
Discovery of NFκB2-Coordinated Dual Regulation of Mitochondrial and Nuclear Genomes Leads to an Effective Therapy for Acute Myeloid Leukemia

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Oligomycin-Inhibiting ATP Synthase (Complex V) Can Sensitize Blasts to TKI-Treatment; However, Refractory Blasts Underwent the Genomic Reprogramming Required for Metabolic Plasticity
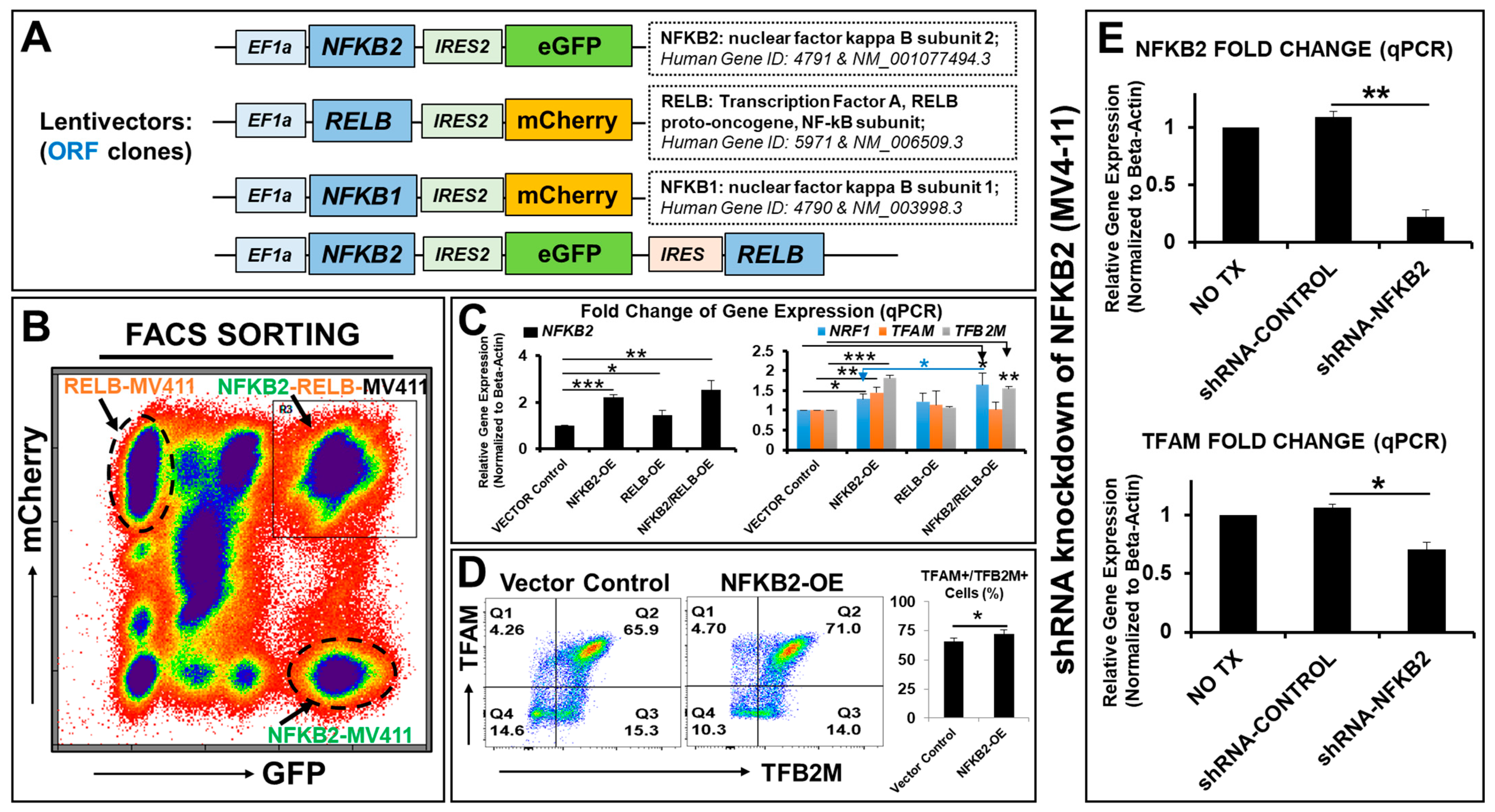
2.2. NFκB2 Regulates the Expression of Essential Nuclear Transcription Factors Responsible for Metabolic Plasticity in AML Blasts
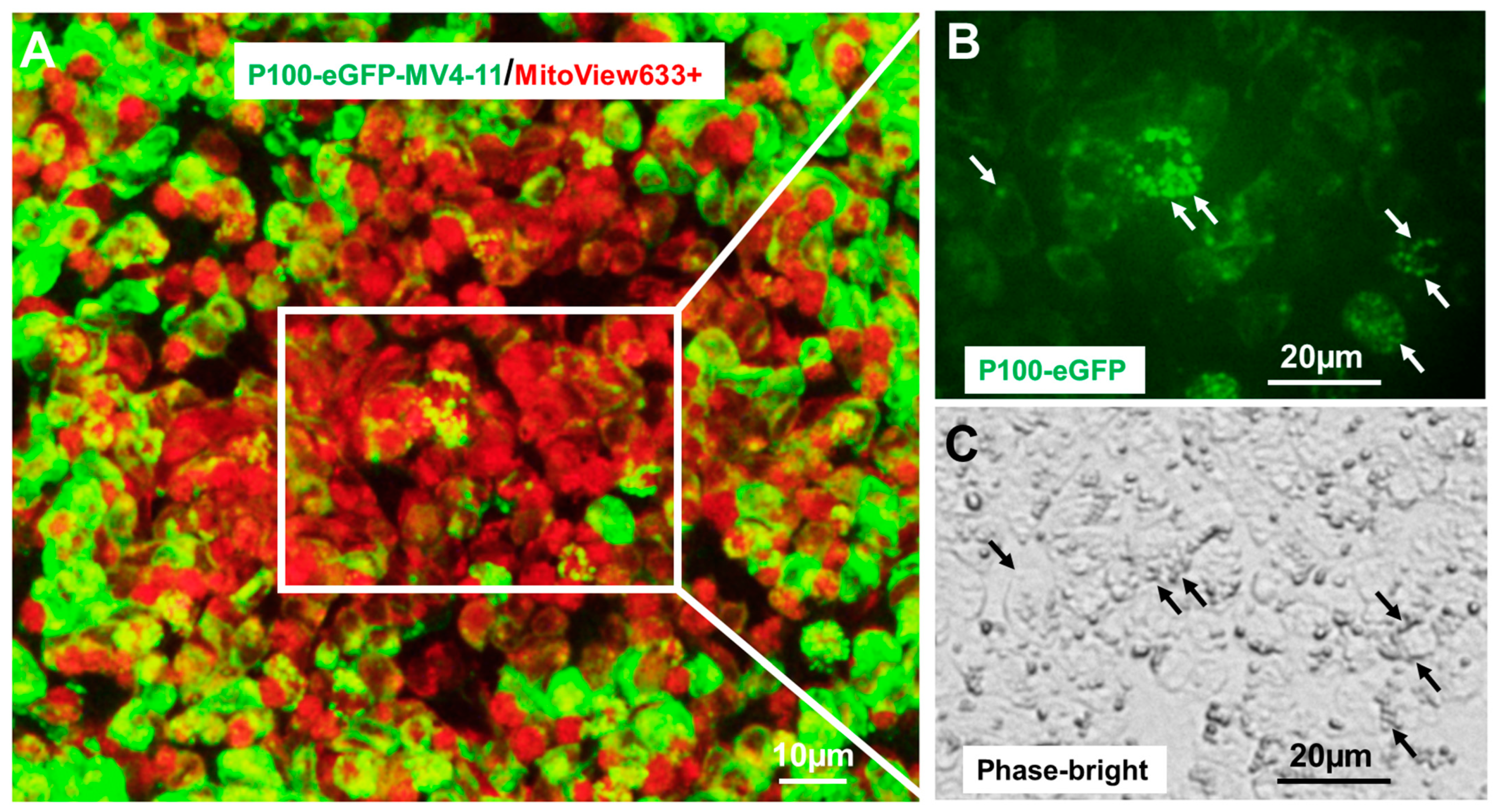
2.3. NFκB2 Was Localized in Human Mitochondria
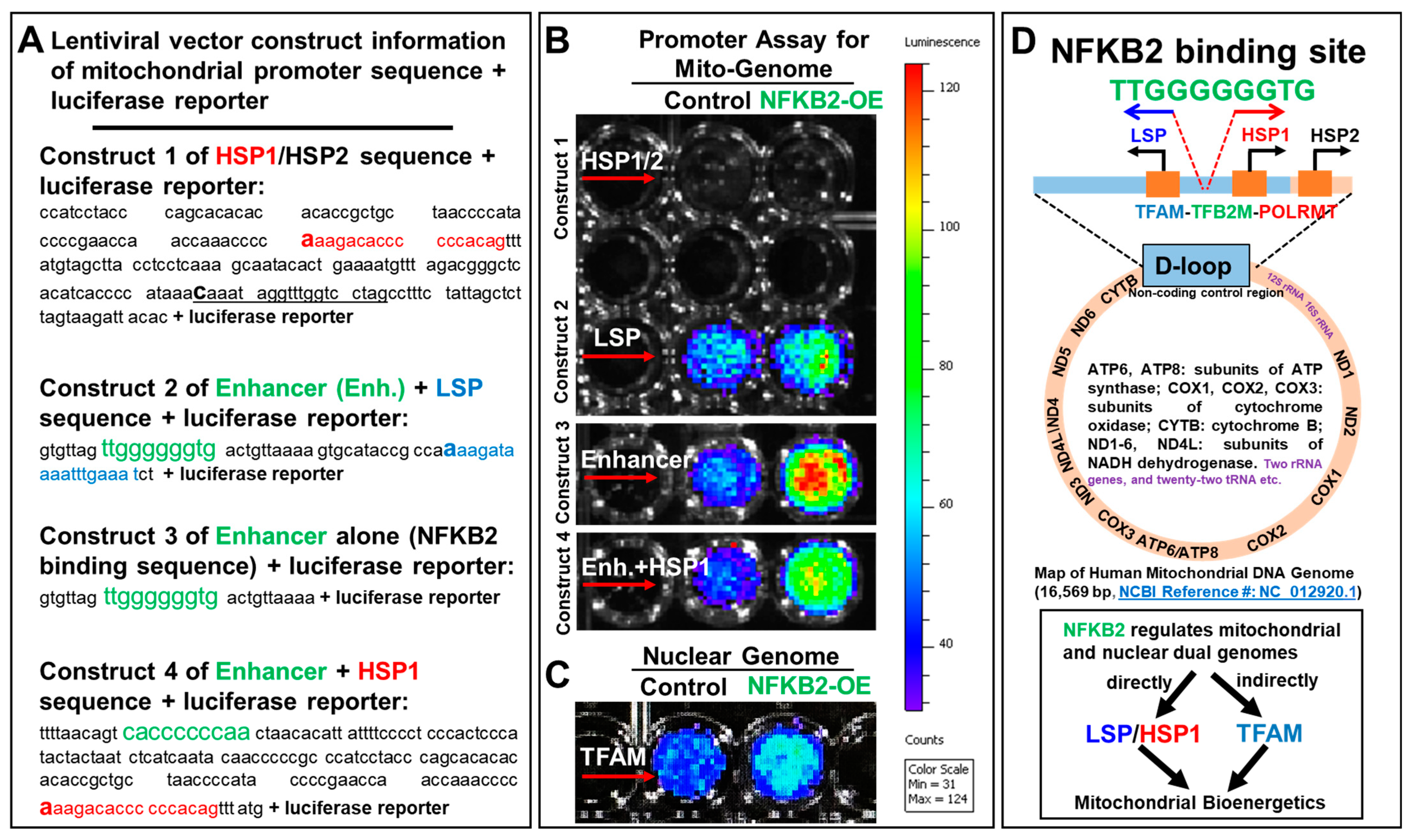
2.4. NFκB2 Activated the Promoters of Blast Mitochondrial–Nuclear Dual Genomes
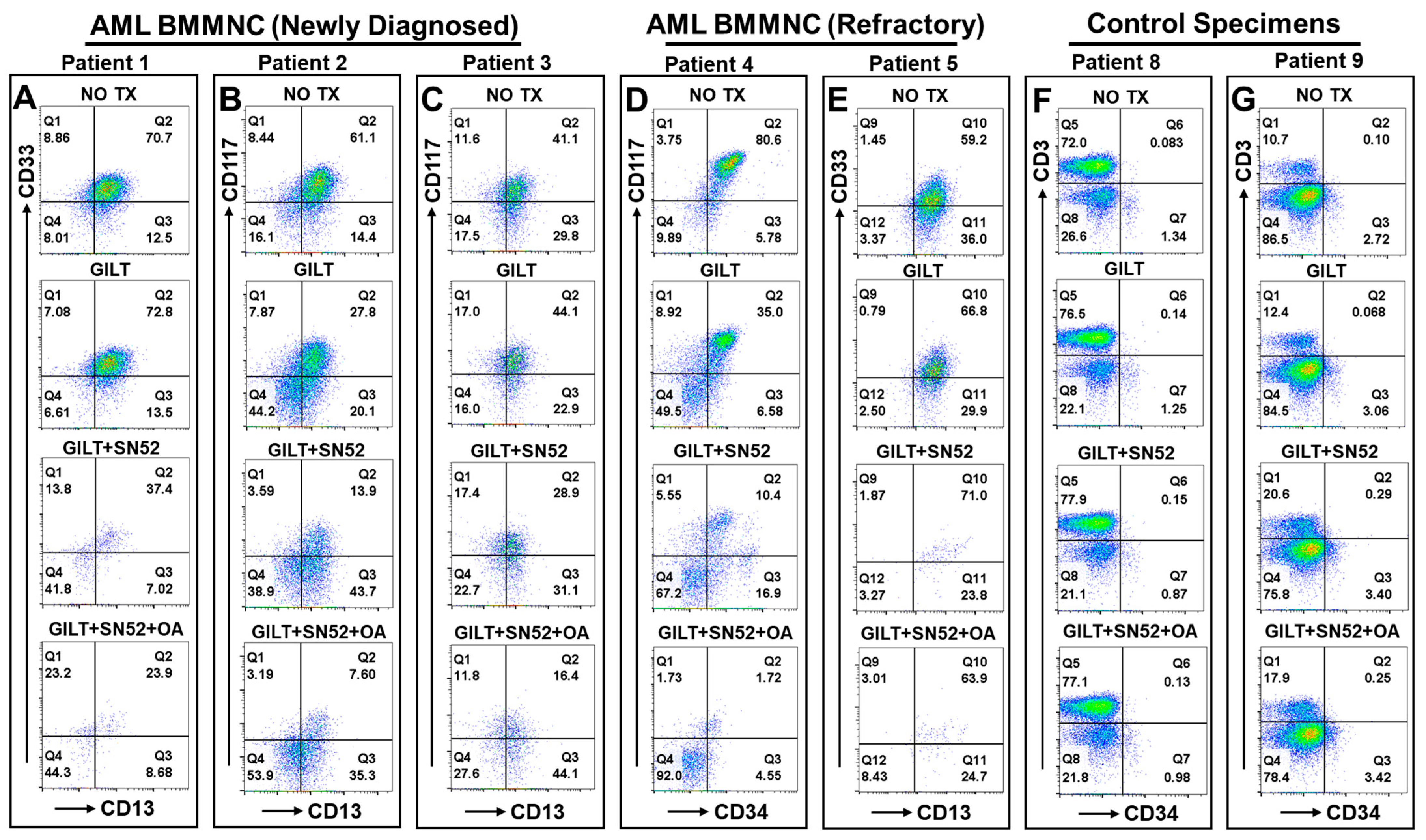
2.5. The Triple Combination of Gilteritinib + SN52 + Oligomycin Effectively Eliminated AML Blasts by Inhibiting Their Mitochondrial Biogenesis and Functions
2.6. The Triplet Therapy Exhibited a Robust Antileukemia Effect against Primary AML Blasts with Minimum Toxicity to the Control Cells of 10 Patients Ex Vivo
3. Discussion
3.1. Noncanonical NFκB2 Is a Single Transcription Factor Individually Regulating the Dual Mitochondrial–Nuclear Genomes Essential for Mitochondrial Biogenesis and Metabolic Reprogramming in AML
3.2. Inhibition of the Key Transcriptional Regulator of Dual Genomes Responsible for Metabolic Plasticity Is a Fundamental Strategy toward Eliminating AML Blasts
4. Materials and Methods
4.1. Primary Patient Specimens
4.2. Cell Culture
4.3. In Vitro and Ex Vivo Treatment of AML Blasts
