
Molecular and Pathological Features of Paediatric High-Grade Gliomas


Abstract
1. Introduction
2. Diffuse Midline Glioma, H3K27-Altered
2.1. Definition and Grade
2.2. Molecular Pathology
2.3. Epidemiology
- −
- −
2.4. Imaging
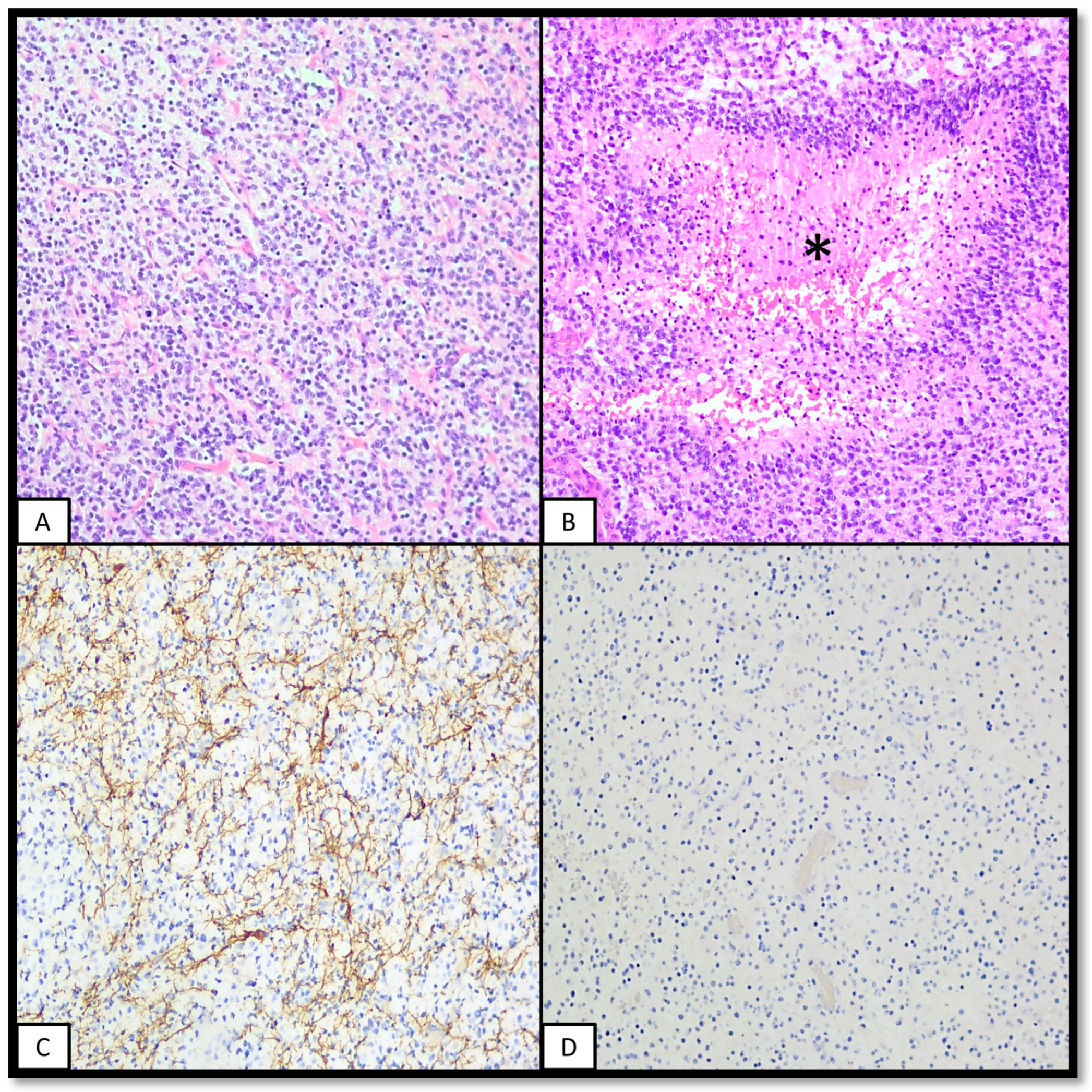
2.5. Pathology
2.6. Differential Diagnosis
2.7. Prognosis
2.8. Non-Diffuse H3K27-Altered Neuroepithelial Tumours
2.9. Bithalamic Glioma (Diffuse Midline Glioma, EGFR-Altered)
2.10. WHO Diagnostic Criteria
- A diffuse cellular glioma;
- Loss of H3 p. K28me3 (K27me3 IHC);
- Midline location;
- One of the following:
- Presence of an H3 p.K28M (K27M) or p.K28I (K27I) mutation (for H3K27-mutant subtypes);
- Presence of a pathogenic mutation or amplification of EGFR (for the EGFR-mutant subtype);
- Overexpression of EZHIP (for the H3-wildtype with EZHIP overexpression subtype);
- Methylation profile of one of the subtypes of diffuse midline gliomas.
3. Diffuse Hemispheric Glioma, H3G34-Altered
3.1. Definition and Grade
3.2. Molecular Pathology
3.3. Epidemiology
3.4. Imaging Features
3.5. Pathological Features
- -
- Approximately three quarters of tumours show a typical malignant glioma morphology (GBM-like), with hypercellularity, brisk mitotic activity, microvascular proliferation, and necrosis. Intense positivity for GFAP is usually found [64]. Some cases may show a dysplastic neuronal component, with bi- or multinucleation and the expression of CD34 [81];
- -
- One quarter show a high-grade small-cell monomorphic proliferation reminiscent of embryonal tumours of the CNS (PNET-like histology). Structures resembling Homer–Wright rosettes may be identified. Intense positivity for synaptophysin and MAP2 is typical of this variant. Focal GFAP positivity may also be seen [64].
3.6. Differential Diagnosis
3.7. Prognosis
3.8. WHO Diagnostic Criteria
- A diffuse cellular glioma with mitotic activity;
- H3.3 p.G35R (G34R) or p.G35V (G34V) mutation (H3-3A [H3F3A] c.103G>A, c.103G>C, or c.104G>T);
- Hemispheric location;
- In unresolved lesions, a methylome profile of diffuse hemispheric glioma, H3G34-mutant;
- Desirable criteria include negativity for OLIG2, loss of ATRX expression, and diffuse p53 immunopositivity
4. Diffuse Paediatric-Type High-Grade Glioma, H3-Wildtype and IDH-Wildtype
4.1. Definition and Grade
4.2. Molecular Pathology
- RTK1 tumours usually show amplification of PDGFRA (33%). Gliomas arising after radiotherapy show similar molecular features, with mutations in TP53 and amplifications of or mutations in PDGFRA, among other alterations [84,85]. Gliomas arising in the context of Lynch syndrome and constitutive mismatch repair deficiency (CMMRD) typically fall in this category [4].
