Abstract
Obesity-related ciliopathies, as a group of ciliopathies including Alström Syndrome and Bardet–Biedl Syndrome, exhibit distinct genetic and phenotypic variability. The understanding of these diseases is highly significant for understanding the functions of primary cilia in the human body, particularly regarding the relationship between obesity and primary cilia. The diagnosis of these diseases primarily relies on clinical presentation and genetic testing. However, there is a significant lack of research on biomarkers to elucidate the variability in clinical manifestations, disease progression, prognosis, and treatment responses. Through an extensive literature review, the paper focuses on obesity-related ciliopathies, reviewing the advancements in the field and highlighting the potential roles of biomarkers in the clinical presentation, diagnosis, and prognosis of these diseases.
1. Introduction
1.1. Primary Cilia and Obesity
Primary cilia, also known as sensory cilia, are highly conserved hair-like organelles [1]. Cilia consist of a microtubule core called the axoneme, which extends from a modified centriole known as the basal body. A variety of receptors and ion channels are embedded within the ciliary membranes, facilitating the detection and transmission of stimuli from the extracellular environment, while also being capable of dispatching signals outward [2].
Obesity prevention and control is a global public health challenge. The 2023 World Obesity Map predicts that the worldwide prevalence of obesity will rise to 24% in 2035, with a total of nearly two billion people, and the obese population is also showing a trend in younger groups [3,4].
There is a strong link between primary cilia and obesity. A series of obesity-associated loci identified by genome-wide association studies (GWAS) are confirmed to be related to hypothalamic cilia, including adenylate cyclase 3 (ADCY3) and the melanocortin-4 receptor (MC4R). ADCY3 has been seen the marker of cilia in neurons, and MC4R is found to be specifically located on primary cilia in the hypothalamic paraventricular nucleus of mice [5].
Meanwhile, the causative genes associated with morbid obesity in humans were later verified to be associated with primary cilia, suggesting that primary cilia may have an important role in obesity and energy metabolism [6].
1.2. Obesity-Related Ciliopathies
Ciliopathies are a class of genetic disorders whose etiology is related to ciliary dysfunction [7]. The proteins these genes encode are localized in the cilia–centromere complex, affecting the assembly, maintenance or function of the centrioles or cilia [8,9,10]. Cilia are expressed and play roles in most mature cells of the human body; thus, ciliary defects affect nearly all organs and tissues, leading to complex symptoms. Common clinical manifestations include obesity, kidney abnormalities, vision and hearing impairments, heart defects, and insulin resistance [8,10,11,12]. In total, five ciliopathies are characterized with obesity, including Bardet–Biedl syndrome (BBS, OMIM #209900), Alström syndrome (ALMS, OMIM #203800), Carpenter syndrome (CRPT, OMIM #201000), mental retardation, truncal obesity, retinal dystrophy, and micropenis syndrome (MORMS, OMIM #610156), and morbid obesity and spermatogenic failure (MOSPGF, OMIM #615703) [8,13,14,15,16,17]. Most of these diseases are inherited in an autosomal recessive pattern and the pathogenic mutations usually lead to the loss of the protein or the production of a non-functional or truncated protein. BBS has also been described to have a triallelic pattern of inheritance [18].
ALMS is caused by mutations in the ALMS1 gene, with an incidence of 1–9 per 1,000,000 [13]. Defect ALMS1 can lead to multi-organ system damage, with primary features including early childhood obesity, insulin resistance and hyperinsulinemia, retinal cone dystrophy, and hearing impairment [19]. The clinical phenotype, onset time, and severity vary significantly among patients [8,13,20,21,22].
BBS is also a ciliopathy affecting multiple organ systems, with the estimated incidence of 1 per 160,000 in northern European populations [14]. Unlike ALMS, it could be caused by mutations in many genes, including BBS1-22, IFT74, SCLT1, SCAPER, and NPHP genes [14,23]. Major clinical manifestations include retinal dystrophy, polydactyly, early-onset obesity, hypogonadism, intellectual disability, and renal abnormalities [23,24]. Secondary clinical manifestations include congenital heart disease, liver involvement (such as liver fibrosis and liver cysts), endocrine disorders, like diabetes, hypothyroidism, and hypercholesterolemia, enamel hypoplasia, ataxia, delayed speech and growth, craniofacial anomalies, and olfactory abnormalities [23,24].
CRPT, with an incidence estimated at 1 per 1 million births [25], is characterized by craniosynostosis, polydactyly, cardiac defects, and obesity [26,27,28]. It can be divided into two types. Carpenter syndrome-1 (CRPT1) is caused by homozygous mutations in the RAB23 gene [28]. According to the Human Gene Mutation Database (HGMD), only 17 pathogenic RAB23 variants have been described in patients with CRPT1 [29]. Carpenter syndrome-2 (CRPT2) is caused by mutations in the MEGF8 gene [15].
MORMS is caused by homozygous mutations in the INPP5E gene [30]. The primary clinical features of MORMS include intellectual disability, truncal obesity, retinal dystrophy, and a micropenis [16,30]. To date, only one family has been reported with this syndrome [16]. Obesity development is one of the hallmark features of MORMS. The case described by Torkar et al. [31] highlighted prominent phenotypic characteristics, including early-onset and severe obesity, accompanied by the development of metabolic syndrome.
MOSPGF is caused by mutations in the CEP19 gene [17]. The clinical symptom of this disease is much less complicated compared to other ciliopathies. The primary features of MOSPGF include morbid obesity and spermatogenic failure [17].
In addition, Joubert syndrome (JBTS, OMIM #213300), as a ciliopathy, is not associated with obesity generally, but, recently, Sophie et al. [32] found that a patient with JBTS caused by mutations in the ARL13B gene exhibited an obesity phenotype, expanding the spectrum of JBTS.
1.2.1. Localization and Functions in Cilia
These proteins encoded by the genes of obesity-related ciliopathies are located in the different accessory substructures of the primary cilia and have unique functions (Figure 1). ALMS1 and CEP19 are located in the basal body [33,34]. CEP19 participates in the process of triggering the entry of intraflagellar transport (IFT) into the cilium [34]. RAB23 localizes to the basal region of the cilium adjacent to one of the centrioles, promoting cilium formation [35]. Eight of the BBS proteins (BBS1, BBS2, BBS4, BBS5, BBS7, BBS8/TTC8, BBS9, and BBS18/BBIP1) form a transport complex called BBSome [36] while proteins BBS6/MKKS, BBS10, and BBS12 are known as chaperone complexes, facilitating the BBSome assembly [37]. In addition, BBS3/ARL6 is a GTPase regulating the BBSome entry to (and exit from) the cilium [38]. ARL13B is also a GTPase ciliary affecting transmembrane protein localizations and anterograde IFT assembly stability though Sonic Hedgehog (Shh) signaling [39]. Proteins encoded by other genes of BBS are believed to be located on the basal body or region, playing roles on recruitment of BBSome [38]. INPP5E encodes a 72 kDa phosphatase localized in the axoneme of primary cilia and plays a crucial role in regulating the PI3K signaling pathway within cilia [40]. Megf8 is now believed to take part in forming a membrane-tethered ubiquitin complex that can fine-tune the strength of Hedgehog (Hh) signaling [41].
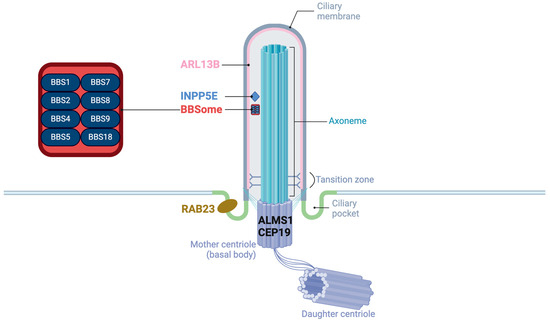
Figure 1.
