibacterium prausnitzii abundance in sarcopenia | [110,121] |
| Peptostreptococcus | Growth of Peptostreptococcaceae with administration of capsules with a mixture of live bacteria Enterococcus faecalis, Bifdobacterium longum, and Lactobacillus acidophilus) in hemodialysis patients | Increased Peptostreptococcus abundance in individuals with physical weakness and sarcopenia | [111,123] |
| Dialister | Dialister abundance decreased with increasing severity of CKD | Increased Dialister abundance in individuals with physical weakness and sarcopenia | [111,124] |
| Pyramidobacter | CKD+AST-120 group showed significant enrichment in Pyramidobacter compared to CKD group without AST-120 treatment | Increased Pyramidobacter abundance in individuals with physical weakness and sarcopenia | [111,125] |
| Eubacterium | CKD+AST-120 group showed significant enrichment in Eubacterium nodatum compared to CKD group without AST-120 treatment | Decreased Eubacterium abundance in individuals with physical weakness and sarcopenia | [111,125] |
| MicroRNA | Expression in Chronic Kidney Disease Conditions | Mechanism Linking Chronic Kidney Disease and Muscle Functionality | References |
|---|---|---|---|
| miR-26a | ↓ | Overexpression of miR-26a inhibits muscle atrophy. | [143,145] |
| miR-29 | ↓ | Overexpression of miR-29 improves C2C12 myoblasts differentiation. Intramuscular injection of miR-29a could reduce muscular atrophy markers. | [142,158] |
| miR-486 | ↓ | Administration of miR-486 mimic into the muscles of CKD mice increased muscle mass and reduced the expression of atrophy markers. | [173] |
| miR-23 | ↓ | Overexpression of miR-23 together with miR-27 increased muscle mass and strength. | [177] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bakinowska, E.; Olejnik-Wojciechowska, J.; Kiełbowski, K.; Skoryk, A.; Pawlik, A. Pathogenesis of Sarcopenia in Chronic Kidney Disease—The Role of Inflammation, Metabolic Dysregulation, Gut Dysbiosis, and microRNA. Int. J. Mol. Sci. 2024, 25, 8474. https://doi.org/10.3390/ijms25158474
Bakinowska E, Olejnik-Wojciechowska J, Kiełbowski K, Skoryk A, Pawlik A. Pathogenesis of Sarcopenia in Chronic Kidney Disease—The Role of Inflammation, Metabolic Dysregulation, Gut Dysbiosis, and microRNA. International Journal of Molecular Sciences. 2024; 25(15):8474. https://doi.org/10.3390/ijms25158474
Chicago/Turabian StyleBakinowska, Estera, Joanna Olejnik-Wojciechowska, Kajetan Kiełbowski, Anastasiia Skoryk, and Andrzej Pawlik. 2024. "Pathogenesis of Sarcopenia in Chronic Kidney Disease—The Role of Inflammation, Metabolic Dysregulation, Gut Dysbiosis, and microRNA" International Journal of Molecular Sciences 25, no. 15: 8474. https://doi.org/10.3390/ijms25158474