- RTK2 tumours usually show molecular features of adult-type IDH-wildtype glioblastomas [86], such as the amplification of EGFR (50%), CDKN2A/B homozygous deletions (72%), mutations of the TERT promoter (64%), the gain of chromosome 7 (28%), and the loss of chromosome 10 (50%). However, although key molecular features are similar, pHHGRTK2 and IDH-wildtype glioblastomas are segregated in methylation studies, arguing that these tumours are different.
- Other alterations include mutations of TP53 (around 50%, and up to 67% in MYCN tumours).
- MGMT promoter methylation is seldom found, more frequently in RTK1 tumours (18%).
4.3. Epidemiology
4.4. Imaging Features
4.5. Pathological Features
4.6. Differential Diagnosis
4.7. Prognosis
4.8. WHO Diagnostic Criteria
- A diffuse glioma with mitotic activity occurring in a child or young adult;
- Absence of mutations in IDH1 or IDH2;
- Absence of mutations in H3 genes;
- Methylation profile aligned with pHGG RTK1, pHGG RTK2, or pHGG MYCN, or key molecular features, such as PDGFRA alteration, EGFR alteration, or MYCN amplification;
- Desirable criteria include the presence of microvascular proliferation, necrosis, and the retention of H3K27 trimethylation.
5. Infant-Type Hemispheric Glioma
5.1. Definition and Grade
5.2. Molecular Pathology
5.3. Epidemiology
5.4. Imaging Features
5.5. Pathological Features
5.6. Differential Diagnosis
5.7. Prognosis
5.8. WHO Diagnostic Criteria
- Cellular astrocytoma;
- Presentation in early childhood;
- Cerebral hemispheric location;
- Presence of a typical Receptor of Tyrosine Kinase abnormality or methylation profile aligned with infant-type hemispheric glioma
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ostrom, Q.T.; Price, M.; Ryan, K.; Edelson, J.; Neff, C.; Cioffi, G.; Waite, K.A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Pediatric brain tumor foundation childhood and adolescent primary brain and other central nervous system tumors diagnosed in the United States in 2014–2018. Neuro-oncology 2022, 24, iii1–iii38. [Google Scholar] [CrossRef]
- Mackay, A.; Burford, A.; Carvalho, D.; Izquierdo, E.; Fazal-Salom, J.; Taylor, K.R.; Bjerke, L.; Clarke, M.; Vinci, M.; Nandhabalan, M.; et al. Integrated molecular meta-analysis of 1000 pediatric high-grade and diffuse intrinsic pontine glioma. Cancer Cell 2017, 32, 520–537. [Google Scholar] [CrossRef]
- Mackay, A.; Burford, A.; Molinari, V.; Jones, D.T.W.; Izquierdo, E.; Brouwer-Visser, J.; Giangaspero, F.; Haberler, C.; Pietsch, T.; Jacques, T.S.; et al. Molecular, pathological, radiological, and immune profiling of non-brainstem pediatric high-grade glioma from the HERBY phase II randomized trial. Cancer Cell 2018, 33, 829–842. [Google Scholar] [CrossRef]
- WHO Classification of Tumours Editorial Board. Central Nervous System Tumours. (WHO Classification of Tumours Series, Vol. 6); International Agency for Research on Cancer: Lyon, France, 2021; pp. 69–82. [Google Scholar]
- Sturm, D.; Orr, B.A.; Toprak, U.H.; Hovestadt, V.; Jones, D.T.W.; Capper, D.; Sill, M.; Buchhalter, I.; Northcott, P.A.; Leis, I.; et al. New brain tumor entities emerge from molecular classification of CNS-PNETs. Cell 2016, 164, 1060–1072. [Google Scholar] [CrossRef]
- Korshunov, A.; Ryzhova, M.; Hovestadt, V.; Bender, S.; Sturm, D.; Capper, D.; Meyer, J.; Schrimpf, D.; Kool, M.; Northcott, P.A.; et al. Integrated analysis of pediatric glioblastoma reveals a subset of biologically favorable tumors with associated molecular prognostic markers. Acta Neuropathol. 2015, 129, 669–678. [Google Scholar] [CrossRef]
- Korshunov, A.; Schrimpf, D.; Ryzhova, M.; Sturm, D.; Chavez, L.; Hovestadt, V.; Sharma, T.; Habel, A.; Burford, A.; Jones, C.; et al. H3-/IDH-wild type pediatric glioblastoma is comprised of molecularly and prognostically distinct subtypes with associated oncogenic drivers. Acta Neuropathol. 2017, 134, 507–516. [Google Scholar] [CrossRef]
- Varlet, P.; Le Teuff, G.; Le Deley, M.C.; Giangaspero, F.; Haberler, C.; Jacques, T.S.; Figarella-Branger, D.; Pietsch, T.; Andreiuolo, F.; Deroulers, C.; et al. WHO grade has no prognostic value in the pediatric high-grade glioma included in the HERBY trial. Neuro-oncology 2020, 22, 116–127. [Google Scholar] [CrossRef]
- Leske, H.; Dalgleish, R.; Lazar, A.J.; Reifenberger, G.; Cree, I.A. A common classification framework for histone sequence alterations in tumours: An expert consensus proposal. J. Pathol. 2021, 254, 109–120. [Google Scholar] [CrossRef]
- Leske, H.; Rushing, E.; Budka, H.; Niehusmann, P.; Pahnke, J.; Panagopoulos, I. K27/G34 versus K28/G35 in histone H3-mutant gliomas: A note of caution. Acta Neuropathol. 2018, 136, 175–176. [Google Scholar] [CrossRef]
- Sievers, P.; Sill, M.; Schrimpf, D.; Stichel, D.; Reuss, D.E.; Sturm, D.; Hench, J.; Frank, S.; Krskova, L.; Vicha, A.; et al. A subset of pediatric-type thalamic gliomas share a distinct DNA methylation profile, H3K27me3 loss and frequent alteration of EGFR. Neuro Oncol. 2021, 23, 34–43. [Google Scholar] [CrossRef]