The structure of primary cilia and the localization of proteins of obesity-related ciliopathies.
1.2.2. Clinical Presentations, Diagnosis, and Treatment
There are similarities as well as differences in the clinical presentations between obesity-related ciliopathies due to the different roles of cilia-related genes in various organs. Table 1 categorizes and lists the confirmed clinical phenotypes of five obesity-related ciliopathies, grouped by systems and organs [8,13,14,15,16,17,19,20,21,22,23,24,25,26,27,28,29,30,38,42,43].

Table 1.
All associated phenotypes of the ciliopathies characterized with obesity.
Apart from the above three systems, the clinical manifestations presented in different obesity-related ciliopathy are different. In the musculoskeletal system, polydactyly is typical for BBS [8,14,23,24,38], while spinal abnormalities, such as scoliosis and kyphosis, are common in ALMS [13,19,20,21,22,42]. The symptoms vary a lot in CRPT [15,25,26,27,28,29].
Patients with BBS, ALMS, and CRPT exhibit urinary system abnormalities, with specific differences observed among these conditions [8,13,14,15,19,20,21,22,23,24,25,26,27,28,29,38,42,44]. Chronic kidney disease is a dominant feature in BBS and could lead to increased morbidity and premature death [45]. The symptom in other diseases is much less severe in comparison with that in BBS [24,45]. Vesicoureteral reflux and nephritis could be observed in patients with ALMS [20], while patients with CRPT usually have hydronephrosis or pyelonephritis [8,26]. In the cardiovascular system, cardiomyopathy is common in ALMS while being rare in other diseases [8,19]. Patients with CRPT are more likely to have congenital heart defects, such as tetralogy of Fallot, patent ductus arteriosus, and atrial septal defect [8]. Abnormalities in the digestive and respiratory systems are usually in BBS and ALMS. Abnormal liver function is common, while non-alcoholic fatty liver disease shows higher incidence in patients with ALMS and BBS. They also exhibit issues related to recurrent respiratory infections [19,23,24]. No abnormalities in these systems were identified in patients with CRPT and MORMS [8,15,16,25,26,27,28,29,30].
The diagnosis of obesity-related ciliopathies primarily consists of two parts: clinical diagnosis and molecular diagnosis [19,24]. There are specific criterial for diagnosis for patients with ALMS and BBS. For example, the diagnosis of BBS can be confirmed if a patient meets four out of six major clinical symptoms or three major symptoms plus two secondary criteria [46]. The clinical diagnosis is made based on the clinical findings (signs and symptoms), medical history, and family history. Genetic sequencing is a powerful tool to confirm a molecular diagnosis. Other strategies, such as PCR and hybridization-based tests, can also be employed, particularly for cascade testing or in populations where there are recurrent hotspot mutations. The number of patients with CRPT, MORMS, and MOSPGF is relatively small, and research on clinical and molecular diagnosis is limited. However, the diagnostic strategy is similar to that for ALMS and BBS.
Currently, symptomatic treatment remains the primary approach for managing obesity-related ciliopathies [47]. For the obesity seen in ALMS and BBS, the MC4R agonist setmelanotide [48] and GLP-1 receptor agonists (GLP-1 RAs) [49] are promising therapeutic options. Notably, the U.S. FDA approved setmelanotide for the chronic management of weight in adults and pediatric patients aged 6 and older with deficiencies in POMC, LEPR, or PCSK1 in 2020 [50]. In June 2022, the indication for setmelanotide was expanded to include patients with BBS [51]. However, the variability in treatment response among patients requires further research to better understand the underlying mechanisms. These new drug targets also have the potential to serve as biomarkers for monitoring treatment efficacy and prognosis.
1.2.3. Characteristics of Obesity
The characteristics of obesity associated with the five ciliopathies are summarized in Table 2. The Table describes the age of onset, prevalence, BMI, and common comorbidities associated with obesity for each condition [14,16,17,19,24,26,28,30]. The onset age and prevalence of obesity vary among the ciliopathies, with BBS, ALMS, and MOSPGF showing early onset obesity [17,19,24], while MORMS typically manifests obesity when patients grow older (5 to 15 years) [16]. The prevalence of obesity is high in ALMS, MOSPGF (91%), and BBS (89%) [17,19,24]. The features of obesity also differ, with central obesity in BBS and truncal obesity in MORMS, whereas MOSPGF is characterized by morbid obesity with BMI > 40.0 kg/m2 [17]. CRPT is notable for high birth weight and obesity present at birth, with a prevalence of 90%, but specific onset range and BMI are not reported [26,28].
Table 2.
Obesity features between BBS, ALMS, CRPT, MORMS, and MOSPGF.
1.2.4. Mechanism of Obesity
Obesity in the human body is actually the result of an imbalance between energy intake and energy expenditure (Figure 2). Most cells, including neurons and glial cells throughout the central nervous system, possess primary cilia. The hypothalamus is especially critical for the regulation of energy homeostasis. The agouti-related protein (AgRP) neurons and the pro-opiomelanocortin (POMC) neurons in the arcuate nucleus of the hypothalamic tuberal region positively and negatively regulate feeding behavior, respectively [52]. They are also regulated by the periphery, mainly by insulin secreted by the pancreas, leptin secreted by adipocytes, ghrelin secreted by stomach, and cholecystokinin secreted by small intestine. MC4R neurons in the paraventricular nucleus, as their downstream neurons, directly participate in the regulation of energy metabolism [52].
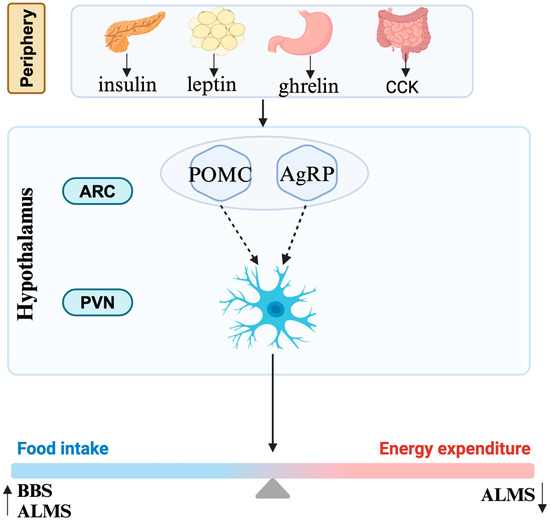
Figure 2.
Mechanism of obesity. CCK, cholecystokinin; ARC, arcuate nucleus; PVN, paraventricular nucleus; POMC, pro-opiomelanocortin; AgRP, agouti-related protein; BBS, Bardet–Biedl syndrome; ALMS, Alström syndrome.
The mechanisms associated with ciliary dysfunction and obesity in these syndromes are complex and only partially understood. It is currently believed that obesity in these diseases is associated through the leptin–melanocortin pathway influencing the hypothalamus [7]. Defect function of cilia in these diseases probably affects signaling between neurons/glial cells in this pathway while the causative mechanisms vary in different diseases. Obesity of BBS is caused by defects in the neurological control of the appetite, although it is unclear whether defective leptin signaling [53] or signaling by anorexigenic GPCR neuropeptide Y family receptors is the primary cause [54]. Similar to the situation in BBS, an increase in energy intake was observed both in human and mice with ALMS [55]. The percentage of ciliated hypothalamic neurons is significantly reduced in Alms1 mutant mice [56]. Apart from that, a significant reduction in energy expenditure also accounts for the process of obesity, though the specific mechanism is still unknown [55]. The obesity in CRPT may result from damaged Hh signaling or RAB23 itself potentially regulating adipogenesis [28,29].
2. Advance of Biomarkers
Due to rarity of patients with obesity-related ciliopathies, the progress in identifying related biomarkers is based predominantly on ALMS and BBS studies [6]. These two syndromes have been the focus of more extensive research, leading to a better understanding of their underlying mechanisms and potential biomarkers for diagnosis, variability in clinical presentation, progress, and prognosis and treatment.