4.4. Generation of New Transgenic Cell Lines In Vitro (See Details in Supplementary Table S3)
- (a)
- Preparation of lentiviral particles in vitro
- (b)
- NFκB family gene-overexpressed MV4-11 cell lines
- (c)
- NFκB2 (P100)-eGFP and (P52)-eGFP reporter cell lines
- (d)
- NFκB2-knockdown MV4-11 cell lines
4.5. Promoter Assays of Mitochondrial Genome (LSP and HSP) and Nuclear Genome (TFAM) In Vitro
4.6. Flow Cytometry (FC)
4.7. Mitochondrial Isolation
4.8. RNA Isolation and Real-Time Polymerase Chain Reaction (qPCR) Analysis
4.9. ATP Assay
4.10. Imaging Acquisition
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Dohner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H. Acute myeloid leukemia--major progress over four decades and glimpses into the future. Am. J. Hematol. 2016, 91, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, V.E.; Smith, C.C. FLT3 Mutations in Acute Myeloid Leukemia: Key Concepts and Emerging Controversies. Front. Oncol. 2020, 10, 612880. [Google Scholar] [CrossRef] [PubMed]
- Perl, A.E.; Martinelli, G.; Cortes, J.E.; Neubauer, A.; Berman, E.; Paolini, S.; Montesinos, P.; Baer, M.R.; Larson, R.A.; Ustun, C.; et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. N. Engl. J. Med. 2019, 381, 1728–1740. [Google Scholar] [CrossRef] [PubMed]
- Ganzel, C.; Sun, Z.; Cripe, L.D.; Fernandez, H.F.; Douer, D.; Rowe, J.M.; Paietta, E.M.; Ketterling, R.; O’Connell, M.J.; Wiernik, P.H.; et al. Very poor long-term survival in past and more recent studies for relapsed AML patients: The ECOG-ACRIN experience. Am. J. Hematol. 2018, 93, 1074–1081. [Google Scholar] [CrossRef] [PubMed]