- Castel, D.; Philippe, C.; Calmon, R.; Le Dret, L.; Truffaux, N.; Boddaert, N.; Pagès, M.; Taylor, K.R.; Saulnier, P.; Lacroix, L.; et al. Histone H3F3A and HIST1H3B K27M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathol. 2015, 130, 815–827. [Google Scholar] [CrossRef]
- Henikoff, S.; Smith, M.M. Histone variants and epigenetics. Cold Spring Harb. Perspect. Biol. 2015, 7, a019364. [Google Scholar] [CrossRef] [PubMed]
- Buczkowicz, P.; Hoeman, C.; Rakopoulos, P.; Pajovic, S.; Letourneau, L.; Dzamba, M.; Morrison, A.; Lewis, P.; Bouffet, E.; Bartels, U.; et al. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat. Genet. 2014, 46, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.M.; Fang, D.; Gan, H.; Hashizume, R.; Yu, C.; Schroeder, M.; Gupta, N.; Mueller, S.; James, C.D.; Jenkins, R.; et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev. 2013, 27, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Bender, S.; Tang, Y.; Lindroth, A.M.; Hovestadt, V.; Jones, D.T.; Kool, M.; Zapatka, M.; Northcott, P.A.; Sturm, D.; Wang, W.; et al. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 2013, 24, 660–672. [Google Scholar] [CrossRef] [PubMed]
- Harutyunyan, A.S.; Krug, B.; Chen, H.; Papillon-Cavanagh, S.; Zeinieh, M.; De Jay, N.; Deshmukh, S.; Chen, C.C.L.; Belle, J.; Mikael, L.G.; et al. H3K27M induces defective chromatin spread of PRC2-mediated repressive H3K27me2/me3 and is essential for glioma tumorigenesis. Nat. Commun. 2019, 10, 1262. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Yu, J.R.; Granat, J.; Saldaña-Meyer, R.; Andrade, J.; LeRoy, G.; Jin, Y.; Lund, P.; Stafford, J.M.; Garcia, B.A.; et al. Automethylation of PRC2 promotes H3K27 methylation and is impaired in H3K27M pediatric glioma. Genes Dev. 2019, 33, 1428–1440. [Google Scholar] [CrossRef]
- Filbin, M.G.; Tirosh, I.; Hovestadt, V.; Shaw, M.L.; Escalante, L.E.; Mathewson, N.D.; Neftel, C.; Frank, N.; Pelton, K.; Hebert, C.M.; et al. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science 2018, 360, 331–335. [Google Scholar] [CrossRef]
- Huang, T.; Garcia, R.; Qi, J.; Lulla, R.; Horbinski, C.; Behdad, A.; Wadhwani, N.; Shilatifard, A.; James, C.; Saratsis, A.M. Detection of histone H3 K27M mutation and post-translational modifications in pediatric diffuse midline glioma via tissue immunohistochemistry informs diagnosis and clinical outcomes. Oncotarget 2018, 9, 37112–37124. [Google Scholar] [CrossRef]
- Venneti, S.; Santi, M.; Felicella, M.M.; Yarilin, D.; Phillips, J.J.; Sullivan, L.M.; Martinez, D.; Perry, A.; Lewis, P.W.; Thompson, C.B.; et al. A sensitive and specific histopathologic prognostic marker for H3F3A K27M mutant pediatric glioblastomas. Acta Neuropathol. 2014, 128, 743–753. [Google Scholar] [CrossRef]
- Wu, G.; Diaz, A.K.; Paugh, B.S.; Rankin, S.L.; Ju, B.; Li, Y.; Zhu, X.; Qu, C.; Chen, X.; Zhang, J.; et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat. Genet. 2014, 46, 444–450. [Google Scholar] [CrossRef]
- Siddaway, R.; Canty, L.; Pajovic, S.; Milos, S.; Coyaud, E.; Sbergio, S.G.; Vadivel Anguraj, A.K.; Lubanszky, E.; Yun, H.Y.; Portante, A.; et al. Oncohistone interactome profiling uncovers contrasting oncogenic mechanisms and identifies potential therapeutic targets in high grade glioma. Acta Neuropathol. 2022, 144, 1027–1048. [Google Scholar] [CrossRef]
- Castel, D.; Kergrohen, T.; Tauziède-Espariat, A.; Mackay, A.; Ghermaoui, S.; Lechapt, E.; Pfister, S.M.; Kramm, C.M.; Boddaert, N.; Blauwblomme, T.; et al. Histone H3 wild-type DIPG/DMG overexpressing EZHIP extend the spectrum diffuse midline gliomas with PRC2 inhibition beyond H3-K27M mutation. Acta Neuropathol. 2020, 139, 1109–1113. [Google Scholar] [CrossRef]
- Nikbakht, H.; Panditharatna, E.; Mikael, L.G.; Li, R.; Gayden, T.; Osmond, M.; Ho, C.Y.; Kambhampati, M.; Hwang, E.I.; Faury, D.; et al. Spatial and temporal homogeneity of driver mutations in diffuse intrinsic pontine glioma. Nat. Commun. 2016, 7, 11185. [Google Scholar] [CrossRef]
- Cordero, F.J.; Huang, Z.; Grenier, C.; He, X.; Hu, G.; McLendon, R.E.; Murphy, S.K.; Hashizume, R.; Becher, O.J. Histone H3.3K27M represses p16 to accelerate gliomagenesis in a murine model of DIPG. Mol. Cancer Res. 2017, 15, 1243–1254. [Google Scholar] [CrossRef]
- Fontebasso, A.M.; Papillon-Cavanagh, S.; Schwartzentruber, J.; Nikbakht, H.; Gerges, N.; Fiset, P.O.; Bechet, D.; Faury, D.; De Jay, N.; Ramkissoon, L.A.; et al. Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat. Genet. 2014, 46, 462–466. [Google Scholar] [CrossRef]
- Taylor, K.R.; Mackay, A.; Truffaux, N.; Butterfield, Y.; Morozova, O.; Philippe, C.; Castel, D.; Grasso, C.S.; Vinci, M.; Carvalho, D.; et al. Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat. Genet. 2014, 46, 457–461. [Google Scholar] [CrossRef]