2.1. Diagnosis and Differential Diagnosis
The diagnosis of obesity-related ciliopathies primarily involves two components: clinical diagnosis and molecular diagnosis [19,24]. For ALMS and BBS, clinical diagnosis is mainly based on characteristic clinical features, while molecular diagnosis, through genetic testing, is considered the gold standard for definitive diagnosis [19,24]. With the advancement of gene sequencing technologies, next-generation sequencing (NGS) has become the primary method for diagnosing monogenic diseases, including obesity-related ciliopathies [57]. These diseases are autosomal recessive disorders, meaning that the diagnosis can be confirmed by detecting two pathogenic variants in the associated genes. However, there are clinical scenarios where the diagnosis may be challenging. In some cases, patients may exhibit high clinical suspicion for a ciliopathy, while the result of genetic testing may be negative, or only one pathogenic variant is detected, which is insufficient to confirm a diagnosis [58].
Gene functional studies can provide new evidence for the ACMG classification of gene variants. Chen et al. [59] conducted a study on patients with a clinical suspicion of ALMS who carried one pathogenic variant and one variant of uncertain significance (VUS). They performed ALMS1 protein expression staining on skin fibroblasts and discovered that ALMS1 expression at the centrosome was severely impaired. This finding provided evidence to prove the pathogenicity of the missense variant of ALMS1 and confirmed the diagnosis of ALMS. In contrast, for patients carrying only VUS, staining results indicated that ALMS1 expression was not impaired, excluding the diagnosis of ALMS; however, further genetic testing confirmed the diagnosis of BBS. Thus, this study suggests that ALMS1 expression detection could be a promising differential diagnostic marker for ALMS.
2.2. Variability in Clinical Presentation, Progress, and Prognosis
Obesity-related ciliopathies exhibit highly complex and variable clinical manifestations. As genetic disorders, genotype–phenotype analyses can partly explain the variability in clinical presentations, disease progression, and prognosis.
2.2.1. Genotype–Phenotype Correlation
The clinical manifestations of obesity-related ciliopathies are highly complex, with the same disease potentially caused by different genes. The clinical presentation of BBS can vary depending on the specific gene involved. In a meta-analysis by Niederlova et al. [38], differences in clinical symptoms caused by various BBS genes were compared (Table 3). Patients with BBS3 mutations tend to have fewer clinical symptoms. Retinal dystrophy is usually milder in patients with BBS1 gene mutations while being severe in patients with BBS2, BBS3, and BBS4 Mutations. Patients with BBS2 mutations have a higher incidence of polydactyly, and BBS10 gene mutations are linked to more pronounced obesity and insulin resistance [38].
Table 3.
Genotype–phenotype correlation in ALMS and BBS.
Unlike BBS, which is associated with many genes, ALMS is a monogenic disease. However, the ALMS1 gene is very large, and different mutation sites within this gene can result in varying severity, progression, and prognosis of clinical manifestations [58]. Patients with variants in exon 16 usually present with early onset (before one-year old) of retinal degeneration, urinary system dysfunction, dilated cardiomyopathy, and diabetes, while those with variants in exon 8 are prone to have milder symptoms or later onset of kidney disease [60]. A recent meta-analysis conducted by Brais Bea-Mascato et al. [22] revealed important insights into the genotype–phenotype, based on data from 227 ALMS patients. Patients with the longest allele of the ALMS1 gene truncated around exon 10 (E10) exhibit a higher prevalence of liver dysfunction and experience worse disease progression. However, no significant differences in the prevalence of dilated cardiomyopathy (DCM), hypertrophic cardiomyopathy (HCM), and type 2 diabetes mellitus (T2DM) are observed among patients grouped by longevity of allele. Savas et al. found that the c.7911dupC (p. Asn2638Glnfs*24) mutation can be related to severe cardiomyopathy in ALMS [61].
2.2.2. Other Biomarkers from Multi-Omics Data
Additionally, advances in multi-omics data have also made great contributions to the discovery of new biomarkers (Table 4).
Fatty liver is a common issue in these diseases and can progress to liver fibrosis in advanced stages. This condition is related to obesity but may also be linked to pathogenic genes. ALMS is currently considered a classic model for non-alcoholic fatty liver disease (NAFLD) [62].
For monitoring fatty liver disease, liver biopsy is considered as the gold standard. However, there is a limit of widespread use in clinic considering its invasiveness and complexity. Non-invasive indexes, such as the alanine aminotransferase (ALT)/aspartate aminotransferase (AST) ratio, the AST-to-platelet ratio index (APRI), and the Fibrosis-4 Index (FIB-4) could predict liver fibrosis in liver disease to some extent [63,64]. Recently, imaging methods, such as transient elastography (TE), FibroScan, and shear-wave elastography (SWE), have advanced. Silvia et al. [62] found that SWE plays an important predictive role in the progression of fatty liver to fibrosis in patients with ALMS.
Cardiomyopathy is another significant clinical manifestation of ALMS. It can be identified and monitored using biomarkers, such as NT-proBNP, high-sensitivity troponin, and T-wave inversion on a 12-lead electrocardiogram [65]. However, Nicola et al. [66] suggested that evidence for using NT-proBNP to monitor cardiomyopathy in ALMS may be insufficient. They thought that extracellular volume (ECV) expansion might be a more robust predictor of adverse cardiovascular outcomes. Additionally, they also proposed that elevated triglycerides could be used as a potential marker for cardiac fibrosis, but more prospective studies are needed to validate this.
Agnieszka et al. [67] conducted a study to detect and analyze microRNA (miRNA) expression in the serum of patients with ALMS, BBS, obese controls, and normal controls. They found that miR-301a-3p expression was significantly reduced in both ALMS and BBS patients. Meanwhile, miR-92b-3p expression was decreased in ALMS but increased in BBS. Additionally, they identified eight miRNAs (miR-30a-5p, miR-92b-3p, miR-99a-5p, miR-122-5p, miR-192-5p, miR-193a-5p, miR-199a-3p, and miR-205-5p) that showed significant correlations with clinical parameters, including lipid profiles, serum creatinine, cystatin C, fasting glucose, insulin, C-peptide levels, HbA1c values, and insulin resistance (HOMA-IR). These findings suggest that miRNAs could serve as valuable biomarkers of disease progression in patients with ALMS and BBS syndromes. However, further research is needed to validate these results and to fully understand the potential of miRNAs as biomarkers for these conditions.
Krzysztof et al. [68] conducted a non-targeted metabolomics analysis on patients with ALMS, BBS, obese controls, and normal controls. They found that metabolic changes in ALMS/BBS patients were similar to those observed in non-syndrome obesity, such as the higher levels of acylcarnitines, and tetrahydroaldosterone-3-glucuronide, which have been correlated with insulin resistance and hypertension, respectively, in numerous studies [69,70,71]. They also found that ALMS/BBS patients were characterized by elevated levels of oxidized phosphatidylcholines (PCs), which are recognized as markers of oxidative stress. These diseases exhibit clinical manifestations that often progress with age. Through further age-related analysis, they further found that lipids, primarily lysophosphatidylethanolamines (LPE), are major markers of disease progression. Interestingly, only one metabolite, long-chain fatty acid (FA 26:1; O2), showed a negative correlation with age in ALMS and BBS patients, while it correlated positively in the other groups, suggesting it may have a unique indicative biomarker role [67].
In another study, Krzysztof et al. conducted serum bone metabolism marker testing on patients with ALMS and BBS. They found serum osteocalcin (OC) and urinary deoxypyridinoline (DPD) levels were negatively correlated with the HOMA-IR index. Additionally, serum receptor activator of nuclear factor kappa-Β ligand (s-RANKL) levels were negatively correlated with fasting blood glucose concentrations [72].