- Janssen, M.; Schmidt, C.; Bruch, P.-M.; Blank, M.F.; Rohde, C.; Waclawiczek, A.; Heid, D.; Renders, S.; Göllner, S.; Vierbaum, L.; et al. Venetoclax synergizes with gilteritinib in FLT3 wild-type high-risk acute myeloid leukemia by suppressing MCL-1. Blood 2022, 140, 2594–2610. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Perl, A.E.; Maly, J.; Levis, M.; Ritchie, E.; Litzow, M.; McCloskey, J.; Smith, C.C.; Schiller, G.; Bradley, T.; et al. Venetoclax Plus Gilteritinib for FLT3-Mutated Relapsed/Refractory Acute Myeloid Leukemia. J. Clin. Oncol. 2022, 40, 4048–4059. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Baylink, D.J.; Chen, C.-S.; Tan, L.; Xiao, J.; Park, B.; Valladares, I.; Reeves, M.E.; Cao, H. Transient TKI-resistant CD44+pBAD+ blasts undergo intrinsic homeostatic adaptation to promote the survival of acute myeloid leukemia in vitro. Front. Oncol. 2023, 13, 1286863. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Tadros, V.; Hiramoto, B.; Leeper, K.; Hino, C.; Xiao, J.; Pham, B.; Kim, D.H.; Reeves, M.E.; Chen, C.-S.; et al. Targeting TKI-Activated NFKB2-MIF/CXCLs-CXCR2 Signaling Pathways in FLT3 Mutated Acute Myeloid Leukemia Reduced Blast Viability. Biomedicines 2022, 10, 1038. [Google Scholar] [CrossRef]
- Hino, C.; Xu, Y.; Xiao, J.; Baylink, D.J.; Reeves, M.E.; Cao, H. The potential role of the thymus in immunotherapies for acute myeloid leukemia. Front. Immunol. 2023, 14, 1102517. [Google Scholar] [CrossRef]