- Messinger, D.; Harris, M.K.; Cummings, J.R.; Thomas, C.; Yang, T.; Sweha, S.R.; Woo, R.; Siddaway, R.; Burkert, M.; Stallard, S.; et al. Therapeutic targeting of prenatal pontine ID1 signaling in diffuse midline glioma. Neuro-oncology 2023, 25, 54–67. [Google Scholar] [CrossRef]
- Castel, D.; Philippe, C.; Kergrohen, T.; Sill, M.; Merlevede, J.; Barret, E.; Puget, S.; Sainte-Rose, C.; Kramm, C.M.; Jones, C.; et al. Transcriptomic and epigenetic profiling of ’diffuse midline gliomas, H3 K27M-mutant’ discriminate two subgroups based on the type of histone H3 mutated and not supratentorial or infratentorial location. Acta Neuropathol. Commun. 2018, 6, 1–13. [Google Scholar] [CrossRef]
- Roux, A.; Pallud, J.; Saffroy, R.; Edjlali-Goujon, M.; Debily, M.A.; Boddaert, N.; Sanson, M.; Puget, S.; Knafo, S.; Adam, C.; et al. High-grade gliomas in adolescents and young adults highlight histomolecular differences from their adult and pediatric counterparts. Neuro-oncology 2020, 22, 1190–1202. [Google Scholar] [CrossRef]
- Mosaab, A.; El-Ayadi, M.; Khorshed, E.N.; Amer, N.; Refaat, A.; El-Beltagy, M.; Hassan, Z.; Soror, S.H.; Zaghloul, M.S.; El-Naggar, S. Histone H3K27M mutation overrides histological grading in pediatric gliomas. Sci. Rep. 2020, 10, 8368. [Google Scholar] [CrossRef]
- Solomon, D.A.; Wood, M.D.; Tihan, T.; Bollen, A.W.; Gupta, N.; Phillips, J.J.; Perry, A. Diffuse midline gliomas with histone H3-K27M mutation: A series of 47 cases assessing the spectrum of morphologic variation and associated genetic alterations. Brain Pathol. 2016, 26, 569–580. [Google Scholar] [CrossRef]
- Chai, R.C.; Zhang, Y.W.; Liu, Y.Q.; Chang, Y.Z.; Pang, B.; Jiang, T.; Jia, W.Q.; Wang, Y.Z. The molecular characteristics of spinal cord gliomas with or without H3 K27M mutation. Acta Neuropathol. Commun. 2020, 8, 1–11. [Google Scholar] [CrossRef]
- Gilbert, A.R.; Zaky, W.; Gokden, M.; Fuller, C.E.; Ocal, E.; Leeds, N.E.; Fuller, G.N. Extending the neuroanatomic territory of diffuse midline glioma, K27M mutant: Pineal region origin. Pediatr. Neurosurg. 2018, 53, 59–63. [Google Scholar] [CrossRef]
- Meyronet, D.; Esteban-Mader, M.; Bonnet, C.; Joly, M.O.; Uro-Coste, E.; Amiel-Benouaich, A.; Forest, F.; Rousselot-Denis, C.; Burel-Vandenbos, F.; Bourg, V.; et al. Characteristics of H3 K27M-mutant gliomas in adults. Neuro-oncology 2017, 19, 1127–1134. [Google Scholar] [CrossRef]
- Sloan, E.A.; Cooney, T.; Oberheim Bush, N.A.; Buerki, R.; Taylor, J.; Clarke, J.L.; Torkildson, J.; Kline, C.; Reddy, A.; Mueller, S.; et al. Recurrent non-canonical histone H3 mutations in spinal cord diffuse gliomas. Acta Neuropathol. 2019, 138, 877–881. [Google Scholar] [CrossRef]
- Yao, J.; Wang, L.; Ge, H.; Yin, H.; Piao, Y. Diffuse midline glioma with H3 K27M mutation of the spinal cord: A series of 33 cases. Neuropathology 2021, 41, 183–190. [Google Scholar] [CrossRef]
- Buczkowicz, P.; Bartels, U.; Bouffet, E.; Becher, O.; Hawkins, C. Histopathological spectrum of paediatric diffuse intrinsic pontine glioma: Diagnostic and therapeutic implications. Acta Neuropathol. 2014, 128, 573–581. [Google Scholar] [CrossRef]
- Hoffman, L.M.; Veldhuijzen van Zanten, S.E.M.; Colditz, N.; Baugh, J.; Chaney, B.; Hoffmann, M.; Lane, A.; Fuller, C.; Miles, L.; Hawkins, C.; et al. Clinical, radiologic, pathologic, and molecular characteristics of long-term survivors of diffuse intrinsic pontine glioma (DIPG): A collaborative report from the International and European Society for Pediatric Oncology DIPG Registries. J. Clin. Oncol. 2018, 36, 1963–1972. [Google Scholar] [CrossRef]
- Lasocki, A.; Abdalla, G.; Chow, G.; Thust, S.C. Imaging features associated with H3 K27-altered and H3 G34-mutant gliomas: A narrative systematic review. Cancer Imaging 2022, 22, 63. [Google Scholar] [CrossRef]
- Hohm, A.; Karremann, M.; Gielen, G.H.; Pietsch, T.; Warmuth-Metz, M.; Vandergrift, L.A.; Bison, B.; Stock, A.; Hoffmann, M.; Pham, M.; et al. Magnetic resonance imaging characteristics of molecular subgroups in pediatric H3 K27M mutant diffuse midline glioma. Clin. Neuroradiol. 2022, 32, 249–258. [Google Scholar] [CrossRef]
- Zhao, J.P.; Liu, X.J.; Lin, H.Z.; Cui, C.X.; Yue, Y.J.; Gao, S.; Xu, H.Z. MRI comparative study of diffuse midline glioma, H3 K27-altered and glioma in the midline without H3 K27-altered. BMC Neurol. 2022, 22, 498. [Google Scholar] [CrossRef]
- Aboian, M.S.; Solomon, D.A.; Felton, E.; Mabray, M.C.; Villanueva-Meyer, J.E.; Mueller, S.; Cha, S. Imaging Characteristics of Pediatric Diffuse Midline Gliomas with Histone H3 K27M Mutation. AJNR Am. J. Neuroradiol. 2017, 38, 795–800. [Google Scholar] [CrossRef]
- Qiu, T.; Chanchotisatien, A.; Qin, Z.; Wu, J.; Du, Z.; Zhang, X.; Gong, F.; Yao, Z.; Chu, S. Imaging characteristics of adult H3 K27M-mutant gliomas. J. Neurosurg. 2019, 133, 1662–1670. [Google Scholar] [CrossRef]