Fibrotic changes have been reported in multiple organs in ALMS. Merlin et al. [73] analyzed miRNA expression on peripheral lymphocytes from six ALMS patients and found that fibrosis-related miR-324-5p was upregulated by 2.1-fold in ALMS males. Additionally, passenger strand members of the same mature miRNA category (miR-27a vs. miR-27a star), along with other miRNAs previously reported to be associated with fibrosis (miR-27a, miR-27b, miR-29b, and miR-25), showed disturbances. These findings suggest that miRNAs could potentially serve as biomarkers for fibrosis in ALMS.
Chronic kidney disease (CKD) is the most common cause of death among BBS patients, with significant variability in severity. Early identification and intervention are crucial for managing CKD in these patients [24]. Magnetic resonance diffusion tensor imaging (DTI) can be used to assess the microstructural integrity of the kidneys. Compared to the control group, BBS patients exhibited lower cortical fractional anisotropy (FA) and axial diffusivity, and higher mean diffusivity and radial diffusivity in the kidneys [74]. The urine protein profile of BBS patients can provide information on the risk and predictive factors for adverse renal outcomes and urinary markers of renal insufficiency. Notably, the abundance of urinary fibronectin (u-FN), CD44 antigen, and lysosomal α-glucosidase significantly correlated with glomerular filtration rate [75]. By comparing the urinary metabolomics of BBS patients and control subjects, they discovered that the excretion of several monocarboxylates, including lactate, was increased in the early and late stages of CKD [76]. Emanuela et al. conducted a targeted serum metabolomics study comparing BBS patients to control subjects. They found that renal insufficiency in BBS patients was associated with abnormal levels of plasma phosphatidylcholines and acylcarnitines. Miriam et al. assessed the renal function of 54 BBS patients. They discovered that maximum urine osmolality (max-Uosm) was correlated with the annual decline in estimated glomerular filtration rate (ΔeGFR), suggesting that a defect in urine concentration may predict disease progression [77].
Table 4.
Other biomarkers from multi-omics data.
Table 4.
Other biomarkers from multi-omics data.
| Clinical Features | Correlation Detection | Biomarkers/Specific Methods | Comment | References | |
|---|---|---|---|---|---|
| Fatty liver | Imaging analysis | SWE, TE | SWE plays an important predictive role in the progression of fatty liver to fibrosis in patients with ALMS | Silvia et al. [62] | |
| Metabolomics | ALT/AST ratio, APRI, FIB-4 | Predicts liver fibrosis in liver disease to some extent | Guangqin et al. [63], Erin et al. [64] | ||
| Cardiomyopathy | Imaging analysis | ECV | ECV expansion might be a more robust predictor of adverse cardiovascular outcomes | Nicola et al. [66] | |
| Metabolomics | NT-proBNP, high-sensitivity troponin, elevated triglycerides | Elevated triglycerides are proposed as a potential marker for cardiac fibrosis | Ashwin et al. [65], Nicola et al. [66] | ||
| Kidney | Proteomics | u-FN, CD44 antigen, lysosomal α-glucosidase | u-FN, CD44 antigen, and lysosomal α-glucosidase significantly correlated with glomerular filtration rate | Marianna et al. [75] | |
| Fibrosis | MiRNA sequencing | miRNAs (miR-324-5p, miR-27a, miR-27b, miR-29b, and miR-25) | MiRNAs could potentially serve as biomarkers for fibrosis in ALMS | Merlin et al. [73] | |
| Obesity and related metabolic syndromes | MiRNA sequencing | miRNAs (miR-30a-5p, miR-92b-3p, miR-99a-5p, miR-122-5p, miR-192-5p, miR-193a-5p, miR-199a-3p, and miR-205-5p) | MiR-301a-3p expression was significantly reduced in both ALMS and BBS patients; miR-92b-3p expression was decreased in ALMS but increased in BBS; miRNAs could serve as valuable biomarkers of disease progression in patients with ALMS and BBS | Agnieszka et al. [67] | |
| Metabolomics | Non-targeted metabolomics analysis | PC, LPE, long-chain fatty acid (FA 26:1; O2), acylcarnitines, tetrahydroaldosterone-3-glucuronide | LPEs are major markers of disease progression; only long-chain fatty acid (FA 26:1; O2) showed a negative correlation with age in ALMS and BBS patients | Krzysztof et al. [68] | |
| Serum bone metabolism marker testing | OC, DPD, s-RANKL | OC and DPD levels were negatively correlated with the HOMA-IR index | Krzysztof et al. [72] | ||
SWE, shear-wave elastography; TE, transient elastography; ECV, extracellular volume; ALMS, Alström syndrome; BBS, Bardet–Biedl syndrome; ALT, alanine aminotransferase; AST, aspartate aminotransferase; APRI, AST-to-platelet ratio index; FIB-4, Fibrosis-4 Index; miRNA, microRNA; PC, oxidized phosphatidylcholine; LPE, lysophosphatidylethanolamines; OC, osteocalcin; DPD, deoxypyridinoline; HOMA-IR, Homeostasis Model Assessment of Insulin Resistance; s-RANKL, serum receptor activator of nuclear factor kappa-Β ligand; u-FN, urinary fibronectin.
3. Discussion
Primary cilia, present in most mature human cells, play crucial roles in various organ systems, and defects in cilia can lead to widespread clinical manifestations. Obesity-related ciliopathies constitute a small part of ciliopathies; however, they provide important clues to the study of the relationship between primary cilia and obesity in humans.
However, obesity-related ciliopathies are very rare, making the discovery of biomarkers much more difficult than other diseases. Regarding the importance of biomarkers in diagnosis, monitoring, and follow-up of these diseases, there is an urgent need for relevant biomarker studies and relevant reviews. Therefore, this paper focuses on the topic of biomarkers for obesity-related ciliopathies and reviews the advances of biomarkers in the clinical presentation, diagnosis, and prognosis of these diseases.
For a long time, the underlying causes of these diseases remained unknown, leading to unclear or delayed diagnoses for many patients. The development of genetic diagnostic technologies, especially NGS, has provided solutions, enabling accurate diagnosis [78]. Trio exome sequencing would be a great option considering the autosomal recessive inheritance pattern. Proband-only medical exome sequencing is considered as a more cost-effective test, especially in China [79], and WES has also proved to be a useful and cost-effective tool in diagnosing ciliopathy [80]. Some countries prefer using targeted gene panels, which focus on a specific set of genes known to be associated with ciliopathies [81]. However, the high cost of genetic testing is a significant barrier to widespread implementation, particularly in remote and low-income areas. The disparity in access to genetic testing in China may contribute to the uneven geographic distribution of ALMS diagnosed cases, with patients in remote and impoverished regions being less likely to receive a diagnosis [42]. RNA sequencing [82] and multiplex PCR [83] could be an optional diagnostic technique for complicated heterogeneous ciliopathies. For physicians with limited literature information of these disease, identifying pathogenic mutations with WES is a promising tool to provide clues to diagnose, although it could lead to overuse and the waste of resources.
For a subset of patients with unclear diagnoses, protein functional studies offer a pathway to resolve uncertainties, thereby providing additional evidence for the classification of variants. This approach not only improves diagnostic rates but also potentially provides new insights into disease severity assessment. Chen et al. [59] conducted immunofluorescence staining for ALMS1 protein expression on skin fibroblasts from 16 ALMS patients. They found that patients with significant ALMS1 expression fluorescence exhibited later onset and milder vision impairments.
The development of multi-omics technologies and analytical methods, including metabolomics, proteomics, imaging analysis, and miRNA sequencing, provide new possibilities for discovering novel biomarkers in these diseases. However, these advancements are still largely focused on explaining the variability in clinical manifestations. Additionally, the majority of research concentrated on AS and BBS, with limited data available for other obesity-related ciliopathies. There is currently a lack of biomarkers that can effectively indicate treatment efficacy. Up to now, only the genotype–phenotype correlation result is powerful enough to be used in clinical practice. For the biomarkers identified from multi-omics data, more evidence should be provided in the future to confirm the correlation and to validate their effectiveness and accuracy in clinical use.