- Cooperrider, J.H.; Shukla, N.; Nawas, M.T.; Patel, A.A. The Cup Runneth Over: Treatment Strategies for Newly Diagnosed Acute Myeloid Leukemia. JCO Oncol. Pract. 2023, 19, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.H.; Zhao, D.; Branciamore, S.; Maestrini, D.; Rodriguez, I.R.; Kuo, Y.-H.; Rockne, R.; Khaled, S.K.; Zhang, B.; Nguyen, L.X.T.; et al. MicroRNA networks in FLT3-ITD acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 2022, 119, e2112482119. [Google Scholar] [CrossRef] [PubMed]
- Weeks, L.D.; Ebert, B.L. Causes and consequences of clonal hematopoiesis. Blood 2023, 142, 2235–2246. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.F.; Latorre-Muro, P.; Puigserver, P. Mechanisms of mitochondrial respiratory adaptation. Nat. Rev. Mol. Cell Biol. 2022, 23, 817–835. [Google Scholar] [CrossRef] [PubMed]
- Missiroli, S.; Perrone, M.; Genovese, I.; Pinton, P.; Giorgi, C. Cancer metabolism and mitochondria: Finding novel mechanisms to fight tumours. EBioMedicine 2020, 59, 102943. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Zhu, J.; Thompson, C.B. The hallmarks of cancer metabolism: Still emerging. Cell Metab. 2022, 34, 355–377. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gindin, M.M.; van der Windt, G.J.W.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Stine, Z.E.; Schug, Z.T.; Salvino, J.M.; Dang, C.V. Targeting cancer metabolism in the era of precision oncology. Nat. Rev. Drug Discov. 2022, 21, 141–162. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, E.H. Cancer interception. Cancer Prev. Res. 2011, 4, 787–792. [Google Scholar] [CrossRef]
- Baum, J.; Lax, H.; Lehmann, N.; Merkel-Jens, A.; Beelen, D.W.; Jöckel, K.-H.; Dührsen, U. Impairment of vocational activities and financial problems are frequent among German blood cancer survivors. Sci. Rep. 2023, 13, 22856. [Google Scholar] [CrossRef]