- Thust, S.; Micallef, C.; Okuchi, S.; Brandner, S.; Kumar, A.; Mankad, K.; Wastling, S.; Mancini, L.; Jäger, H.R.; Shankar, A. Imaging characteristics of H3 K27M histone-mutant diffuse midline glioma in teenagers and adults. Quant. Imaging Med. Surg. 2021, 11, 43–56. [Google Scholar] [CrossRef]
- Chen, H.; Hu, W.; He, H.; Yang, Y.; Wen, G.; Lv, X. Noninvasive assessment of H3 K27M mutational status in diffuse midline gliomas by using apparent diffusion coefficient measurements. Eur. J. Radiol. 2019, 114, 152–159. [Google Scholar] [CrossRef]
- Calmon, R.; Dangouloff-Ros, V.; Varlet, P.; Deroulers, C.; Philippe, C.; Debily, M.A.; Castel, D.; Beccaria, K.; Blauwblomme, T.; Grevent, D.; et al. Radiogenomics of diffuse intrinsic pontine gliomas (DIPGs): Correlation of histological and biological characteristics with multimodal MRI features. Eur. Radiol. 2021, 31, 8913–8924. [Google Scholar] [CrossRef]
- Pratt, D.; Natarajan, S.K.; Banda, A.; Giannini, C.; Vats, P.; Koschmann, C.; Mody, R.; Chinnaiyan, A.; Venneti, S. Circumscribed/non-diffuse histology confers a better prognosis in H3K27M-mutant gliomas. Acta Neuropathol. 2018, 135, 299–301. [Google Scholar] [CrossRef]
- Tauziède-Espariat, A.; Debily, M.A.; Castel, D.; Grill, J.; Puget, S.; Sabel, M.; Blomgren, K.; Gareton, A.; Dangouloff-Ros, V.; Lechapt, E.; et al. An integrative radiological, histopathological and molecular analysis of pediatric pontine histone-wildtype glioma with MYCN amplification (HGG-MYCN). Acta Neuropathol. Commun. 2019, 7, 1–4. [Google Scholar] [CrossRef]
- Pagès, M.; Beccaria, K.; Boddaert, N.; Saffroy, R.; Besnard, A.; Castel, D.; Fina, F.; Barets, D.; Barret, E.; Lacroix, L.; et al. Co-occurrence of histone H3 K27M and BRAF V600E mutations in paediatric midline grade I ganglioglioma. Brain Pathol. 2018, 28, 103–111. [Google Scholar] [CrossRef]
- Ryall, S.; Guzman, M.; Elbabaa, S.K.; Luu, B.; Mack, S.C.; Zapotocky, M.; Taylor, M.D.; Hawkins, C.; Ramaswamy, V. H3 K27M mutations are extremely rare in posterior fossa group A ependymoma. Childs Nerv. Syst. 2017, 33, 1047–1051. [Google Scholar] [CrossRef]
- Yao, K.; Duan, Z.; Wang, Y.; Zhang, M.; Fan, T.; Wu, B.; Qi, X. Detection of H3K27M mutation in cases of brain stem subependymoma. Hum. Pathol. 2019, 84, 262–269. [Google Scholar] [CrossRef]
- Louis, D.N.; Giannini, C.; Capper, D.; Paulus, W.; Figarella-Branger, D.; Lopes, M.B.; Batchelor, T.T.; Cairncross, J.G.; van den Bent, M.; Wick, W.; et al. cIMPACT-NOW update 2: Diagnostic clarifications for diffuse midline glioma, H3 K27M-mutant and diffuse astrocytoma/anaplastic astrocytoma, IDH-mutant. Acta Neuropathol. 2018, 135, 639–642. [Google Scholar] [CrossRef]
- Werbrouck, C.; Evangelista, C.C.S.; Lobón-Iglesias, M.J.; Barret, E.; Le Teuff, G.; Merlevede, J.; Brusini, R.; Kergrohen, T.; Mondini, M.; Bolle, S.; et al. TP53 pathway alterations drive radioresistance in Diffuse Intrinsic Pontine Gliomas (DIPG). Clin. Cancer Res. 2019, 25, 6788–6800. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef]
- Vuong, H.G.; Ngo, T.N.M.; Le, H.T.; Jea, A.; Hrachova, M.; Battiste, J.; McNall-Knapp, R.; Dunn, I.F. Prognostic implication of patient age in H3K27M-mutant midline gliomas. Front. Oncol. 2022, 12, 858148. [Google Scholar] [CrossRef]
- Banan, R.; Christians, A.; Bartels, S.; Lehmann, U.; Hartmann, C. Absence of MGMT promoter methylation in diffuse midline glioma, H3 K27M-mutant. Acta Neuropathol. Commun. 2017, 5, 98. [Google Scholar] [CrossRef]
- Auffret, L.; Ajlil, Y.; Tauziède-Espariat, A.; Kergrohen, T.; Puiseux, C.; Riffaud, L.; Blouin, P.; Bertozzi, A.I.; Leblond, P.; Blomgren, K.; et al. A new subtype of diffuse midline glioma, H3 K27 and BRAF/FGFR1 co-altered: A clinico-radiological and histomolecular characterisation. Acta Neuropathol. 2023, 147, 2. [Google Scholar] [CrossRef]
- López, G.; Oberheim Bush, N.A.; Berger, M.S.; Perry, A.; Solomon, D.A. Diffuse non-midline glioma with H3F3A K27M mutation: A prognostic and treatment dilemma. Acta Neuropathol. Commun. 2017, 5, 38. [Google Scholar] [CrossRef]
- Broniscer, A.; Hwang, S.N.; Chamdine, O.; Lin, T.; Pounds, S.; Onar-Thomas, A.; Chi, L.; Shurtleff, S.; Allen, S.; Gajjar, A.; et al. Bithalamic gliomas may be molecularly distinct from their unilateral high-grade counterparts. Brain Pathol. 2018, 28, 112–120. [Google Scholar] [CrossRef]
- Mondal, G.; Lee, J.C.; Ravindranathan, A.; Villanueva-Meyer, J.E.; Tran, Q.T.; Allen, S.J.; Barreto, J.; Gupta, R.; Doo, P.; Van Ziffle, J.; et al. Pediatric bithalamic gliomas have a distinct epigenetic signature and frequent EGFR exon 20 insertions resulting in potential sensitivity to targeted kinase inhibition. Acta Neuropathol. 2020, 139, 1071–1088. [Google Scholar] [CrossRef]
- Niu, X.; Wang, T.; Yang, Y.; Gan, Y.; Li, J.; Liu, Y.; Mao, Q. Prognostic factors for the survival outcome of bilateral thalamic glioma: An integrated survival analysis. World Neurosurg. 2018, 110, e222–e230. [Google Scholar] [CrossRef]