A significant challenge in discovering biomarkers for obesity-related ciliopathies is the limited number of patients, which restricts the statistical power for identifying genotype–phenotype correlations and other omics biomarkers. For example, due to the limited number of patients, Krzysztof et al. did not separately conduct non-targeted metabolomics analysis for BBS and ALMS, which is a noted limitation [68].
Understanding these diseases is not only beneficial for the diagnosis and treatment of the disease itself, but also for the discovery of the role of cilia-related genes in obesity, which is expected to lead to the discovery of new therapeutic targets for obesity. For instance, ALMS1 is considered a biomarker for the diagnosis and prognosis of acute myocardial infarction (AMI) [84], with specific variants, such as G/A variant (rs674804) and glutamic acid repeat polymorphism, being markers for early-onset myocardial infarction [85,86]. Furthermore, Edwige et al. [87] identified a crucial protein–protein interaction between ALMS1 and protein kinase C-α (PKCα), which could be a new pharmacological class for treating IR and its numerous comorbidities, such as type 2 diabetes and CVD, in large populations.
4. Conclusions
Obesity-related ciliopathy is rare. Identifying the pathogenic gene with genetic sequencing is the main method to diagnose these diseases, making it a dominant biomarker. Genotype–phenotype correlation could also explain some variability in clinical presentation. Biomarkers identified with multi-omics are restricted to ALMS and BBS patients. There is currently a lack of biomarkers that can effectively indicate treatment efficacy.
5. Future Directions
More cost-effective and convenient genetic testing is crucial for enhancing the early diagnosis and management of obesity-related ciliopathies.
It is crucial to conduct international multi-center studies, pooling patient resources to carry out comprehensive analyses and particularly conducting lipidomics analysis in ALMS [68] and genotype–phenotype analyses focusing on different pathogenic variants within the same BBS gene.
Building on these efforts, integrating multi-omics data with longitudinal cohort studies is essential. Prospective studies that follow patients over time can provide valuable insights into the progression of diseases, prognosis, and treatment efficacy. By combining genomic, transcriptomic, proteomic, and metabolomic data, researchers can uncover new biomarkers and better understand their roles in disease mechanisms.
Author Contributions
Conceptualization, Q.Z., Y.H., S.G. and X.W.; writing—original draft preparation, Q.Z., Y.H. and S.G.; writing—review and editing, Q.Z., Y.H., S.G., Y.D., H.Z., G.C. and X.W. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Key Research and Development Program of China (2022YFC2703102) and National Nature Science Foundation of China (82170910).
Institutional Review Board Statement
This study did not require ethical approval because it reports other studies.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| GWAS | Genome-wide association studies |
| ADCY3 | Adenylate cyclase 3 |
| MC4R | Melanocortin 4 Receptor |
| GLP-1 | Glucagon-like Peptide-1 |
| BBS | Bardet–Biedl syndrome |
| ALMS | Alström syndrome |
| CRPT | Carpenter syndrome |
| MORMS | Mental retardation, truncal obesity, retinal dystrophy, and micropenis syndrome |
| MOSPGF | Morbid obesity and spermatogenic failure |
| JBTS | Joubert syndrome |
| HGMD | Human Gene Mutation Database |
| IFT | Intraflagellar transport |
| Shh | Sonic Hedgehog |
| Hh | Hedgehog |
| BMI | Body mass index |
| AgRP | Agouti-related protein |
| POMC | Pro-opiomelanocortin |
| NGS | Next-generation sequencing |
| ACMG | American College of Medical Genetics and Genomics |
| VUS | Variant of uncertain significance |
| DCM | Dilated cardiomyopathy |
| HCM | Hypertrophic cardiomyopathy |
| T2DM | Type 2 diabetes mellitus |
| NAFLD | Non-alcoholic fatty liver disease |
| ALT | Alanine aminotransferase |
| AST | Aspartate aminotransferase |
| APRI | AST-to-platelet ratio index |
| FIB-4 | Fibrosis-4 Index |
| TE | Transient elastography |
| SWE | Shear-wave elastography |
| NT-proBNP | N-terminal pro-brain natriuretic peptide |
| ECV | Extracellular volume |
| miRNA | MicroRNA |
| HOMA-IR | Homeostasis Model Assessment of Insulin Resistance |
| HbA1c | Glycated Hemoglobin A1c |
| PC | Phosphatidylcholine |
| LPE | Lysophosphatidylethanolamine |
| FA | Fatty acid |
| OC | Osteocalcin |
| DPD | Deoxypyridinoline |
| s-RANKL | Serum receptor activator of nuclear factor kappa-Β ligand |
| CKD | Chronic kidney disease |
| DTI | Diffusion tensor imaging |
| u-FN | Urinary fibronectin |
| GFR | Glomerular filtration rate |
| max-Uosm | Maximum urine osmolality |
| ΔeGFR | Estimated glomerular filtration rate |
| WES | Whole-exome sequencing |
| PCR | Polymerase chain reaction |
| AMI | Acute myocardial infarction |
| CCK | Cholecystokinin |
| ARC | Arcuate nucleus |
| PVN | Paraventricular nucleus |
References
- Wallmeier, J.; Nielsen, K.G.; Kuehni, C.E.; Lucas, J.S.; Leigh, M.W.; Zariwala, M.A.; Omran, H. Motile ciliopathies. Nat. Rev. Dis. Primers 2020, 6, 77. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wen, X.; Wang, Z.; Lin, Z.; Li, C.; Zhou, H.; Yu, H.; Li, Y.; Cheng, Y.; Chen, Y.; et al. Ciliary transition zone proteins coordinate ciliary protein composition and ectosome shedding. Nat. Commun. 2022, 13, 3997. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Ullah, R.; Wang, J.B.; Fu, J.F. Trends of obesity and overweight among children and adolescents in China. World J. Pediatr. 2023, 19, 1115–1126. [Google Scholar] [CrossRef] [PubMed]
- World Obesity Atlas 2023. Available online: https://www.worldobesityday.org/assets/downloads/ (accessed on 31 July 2024).