- Pratz, K.W.; Panayiotidis, P.; Recher, C.; Wei, X.; Jonas, B.A.; Montesinos, P.; Ivanov, V.; Schuh, A.C.; DiNardo, C.D.; Novak, J.; et al. Venetoclax combinations delay the time to deterioration of HRQoL in unfit patients with acute myeloid leukemia. Blood Cancer J. 2022, 12, 71. [Google Scholar] [CrossRef] [PubMed]
- Vafai, S.B.; Mootha, V.K. Mitochondrial disorders as windows into an ancient organelle. Nature 2012, 491, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Koopman, W.J.; Willems, P.H.; Smeitink, J.A. Monogenic mitochondrial disorders. N. Engl. J. Med. 2012, 366, 1132–1141. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, R.P.; Chandel, N.S. Beyond ATP, new roles of mitochondria. Biochemist 2022, 44, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Barshad, G.; Marom, S.; Cohen, T.; Mishmar, D. Mitochondrial DNA Transcription and Its Regulation: An Evolutionary Perspective. Trends Genet. 2018, 34, 682–692. [Google Scholar] [CrossRef] [PubMed]
- Basu, U.; Bostwick, A.M.; Das, K.; Dittenhafer-Reed, K.E.; Patel, S.S. Structure, mechanism, and regulation of mitochondrial DNA transcription initiation. J. Biol. Chem. 2020, 295, 18406–18425. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Kepp, O.; Trojel-Hansen, C.; Kroemer, G. Mitochondrial control of cellular life, stress, and death. Circ. Res. 2012, 111, 1198–1207. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, M.A.; Sullivan, E.D.; Copeland, W.C. Consequences of compromised mitochondrial genome integrity. DNA Repair 2020, 93, 102916. [Google Scholar] [CrossRef]