- Korshunov, A.; Capper, D.; Reuss, D.; Schrimpf, D.; Ryzhova, M.; Hovestadt, V.; Sturm, D.; Meyer, J.; Jones, C.; Zheludkova, O.; et al. Histologically distinct neuroepithelial tumors with histone 3 G34 mutation are molecularly similar and comprise a single nosologic entity. Acta Neuropathol. 2016, 131, 137–146. [Google Scholar] [CrossRef]
- Cheng, Z.; Cheung, P.; Kuo, A.J.; Yukl, E.T.; Wilmot, C.M.; Gozani, O.; Patel, D.J. A molecular threading mechanism underlies Jumonji lysine demethylase KDM2A regulation of methylated H3K36. Genes Dev. 2014, 28, 1758–1771. [Google Scholar] [CrossRef]
- Fang, J.; Huang, Y.; Mao, G.; Yang, S.; Rennert, G.; Gu, L.; Li, H.; Li, G.M. Cancer-driving H3G34V/R/D mutations block H3K36 methylation and H3K36me3-MutSα interaction. Proc. Natl. Acad. Sci. USA 2018, 115, 9598–9603. [Google Scholar] [CrossRef]
- Jain, S.U.; Khazaei, S.; Marchione, D.M.; Lundgren, S.M.; Wang, X.; Weinberg, D.N.; Deshmukh, S.; Juretic, N.; Lu, C.; Allis, C.D.; et al. Histone H3.3 G34 mutations promote aberrant PRC2 activity and drive tumor progression. Proc. Natl. Acad. Sci. USA 2020, 117, 27354–27364. [Google Scholar] [CrossRef]
- Yang, S.; Zheng, X.; Lu, C.; Li, G.M.; Allis, C.D.; Li, H. Molecular basis for oncohistone H3 recognition by SETD2 methyltransferase. Genes Dev. 2016, 30, 1611–1616. [Google Scholar] [CrossRef]
- Zhang, Y.; Shan, C.M.; Wang, J.; Bao, K.; Tong, L.; Jia, S. Molecular basis for the role of oncogenic histone mutations in modulating H3K36 methylation. Sci. Rep. 2017, 7, 43906. [Google Scholar] [CrossRef]
- Bressan, R.B.; Southgate, B.; Ferguson, K.M.; Blin, C.; Grant, V.; Alfazema, N.; Wills, J.C.; Marques-Torrejon, M.A.; Morrison, G.M.; Ashmore, J.; et al. Regional identity of human neural stem cells determines oncogenic responses to histone H3.3 mutants. Cell Stem Cell 2021, 28, 877–893. [Google Scholar] [CrossRef]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.A.; Jones, D.T.; Konermann, C.; Pfaff, E.; Tönjes, M.; Sill, M.; Bender, S.; et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 2012, 22, 425–437. [Google Scholar] [CrossRef]
- Bjerke, L.; Mackay, A.; Nandhabalan, M.; Burford, A.; Jury, A.; Popov, S.; Bax, D.A.; Carvalho, D.; Taylor, K.R.; Vinci, M.; et al. Histone H3.3. mutations drive pediatric glioblastoma through upregulation of MYCN. Cancer Discov. 2013, 3, 512–519. [Google Scholar] [CrossRef]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tönjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef]
- Sato, M.; Rodriguez-Barrueco, R.; Yu, J.; Do, C.; Silva, J.M.; Gautier, J. MYC is a critical target of FBXW7. Oncotarget 2015, 6, 3292–3305. [Google Scholar] [CrossRef]
- Gianno, F.; Antonelli, M.; Ferretti, E.; Massimino, M.; Arcella, A.; Giangaspero, F. Pediatric high-grade glioma: A heterogeneous group of neplasms with different molecular drivers. Glioma 2018, 1, 117–124. [Google Scholar] [CrossRef]
- Kasper, L.H.; Baker, S.J. Invited Review: Emerging functions of histone H3 mutations in paediatric diffuse high-grade gliomas. Neuropathol. Appl. Neurobiol. 2020, 46, 73–85. [Google Scholar] [CrossRef]
- Nguyen, A.V.; Soto, J.M.; Gonzalez, S.M.; Murillo, J.; Trumble, E.R.; Shan, F.Y.; Huang, J.H. H3G34-mutant gliomas—A review of molecular pathogenesis and therapeutic options. Biomedicines 2023, 11, 2002. [Google Scholar] [CrossRef]
- Picart, T.; Barritault, M.; Poncet, D.; Berner, L.P.; Izquierdo, C.; Tabouret, E.; Figarella-Branger, D.; Idbaïh, A.; Bielle, F.; Bourg, V.; et al. Characteristics of diffuse hemispheric gliomas, H3 G34-mutant in adults. Neurooncol. Adv. 2021, 3, vdab061. [Google Scholar] [CrossRef]
- Vettermann, F.J.; Felsberg, J.; Reifenberger, G.; Hasselblatt, M.; Forbrig, R.; Berding, G.; la Fougère, C.; Galldiks, N.; Schittenhelm, J.; Weis, J.; et al. Characterization of diffuse gliomas with histone H3-G34 mutation by MRI and dynamic 18F-FET PET. Clin. Nucl. Med. 2018, 43, 895–898. [Google Scholar] [CrossRef]
- Puntonet, J.; Dangouloff-Ros, V.; Saffroy, R.; Pagès, M.; Andreiuolo, F.; Grill, J.; Puget, S.; Boddaert, N.; Varlet, P. Historadiological correlations in high-grade glioma with the histone 3.3 G34R mutation. J. Neuroradiol. 2018, 45, 316–322. [Google Scholar] [CrossRef]
- Andreiuolo, F.; Lisner, T.; Zlocha, J.; Kramm, C.; Koch, A.; Bison, B.; Gareton, A.; Zanello, M.; Waha, A.; Varlet, P.; et al. H3F3A-G34R mutant high grade neuroepithelial neoplasms with glial and dysplastic ganglion cell components. Acta Neuropathol. Commun. 2019, 7, 78. [Google Scholar] [CrossRef]
- Gianno, F.; Antonelli, M.; Di Dio, T.; Minasi, S.; Donofrio, V.; Buccoliero, A.M.; Gardiman, M.P.; Pollo, B.; DiomediCamassei, F.; Rossi, S.; et al. Correlation between immunohistochemistry and sequencing in H3G34-mutant gliomas. Am. J. Surg. Pathol. 2021, 45, 200–204. [Google Scholar] [CrossRef]
- Schäfer, S.; Behling, F.; Skardelly, M.; Koch, M.; Ott, I.; Paulsen, F.; Tabatabai, G.; Schittenhelm, J. Low FoxG1 and high Olig-2 labelling indices define a prognostically favourable subset in isocitrate dehydrogenase (IDH)-mutant gliomas. Neuropathol. Appl Neurobiol. 2018, 44, 207–223. [Google Scholar] [CrossRef]