- Siljee, J.E.; Wang, Y.; Bernard, A.A.; Ersoy, B.A.; Zhang, S.; Marley, A.; Von Zastrow, M.; Reiter, J.F.; Vaisse, C. Subcellular localization of MC4R with ADCY3 at neuronal primary cilia underlies a common pathway for genetic predisposition to obesity. Nat. Genet. 2018, 50, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Vaisse, C.; Reiter, J.F.; Berbari, N.F. Cilia and Obesity. Cold Spring Harb. Perspect. Biol. 2017, 9, a028217. [Google Scholar] [CrossRef] [PubMed]
- Engle, S.E.; Bansal, R.; Antonellis, P.J.; Berbari, N.F. Cilia signaling and obesity. Semin. Cell Dev. Biol. 2021, 110, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Lovera, M.; Lüders, J. The ciliary impact of nonciliary gene mutations. Trends Cell Biol. 2021, 31, 876–887. [Google Scholar] [CrossRef] [PubMed]
- Reiter, J.F.; Leroux, M.R. Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell Biol. 2017, 18, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, F.; Benzing, T.; Katsanis, N. Ciliopathies. N. Engl. J. Med. 2011, 364, 1533–1543. [Google Scholar] [CrossRef]
- Tobin, J.L.; Beales, P.L. The nonmotile ciliopathies. Genet. Med. 2009, 11, 386–402. [Google Scholar] [CrossRef]
- Legendre, M.; Zaragosi, L.-E.; Mitchison, H.M. Motile cilia and airway disease. Semin. Cell Dev. Biol. 2021, 110, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.D.; Beck, S.; Maffei, P.; Naggert, J.K. Alström Syndrome. Eur. J. Hum. Genet. 2007, 15, 1193–1202. [Google Scholar] [CrossRef] [PubMed]
- Forsythe, E.; Beales, P.L. Bardet-Biedl syndrome. Eur. J. Hum. Genet. 2013, 21, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Twigg, S.R.F.; Lloyd, D.; Jenkins, D.; Elçioglu, N.E.; Cooper, C.D.O.; Al-Sannaa, N.; Annagür, A.; Gillessen-Kaesbach, G.; Hüning, I.; Knight, S.J.L.; et al. Mutations in multidomain protein MEGF8 identify a Carpenter syndrome subtype associated with defective lateralization. Am. J. Hum. Genet. 2012, 91, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Hampshire, D.J.; Ayub, M.; Springell, K.; Roberts, E.; Jafri, H.; Rashid, Y.; Bond, J.; Riley, J.H.; Woods, C.G. MORM syndrome (mental retardation, truncal obesity, retinal dystrophy and micropenis), a new autosomal recessive disorder, links to 9q34. Eur. J. Hum. Genet. 2006, 14, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Shalata, A.; Ramirez, M.C.; Desnick, R.J.; Priedigkeit, N.; Buettner, C.; Lindtner, C.; Mahroum, M.; Abdul-Ghani, M.; Dong, F.; Arar, N.; et al. Morbid obesity resulting from inactivation of the ciliary protein CEP19 in humans and mice. Am. J. Hum. Genet. 2013, 93, 1061–1071. [Google Scholar] [CrossRef] [PubMed]
- Katsanis, N.; Ansley, S.J.; Badano, J.L.; Eichers, E.R.; Lewis, R.A.; Hoskins, B.E.; Scambler, P.J.; Davidson, W.S.; Beales, P.L.; Lupski, J.R. Triallelic inheritance in Bardet-Biedl syndrome, a Mendelian recessive disorder. Science 2001, 293, 2256–2259. [Google Scholar] [CrossRef] [PubMed]
- Paisey, R.B.; Steeds, R.; Barrett, T.; Williams, D.; Geberhiwot, T.; Gunay-Aygun, M. Alström Syndrome. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. Available online: http://www.ncbi.nlm.nih.gov/books/NBK1267/ (accessed on 25 April 2024).
- Marshall, J.D.; Maffei, P.; Collin, G.B.; Naggert, J.K. Alström Syndrome: Genetics and Clinical Overview. Curr. Genom. 2011, 12, 225–235. [Google Scholar] [CrossRef]
- Marshall, J.D.; Bronson, R.T.; Collin, G.B.; Nordstrom, A.D.; Maffei, P.; Paisey, R.B.; Carey, C.; MacDermott, S.; Russell-Eggitt, I.; Shea, S.E.; et al. New Alström Syndrome Phenotypes Based on the Evaluation of 182 Cases. Arch. Intern. Med. 2005, 165, 675–683. [Google Scholar] [CrossRef]
- Bea-Mascato, B.; Valverde, D. Genotype–phenotype associations in Alström syndrome: A systematic review and meta-analysis. J. Med. Genet. 2024, 61, 18–26. [Google Scholar] [CrossRef]
- Melluso, A.; Secondulfo, F.; Capolongo, G.; Capasso, G.; Zacchia, M. Bardet-Biedl Syndrome: Current Perspectives and Clinical Outlook. Ther. Clin. Risk Manag. 2023, 19, 115–132. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, R.; Gunay-Aygun, M. Bardet-Biedl Syndrome Overview. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. Available online: http://www.ncbi.nlm.nih.gov/books/NBK1363/ (accessed on 27 April 2024).
- Bouaré, F.; Noureldine, M.H.A.; Hajhouji, F.; Ghannane, H.; Jallo, G.I.; Benali, S.A. Complex craniosynostosis in the context of Carpenter’s syndrome. Childs Nerv. Syst. ChNS Off. J. Int. Soc. Pediatr. Neurosurg. 2022, 38, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Hidestrand, P.; Vasconez, H.; Cottrill, C. Carpenter syndrome. J. Craniofac. Surg. 2009, 20, 254–256. [Google Scholar] [CrossRef] [PubMed]
- Temtamy, S.A. Carpenter’s syndrome: Acrocephalopolysyndactyly. An autosomal recessive syndrome. J. Pediatr. 1966, 69, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, D.; Seelow, D.; Jehee, F.S.; Perlyn, C.A.; Alonso, L.G.; Bueno, D.F.; Donnai, D.; Josifova, D.; Mathijssen, I.M.J.; Morton, J.E.V.; et al. RAB23 Mutations in Carpenter Syndrome Imply an Unexpected Role for Hedgehog Signaling in Cranial-Suture Development and Obesity. Am. J. Hum. Genet. 2007, 80, 1162–1170. [Google Scholar] [CrossRef] [PubMed]
- Khairat, R.; Elhossini, R.; Sobreira, N.; Wohler, E.; Otaify, G.; Mohamed, A.M.; Raouf, E.R.A.; Sayed, I.; Aglan, M.; Ismail, S.; et al. Expansion of the phenotypic and mutational spectrum of Carpenter syndrome. Eur. J. Med. Genet. 2022, 65, 104377. [Google Scholar] [CrossRef]
- Jacoby, M.; Cox, J.J.; Gayral, S.; Hampshire, D.J.; Ayub, M.; Blockmans, M.; Pernot, E.; Kisseleva, M.V.; Compère, P.; Schiffmann, S.N.; et al. INPP5E mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse. Nat. Genet. 2009, 41, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Drole Torkar, A.; Avbelj Stefanija, M.; Bertok, S.; Podkrajšek, K.T.; Debeljak, M.; Kranjc, B.S.; Battelino, T.; Kotnik, P. Novel Insights Into Monogenic Obesity Syndrome Due to INPP5E Gene Variant: A Case Report of a Female Patient. Front. Endocrinol. 2021, 12, 581134. [Google Scholar] [CrossRef]
- Thomas, S.; Cantagrel, V.; Mariani, L.; Serre, V.; Lee, J.-E.; Elkhartoufi, N.; de Lonlay, P.; Desguerre, I.; Munnich, A.; Boddaert, N.; et al. Identification of a novel ARL13B variant in a Joubert syndrome-affected patient with retinal impairment and obesity. Eur. J. Hum. Genet. 2015, 23, 621–627. [Google Scholar] [CrossRef]
- Hearn, T.; Spalluto, C.; Phillips, V.J.; Renforth, G.L.; Copin, N.; Hanley, N.A.; Wilson, D.I. Subcellular localization of ALMS1 supports involvement of centrosome and basal body dysfunction in the pathogenesis of obesity, insulin resistance, and type 2 diabetes. Diabetes 2005, 54, 1581–1587. [Google Scholar] [CrossRef]
- The CEP19-RABL2 GTPase Complex Binds IFT-B to Initiate Intraflagellar Transport at the Ciliary Base—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/28625565/ (accessed on 29 June 2024).
- Gerondopoulos, A.; Strutt, H.; Stevenson, N.L.; Sobajima, T.; Levine, T.P.; Stephens, D.J.; Strutt, D.; Barr, F.A. Planar Cell Polarity Effector Proteins Inturned and Fuzzy Form a Rab23 GEF Complex. Curr. Biol. 2019, 29, 3323–3330.e8. [Google Scholar] [CrossRef] [PubMed]
- Bardet-Biedl Syndrome: Genetics, Molecular Pathophysiology, and Disease Management—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/27853007/ (accessed on 21 July 2024).