- Begum, H.M.; Shen, K. Intracellular and microenvironmental regulation of mitochondrial membrane potential in cancer cells. WIREs Mech. Dis. 2023, 15, e1595. [Google Scholar] [CrossRef]
- Gammage, P.A.; Frezza, C. Mitochondrial DNA: The overlooked oncogenome? BMC Biol. 2019, 17, 53. [Google Scholar] [CrossRef]
- Rath, S.; Sharma, R.; Gupta, R.; Ast, T.; Chan, C.; Durham, T.J.; Goodman, R.P.; Grabarek, Z.; Haas, M.E.; Hung, W.H.W.; et al. MitoCarta3.0: An updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 2021, 49, D1541–D1547. [Google Scholar] [CrossRef] [PubMed]
- Litonin, D.; Sologub, M.; Shi, Y.; Savkina, M.; Anikin, M.; Falkenberg, M.; Gustafsson, C.M.; Temiakov, D. Human mitochondrial transcription revisited: Only TFAM and TFB2M are required for transcription of the mitochondrial genes in vitro. J. Biol. Chem. 2010, 285, 18129–18133. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.T.; Hoogenraad, N.J. Mitochondrial-nuclear communications. Annu. Rev. Biochem. 2007, 76, 701–722. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.P.; Scarpulla, R.C. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004, 18, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Kleine, T.; Leister, D. Retrograde signaling: Organelles go networking. Biochim. Biophys. Acta 2016, 1857, 1313–1325. [Google Scholar] [CrossRef] [PubMed]
- Calvo, S.E.; Mootha, V.K. The mitochondrial proteome and human disease. Annu. Rev. Genom. Hum. Genet. 2010, 11, 25–44. [Google Scholar] [CrossRef]