- López, G.Y.; Van Ziffle, J.; Onodera, C.; Grenert, J.P.; Yeh, I.; Bastian, B.C.; Clarke, J.; Oberheim Bush, N.A.; Taylor, J.; Chang, S.; et al. The genetic landscape of gliomas arising after therapeutic radiation. Acta Neuropathol. 2019, 137, 139–150. [Google Scholar] [CrossRef]
- Phi, J.H.; Park, A.K.; Lee, S.; Choi, S.A.; Baek, I.P.; Kim, P.; Kim, E.H.; Park, H.C.; Kim, B.C.; Bhak, J.; et al. Genomic analysis reveals secondary glioblastoma after radiotherapy in a subset of recurrent medulloblastomas. Acta Neuropathol. 2018, 135, 939–953. [Google Scholar] [CrossRef]
- Stichel, D.; Ebrahimi, A.; Reuss, D.; Schrimpf, D.; Ono, T.; Shirahata, M.; Reifenberger, G.; Weller, M.; Hänggi, D.; Wick, W.; et al. Distribution of EGFR amplification, combined chromosome 7 gain and chromosome 10 loss, and TERT promoter mutation in brain tumors and their potential for the reclassification of IDHwt astrocytoma to glioblastoma. Acta Neuropathol. 2018, 136, 793–803. [Google Scholar] [CrossRef]
- Tauziède-Espariat, A.; Debily, M.A.; Castel, D.; Grill, J.; Puget, S.; Roux, A.; Saffroy, R.; Pagès, M.; Gareton, A.; Chrétien, F.; et al. The pediatric supratentorial MYCN-amplified high-grade gliomas methylation class presents the same radiological, histopathological and molecular features as their pontine counterparts. Acta Neuropathol. Commun. 2020, 8, 104. [Google Scholar] [CrossRef]
- Bouffet, E.; Larouche, V.; Campbell, B.B.; Merico, D.; de Borja, R.; Aronson, M.; Durno, C.; Krueger, J.; Cabric, V.; Ramaswamy, V.; et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J. Clin. Oncol. 2016, 34, 2206–2211. [Google Scholar] [CrossRef]
- Clarke, M.; Mackay, A.; Ismer, B.; Pickles, J.C.; Tatevossian, R.G.; Newman, S.; Bale, T.A.; Stoler, I.; Izquierdo, E.; Temelso, S.; et al. Infant high-grade gliomas comprise multiple subgroups characterized by novel targetable gene fusions and favorable outcomes. Cancer Discov. 2020, 10, 942–963. [Google Scholar] [CrossRef]
- Di Ruscio, V.; Carai, A.; Del Baldo, G.; Vinci, M.; Cacchione, A.; Miele, E.; Rossi, S.; Antonelli, M.; Barresi, S.; Caulo, M.; et al. Molecular landscape in infant high-grade gliomas: A single center experience. Diagnostics 2022, 12, 372. [Google Scholar] [CrossRef]
- Ziegler, D.S.; Wong, M.; Mayoh, C.; Kumar, A.; Tsoli, M.; Mould, E.; Tyrrell, V.; Khuong-Quang, D.A.; Pinese, M.; Gayevskiy, V.; et al. Brief Report: Potent clinical and radiological response to larotrectinib in TRK fusion-driven high-grade glioma. Br. J. Cancer 2018, 119, 693–696. [Google Scholar] [CrossRef]
- Aghajan, Y.; Levy, M.L.; Malicki, D.M.; Crawford, J.R. Novel PPP1CB-ALK fusion protein in a high-grade glioma of infancy. BMJ Case Rep. 2016, 2016, bcr2016217189. [Google Scholar] [CrossRef]
- Coccé, M.C.; Mardin, B.R.; Bens, S.; Stütz, A.M.; Lubieniecki, F.; Vater, I.; Korbel, J.O.; Siebert, R.; Alonso, C.N.; Gallego, M.S. Identification of ZCCHC8 as fusion partner of ROS1 in a case of congenital glioblastoma multiforme with a t(6;12)(q21;q24.3). Genes Chromosomes Cancer 2016, 55, 677–687. [Google Scholar] [CrossRef]
- Guerreiro Stucklin, A.S.; Ryall, S.; Fukuoka, K.; Zapotocky, M.; Lassaletta, A.; Li, C.; Bridge, T.; Kim, B.; Arnoldo, A.; Kowalski, P.E.; et al. Alterations in ALK/ROS1/NTRK/MET drive a group of infantile hemispheric gliomas. Nat. Commun. 2019, 10, 4343. [Google Scholar] [CrossRef]
- Ng, A.; Levy, M.L.; Malicki, D.M.; Crawford, J.R. Unusual high-grade and low-grade glioma in an infant with PPP1CB-ALK gene fusion. BMJ Case Rep. 2019, 12, e228248. [Google Scholar] [CrossRef]
- Olsen, T.K.; Panagopoulos, I.; Meling, T.R.; Micci, F.; Gorunova, L.; Thorsen, J.; Due-Tønnessen, B.; Scheie, D.; Lund-Iversen, M.; Krossnes, B.; et al. Fusion genes with ALK as recurrent partner in ependymoma-like gliomas: A new brain tumor entity? Neuro-oncology 2015, 17, 1365–1373. [Google Scholar] [CrossRef]
- Valera, E.T.; Neder, L.; Queiroz, R.G.; Santos, A.C.; Sousa, G.R.; Oliveira, R.S.; Santos, M.V.; Machado, H.R.; Tone, L.G. Perinatal complex low- and high-grade glial tumor harboring a novel GIGYF2-ALK fusion. Pediatr. Blood Cancer 2020, 67, e28015. [Google Scholar] [CrossRef]
- Duffner, P.K.; Krischer, J.P.; Burger, P.C.; Cohen, M.E.; Backstrom, J.W.; Horowitz, M.E.; Sanford, R.A.; Friedman, H.S.; Kun, L.E. Treatment of infants with malignant gliomas: The Pediatric Oncology Group experience. J. Neurooncol. 1996, 28, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Torre, M.; Vasudevaraja, V.; Serrano, J.; DeLorenzo, M.; Malinowski, S.; Blandin, A.F.; Pages, M.; Ligon, A.H.; Dong, F.; Meredith, D.M.; et al. Molecular and clinicopathologic features of gliomas harboring NTRK fusions. Acta Neuropathol. Commun. 2020, 8, 107. [Google Scholar] [CrossRef]