- Gupta, N.; D’Acierno, M.; Zona, E.; Capasso, G.; Zacchia, M. Bardet–Biedl syndrome: The pleiotropic role of the chaperonin-like BBS6, 10, and 12. Proteins 2022, 190, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Niederlova, V.; Modrak, M.; Tsyklauri, O.; Huranova, M.; Stepanek, O. Meta-analysis of genotype-phenotype associations in Bardet-Biedl syndrome uncovers differences among causative genes. Hum. Mutat. 2019, 40, 2068–2087. [Google Scholar] [CrossRef] [PubMed]
- Ferent, J.; Constable, S.; Gigante, E.D.; Yam, P.T.; Mariani, L.E.; Legué, E.; Liem, K.F., Jr.; Caspary, T.; Charron, F. The Ciliary Protein Arl13b Functions Outside of the Primary Cilium in Shh-Mediated Axon Guidance. Cell Rep. 2019, 29, 3356–3366.e3. [Google Scholar] [CrossRef] [PubMed]
- Whiting, K.R.; Haer-Wigman, L.; Florijn, R.J.; van Beek, R.; Oud, M.M.; Plomp, A.S.; Boon, C.J.F.; Kroes, H.Y.; Roepman, R. Utilization of automated cilia analysis to characterize novel INPP5E variants in patients with non-syndromic retinitis pigmentosa. Eur. J. Hum. Genet. 2024. [Google Scholar] [CrossRef] [PubMed]
- Kopinke, D.; Norris, A.M.; Mukhopadhyay, S. Developmental and regenerative paradigms of cilia regulated hedgehog signaling. Semin. Cell Dev. Biol. 2021, 110, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Ding, Y.; Feng, B.; Tang, Y.; Chen, Y.; Wang, Y.; Chang, G.; Liu, S.; Wang, J.; Li, Q.; et al. Molecular and Phenotypic Expansion of Alström Syndrome in Chinese Patients. Front. Genet. 2022, 13, 808919. [Google Scholar] [CrossRef]
- Mujahid, S.; Hunt, K.F.; Cheah, Y.S.; Forsythe, E.; Hazlehurst, J.M.; Sparks, K.; Mohammed, S.; Tomlinson, J.W.; Amiel, S.A.; Carroll, P.V.; et al. The Endocrine and Metabolic Characteristics of a Large Bardet-Biedl Syndrome Clinic Population. J. Clin. Endocrinol. Metab. 2018, 103, 1834–1841. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.W. The characterization and comorbidities of heterozygous Bardet-Biedl syndrome carriers. Int. J. Med. Sci. 2024, 21, 784–794. [Google Scholar] [CrossRef]
- Meyer, J.R.; Krentz, A.D.; Berg, R.L.; Richardson, J.G.; Pomeroy, J.; Hebbring, S.J.; Haws, R.M. Kidney failure in Bardet–Biedl syndrome. Clin. Genet. 2022, 101, 429–441. [Google Scholar] [CrossRef]
- Forsythe, E.; Kenny, J.; Bacchelli, C.; Beales, P.L. Managing Bardet-Biedl Syndrome-Now and in the Future. Front. Pediatr. 2018, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Faccioli, N.; Poitou, C.; Clément, K.; Dubern, B. Current Treatments for Patients with Genetic Obesity. J. Clin. Res. Pediatr. Endocrinol. 2023, 15, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Haws, R.M.; Gordon, G.; Han, J.C.; Yanovski, J.A.; Yuan, G.; Stewart, M.W. The efficacy and safety of setmelanotide in individuals with Bardet-Biedl syndrome or Alström syndrome: Phase 3 trial design. Contemp. Clin. Trials Commun. 2021, 22, 100780. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Baig, S.; Wanninayake, S.; da Silva Xavier, G.; Dawson, C.; Paisey, R.; Geberhiwot, T. Glucagon-like peptide-1 analogues in monogenic syndromic obesity: Real-world data from a large cohort of Alström syndrome patients. Diabetes Obes. Metab. 2024, 26, 989–996. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Setmelanotide: First Approval. Drugs 2021, 81, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Trapp, C.M.; Censani, M. Setmelanotide: A promising advancement for pediatric patients with rare forms of genetic obesity. Curr. Opin. Endocrinol. Diabetes Obes. 2023, 30, 136–140. [Google Scholar] [CrossRef]
- Brüning, J.C.; Fenselau, H. Integrative neurocircuits that control metabolism and food intake. Science 2023, 381, eabl7398. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Lechtreck, K.F. The Bardet-Biedl syndrome protein complex is an adapter expanding the cargo range of intraflagellar transport trains for ciliary export. Proc. Natl. Acad. Sci. USA 2018, 115, E934–E943. [Google Scholar] [CrossRef] [PubMed]
- Loktev, A.V.; Jackson, P.K. Neuropeptide Y family receptors traffic via the Bardet-Biedl syndrome pathway to signal in neuronal primary cilia. Cell Rep. 2013, 5, 1316–1329. [Google Scholar] [CrossRef]
- Stephenson, E.J.; Kinney, C.E.; Stayton, A.S.; Han, J.C. Energy expenditure deficits drive obesity in a mouse model of Alström syndrome. Obesity 2023, 31, 2786–2798. [Google Scholar] [CrossRef]
- Hearn, T. ALMS1 and Alström syndrome: A recessive form of metabolic, neurosensory and cardiac deficits. J. Mol. Med. 2019, 97, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Xuan, J.; Yu, Y.; Qing, T.; Guo, L.; Shi, L. Next-generation sequencing in the clinic: Promises and challenges. Cancer Lett. 2013, 340, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.D.; Muller, J.; Collin, G.B.; Milan, G.; Kingsmore, S.F.; Dinwiddie, D.; Farrow, E.G.; Miller, N.A.; Favaretto, F.; Maffei, P.; et al. Alström Syndrome: Mutation spectrum of ALMS1. Hum. Mutat. 2015, 36, 660–668. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; Geberhiwot, T.; Barrett, T.G.; Paisey, R.; Semple, R.K. Refining genotype-phenotype correlation in Alström syndrome through study of primary human fibroblasts. Mol. Genet. Genomic. Med. 2017, 5, 390–404. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.D.; Hinman, E.G.; Collin, G.B.; Beck, S.; Cerqueira, R.; Maffei, P.; Milan, G.; Zhang, W.; Wilson, D.I.; Hearn, T.; et al. Spectrum of ALMS1 variants and evaluation of genotype-phenotype correlations in Alström syndrome. Hum. Mutat. 2007, 28, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- Dedeoglu, S.; Dede, E.; Oztunc, F.; Gedikbasi, A.; Yesil, G.; Dedeoglu, R. Mutation identification and prediction for severe cardiomyopathy in Alström syndrome, and review of the literature for cardiomyopathy. Orphanet. J. Rare Dis. 2022, 17, 359. [Google Scholar] [CrossRef] [PubMed]
- Bettini, S.; Bombonato, G.; Dassie, F.; Favaretto, F.; Piffer, L.; Bizzotto, P.; Busetto, L.; Chemello, L.; Senzolo, M.; Merkel, C.; et al. Liver Fibrosis and Steatosis in Alström Syndrome: A Genetic Model for Metabolic Syndrome. Diagnostics 2021, 11, 797. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.; Zhu, S.; Xiao, X.; Yan, L.; Yang, J.; Wu, G. Comparison of laboratory tests, ultrasound, or magnetic resonance elastography to detect fibrosis in patients with nonalcoholic fatty liver disease: A meta-analysis. Hepatology 2017, 66, 1486–1501. [Google Scholar] [CrossRef]
- Cleveland, E.; Bandy, A.; VanWagner, L.B. Diagnostic challenges of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Clin. Liver. Dis. 2018, 11, 98–104. [Google Scholar] [CrossRef]