- Wasilewski, M.; Draczkowski, P.; Chacinska, A. Protein import into mitochondria—A new path through the membranes. Nat. Struct. Mol. Biol. 2023, 30, 1831–1833. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.; Rutter, J.C.; Li, Y.D.; Ebert, B.L. Induced protein degradation for therapeutics: Past, present, and future. J. Clin. Investig. 2024, 134, e175265. [Google Scholar] [CrossRef]
- Latchman, D.S. Transcription factors: An overview. Int. J. Biochem. Cell Biol. 1997, 29, 1305–1312. [Google Scholar] [CrossRef]
- Libermann, T.A.; Zerbini, L.F. Targeting transcription factors for cancer gene therapy. Curr. Gene Ther. 2006, 6, 17–33. [Google Scholar] [CrossRef]
- Overington, J.P.; Al-Lazikani, B.; Hopkins, A.L. How many drug targets are there? Nat. Rev. Drug Discov. 2006, 5, 993–996. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2017, 16, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Miranda, M.; Bonekamp, N.A.; Kuhl, I. Starting the engine of the powerhouse: Mitochondrial transcription and beyond. Biol. Chem. 2022, 403, 779–805. [Google Scholar] [CrossRef] [PubMed]
- Panina, S.B.; Pei, J.; Kirienko, N.V. Mitochondrial metabolism as a target for acute myeloid leukemia treatment. Cancer Metab. 2021, 9, 17. [Google Scholar] [CrossRef]
- Yu, H.; Lin, L.; Zhang, Z.; Zhang, H.; Hu, H. Targeting NF-kappaB pathway for the therapy of diseases: Mechanism and clinical study. Signal Transduct. Target. Ther. 2020, 5, 209. [Google Scholar] [CrossRef]
- Cildir, G.; Low, K.C.; Tergaonkar, V. Noncanonical NF-kappaB Signaling in Health and Disease. Trends Mol. Med. 2016, 22, 414–429. [Google Scholar] [CrossRef]
- Boulanger, M.; Aqrouq, M.; Tempé, D.; Kifagi, C.; Ristic, M.; Akl, D.; Hallal, R.; Carusi, A.; Gabellier, L.; de Toledo, M.; et al. DeSUMOylation of chromatin-bound proteins limits the rapid transcriptional reprogramming induced by daunorubicin in acute myeloid leukemias. Nucleic Acids Res. 2023, 51, 8413–8433. [Google Scholar] [CrossRef]