- Duffner, P.K.; Horowitz, M.E.; Krischer, J.P.; Burger, P.C.; Cohen, M.E.; Sanford, R.A.; Friedman, H.S.; Kun, L.E. The treatment of malignant brain tumors in infants and very young children: An update of the Pediatric Oncology Group experience. Neuro-oncology 1999, 1, 152–161. [Google Scholar] [CrossRef]
- Drilon, A.; Siena, S.; Ou, S.I.; Patel, M.; Ahn, M.J.; Lee, J.; Bauer, T.M.; Farago, A.F.; Wheler, J.J.; Liu, S.V.; et al. Safety and antitumor activity of the multitargeted pan-TRK, ROS1, and ALK inhibitor entrectinib: Combined results from two phase I trials (ALKA-372-001 and STARTRK-1). Cancer Discov. 2017, 7, 400–409. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diffuse Midline Glioma H3K27-Altered | Diffuse Hemispheric Glioma, H3G34-Altered | Diffuse Paediatric-Type High-Grade Glioma, H3-Wildtype and IDH-Wildtype | Infant-Type Hemispheric Glioma | |
|---|---|---|---|---|
| Driver Molecular Events | Loss of H3 trimethylation
| H3.3 G34R or G34V point mutations
| IDH and H3 Wildtype Methylation subtypes:
| May harbour fusions with NTRK, ALK, ROS1 or MET |
| CNS WHO grade | Grade 4 | Grade 4 | Grade 4 | Not assigned |
| Epidemiology | Most patients are children and adolescents H3.1-altered cases are younger than H3.3-altered cases (which may affect adults) | Most are adolescents and young adults Usually from 11 to 30 years | MYCN cases show a median age of 8–9 years RTK1 and RTK2 are older, with a median age of 10–11 years | Most cases below 1 year of age May be congenital |
| Location | Midline structures
| Hemispheric, with preference for temporal and parietal lobes | Most cases are supratentorial masses RTK1 (18%) or MYCN (14%) tumours may appear in the brainstem (both) or cerebellum (RTK1) | Cerebral hemispheres |
| Histopathology | Diffuse pattern of infiltration Usually astrocytic features High-grade features may not be present | Two patterns, usually with high-grade features:
| Presence of high-grade features May be circumscribed May show biphasic pattern (spindle and epithelioid) or PNET-like histology | Hypercellular, usually high-grade features |
| IHC | H3K27me3 loss is essential in all subtypes H3K27M or EZHIP positivity depends on the subtype Most cases are GFAP+, OLIG2+, and MAP2+ Bithalamic gliomas are usually GFAP+, OLIG2-, and MAP2- | Usually GFAP+ and OLIG2- Loss of ATRX IHC and diffuse p53 staining are typical PNET-like cases may stain with neuronal markers IHC for H3G34R or H3G34V may be of help | Glial markers are usually positive (GFAP+, OLIG2+) MYCN cases may express neuronal markers (NeuN, Neurofilaments, CD56) H3KK27me3 should be retained IDH1 and H3K27M must be negative | Usually positive for glial markers ALK rearranged cases may show IHC positivity |
| Differential Diagnosis | Consider DMG H3K27-altered in midline tumours pHGG H3- and IDH-wildtype, MYCN subtype, may also affect the midline (consider FISH for MYCN) Consider low-grade tumours if high-grade features are absent | Essential to prove presence of an H3G34 point mutation in hemispheric neoplasms Consider IDH-altered gliomas in adolescents and adults If PNET-like histology, also consider embryonal or ependymal neoplasms | Dependent on location and morphology Essentially excluding H3-altered or IDH-altered gliomas PNET-like histology may require differential diagnosis with embryonal neoplasms | Dependent on morphology May include embryonal neoplasms (medulloblastoma), ependymomas, or gliomas Testing for RTK fusions is useful for diagnosis |
| Prognosis | Dismal, median OS of 11 months Longer survival in H3.1-altered cases (median OS 15 months) Bithalamic glioma has equally bad prognosis (median OS of 8 to 12 months) | Bad prognosis, but better (median OS 22 months) than DMG H3K27-altered Methylation of MGMT promoter may be a factor in survival | Survival is bad, but depends on methylation subtypes: MYCN subtype is comparable to DMG H3K27-altered (median OS: 14 months); RTK1 and RTK2 show longer survival (median OS of 21 and 44 months) | Better than other pHGGs Consider Tyrosine Kinase inhibition if possible |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blasco-Santana, L.; Colmenero, I. Molecular and Pathological Features of Paediatric High-Grade Gliomas. Int. J. Mol. Sci. 2024, 25, 8498. https://doi.org/10.3390/ijms25158498
Blasco-Santana L, Colmenero I. Molecular and Pathological Features of Paediatric High-Grade Gliomas. International Journal of Molecular Sciences. 2024; 25(15):8498. https://doi.org/10.3390/ijms25158498
Chicago/Turabian StyleBlasco-Santana, Luis, and Isabel Colmenero. 2024. "Molecular and Pathological Features of Paediatric High-Grade Gliomas" International Journal of Molecular Sciences 25, no. 15: 8498. https://doi.org/10.3390/ijms25158498
APA StyleBlasco-Santana, L., & Colmenero, I. (2024). Molecular and Pathological Features of Paediatric High-Grade Gliomas. International Journal of Molecular Sciences, 25(15), 8498. https://doi.org/10.3390/ijms25158498