- Roy, A.; Patel, L.; Yuan, M.; O'Shea, C.; Alvior, A.M.B.; Charalambides, M.; Moxon, D.; Baig, S.; Bunting, K.V.; Gehmlich, K.; et al. Defining the cardiovascular phenotype of adults with Alström syndrome. Int. J. Cardiol. 2024, 409, 132212. [Google Scholar] [CrossRef]
- Edwards, N.C.; Moody, W.E.; Yuan, M.; Warfield, A.T.; Cramb, R.; Paisey, R.B.; Geberhiwot, T.; Steeds, R.P. Diffuse Left Ventricular Interstitial Fibrosis Is Associated With Sub-Clinical Myocardial Dysfunction in Alström Syndrome: An Observational Study. Orphanet J. Rare Dis. 2015, 10, 83. [Google Scholar] [CrossRef] [PubMed]
- Zmyslowska, A.; Smyczynska, U.; Stanczak, M.; Jeziorny, K.; Szadkowska, A.; Fendler, W.; Borowiec, M. Association of circulating miRNAS in patients with Alstrőm and Bardet-Biedl syndromes with clinical course parameters. Front. Endocrinol. 2022, 13, 1057056. [Google Scholar] [CrossRef] [PubMed]
- Jeziorny, K.; Pietrowska, K.; Sieminska, J.; Zmysłowska-Polakowska, E.; Kretowski, A.; Ciborowski, M.; Zmyslowska, A. Serum metabolomics identified specific lipid compounds which may serve as markers of disease progression in patients with Alström and Bardet-Biedl syndromes. Front. Mol. Biosci. 2023, 10, 1251905. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Han, S.; Klier, K.; Fobo, G.; Montrone, C.; Yu, S.; Harada, M.; Henning, A.-K.; Friedrich, N.; Bahls, M.; et al. Identification of candidate metabolite biomarkers for metabolic syndrome and its five components in population-based human cohorts. Cardiovasc. Diabetol. 2023, 22, 141. [Google Scholar] [CrossRef] [PubMed]
- Mihalik, S.J.; Goodpaster, B.H.; Kelley, D.E.; Chace, D.H.; Vockley, J.; Toledo, F.G.S.; DeLany, J.P. Increased levels of plasma acylcarnitines in obesity and type 2 diabetes and identification of a marker of glucolipotoxicity. Obesity 2010, 18, 1695–1700. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.R.; Ning, Y.; Yu, H.; Tang, N.J. A HPLC-Q-TOF-MS-based urinary metabolomic approach to identification of potential biomarkers of metabolic syndrome. J. Huazhong Univ. Sci Technolog. Med. Sci. 2014, 34, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Jeziorny, K.; Zmyslowska-Polakowska, E.; Wyka, K.; Pyziak-Skupień, A.; Borowiec, M.; Szadkowska, A.; Zmysłowska, A. Identification of bone metabolism disorders in patients with Alström and Bardet-Biedl syndromes based on markers of bone turnover and mandibular atrophy. Bone Rep. 2022, 17, 101600. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.G.; Wang, K.; Marshall, J.D.; Rethmeyer, J.; Gunewardena, S.; Manzardo, A. Coding and noncoding expression patterns associated with rare obesity-related disorders: Prader—Willi and Alström syndromes. AGG 2015, 5, 53–75. [Google Scholar] [CrossRef] [PubMed]
- Borrelli, P.; Zacchia, M.; Cavaliere, C.; Basso, L.; Salvatore, M.; Capasso, G.; Aiello, M. Diffusion tensor imaging for the study of early renal dysfunction in patients affected by bardet-biedl syndrome. Sci. Rep. 2021, 11, 20855. [Google Scholar] [CrossRef]
- Caterino, M.; Zacchia, M.; Costanzo, M.; Bruno, G.; Arcaniolo, D.; Trepiccione, F.; Siciliano, R.A.; Mazzeo, M.F.; Ruoppolo, M.; Capasso, G. Urine Proteomics Revealed a Significant Correlation Between Urine-Fibronectin Abundance and Estimated-GFR Decline in Patients with Bardet-Biedl Syndrome. Kidney Blood Press Res. 2018, 43, 389–405. [Google Scholar] [CrossRef]
- Marchese, E.; Caterino, M.; Fedele, R.; Pirozzi, F.; Cevenini, A.; Gupta, N.; Ingrosso, D.; Perna, A.; Capasso, G.; Ruoppolo, M.; et al. Multi-Omics Studies Unveil Extraciliary Functions of BBS10 and Show Metabolic Aberrations Underlying Renal Disease in Bardet-Biedl Syndrome. Int. J. Mol. Sci. 2022, 23, 9420. [Google Scholar] [CrossRef] [PubMed]
- Zacchia, M.; Blanco, F.D.V.; Torella, A.; Raucci, R.; Blasio, G.; Onore, M.E.; Marchese, E.; Trepiccione, F.; Vitagliano, C.; Di Iorio, V.; et al. Urine concentrating defect as presenting sign of progressive renal failure in Bardet-Biedl syndrome patients. Clin. Kidney J. 2021, 14, 1545–1551. [Google Scholar] [CrossRef]
- Fernandez-Marmiesse, A.; Gouveia, S.; Couce, M.L. NGS Technologies as a Turning Point in Rare Disease Research, Diagnosis and Treatment. Curr. Med. Chem. 2018, 25, 404–432. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, N.; Xu, Y.; Li, G.; Yu, T.; Yao, R.-E.; Fu, L.; Wang, J.; Yin, L.; Yin, Y.; et al. Proband-only medical exome sequencing as a cost-effective first-tier genetic diagnostic test for patients without prior molecular tests and clinical diagnosis in a developing country: The China experience. Genet. Med. 2018, 20, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Castro-Sánchez, S.; Álvarez-Satta, M.; Tohamy, M.A.; Beltran, S.; Derdak, S.; Valverde, D. Whole exome sequencing as a diagnostic tool for patients with ciliopathy-like phenotypes. PLoS ONE 2017, 12, e0183081. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, R.; Szymanska, K.; Basu, B.; Patel, N.; Ewida, N.; Faqeih, E.; Al Hashem, A.; Derar, N.; Alsharif, H.; Aldahmesh, M.A. Characterizing the morbid genome of ciliopathies. Genome. Biol. 2016, 17, 242. [Google Scholar] [CrossRef] [PubMed]
- Frésard, L.; Smail, C.; Ferraro, N.M.; Teran, N.A.; Li, X.; Smith, K.S.; Bonner, D.; Kernohan, K.D.; Marwaha, S.; Zappala, Z.; et al. Identification of rare-disease genes using blood transcriptome sequencing and large control cohorts. Nat. Med. 2019, 25, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Modarage, K.; Malik, S.A.; Goggolidou, P. Molecular Diagnostics of Ciliopathies and Insights Into Novel Developments in Diagnosing Rare Diseases. Br. J. Biomed. Sci. 2022, 79, 10221. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Gao, C.; Wang, R.; Xiao, C.; Yang, M. Genes associated with inflammation and the cell cycle may serve as biomarkers for the diagnosis and prognosis of acute myocardial infarction in a Chinese population. Mol. Med. Rep. 2018, 18, 1311–1322. [Google Scholar] [CrossRef]
- Zhang, S.Y.; Xuan, C.; Wang, Y.; Zhang, S.-Q.; Li, H.; He, G.-W.; Tian, Q.-W. Association between ALMS 1 variants and early-onset coronary artery disease: A case-control study in Chinese population. Biosci. Rep. 2020, 40, BSR20193637. [Google Scholar] [CrossRef]
- Xuan, C.; Li, H.; Tian, Q.W.; Guo, J.J.; He, G.W.; Lun, L.M.; Wang, Q. Quantitative Assessment of Serum Amino Acids and Association with Early-Onset Coronary Artery Disease. Clin. Interv. Aging 2021, 16, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Schreyer, E.; Obringer, C.; Messaddeq, N.; Kieffer, B.; Zimmet, P.; Fleming, A.; Geberhiwot, T.; Marion, V. PATAS, a First-in-Class Therapeutic Peptide Biologic, Improves Whole-Body Insulin Resistance and Associated Comorbidities In Vivo. Diabetes 2022, 71, 2034–2047. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).