- Gilmore, T.D. NF-kappaB and Human Cancer: What Have We Learned over the Past 35 Years? Biomedicines 2021, 9, 889. [Google Scholar] [CrossRef]
- Wang, T.; Ma, F.; Qian, H.L. Defueling the cancer: ATP synthase as an emerging target in cancer therapy. Mol. Ther. Oncolytics 2021, 23, 82–95. [Google Scholar] [CrossRef]
- Xu, Y.; Tran, L.; Tang, J.; Nguyen, V.; Sewell, E.; Xiao, J.; Hino, C.; Wasnik, S.; Francis-Boyle, O.L.; Zhang, K.K.; et al. FBP1-Altered Carbohydrate Metabolism Reduces Leukemic Viability through Activating P53 and Modulating the Mitochondrial Quality Control System In Vitro. Int. J. Mol. Sci. 2022, 23, 11387. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.J.; Eling, T. Growth differentiation factor 15 (GDF15): A survival protein with therapeutic potential in metabolic diseases. Pharmacol. Ther. 2019, 198, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Scarpulla, R.C. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev. 2008, 88, 611–638. [Google Scholar] [CrossRef] [PubMed]
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N. Regulation of Mitochondrial Biogenesis as a Way for Active Longevity: Interaction Between the Nrf2 and PGC-1alpha Signaling Pathways. Front. Genet. 2019, 10, 435. [Google Scholar] [CrossRef] [PubMed]
- Portt, L.; Norman, G.; Clapp, C.; Greenwood, M.; Greenwood, M.T. Anti-apoptosis and cell survival: A review. Biochim. Biophys. Acta 2011, 1813, 238–259. [Google Scholar] [CrossRef]
- Psarra, A.M.; Sekeris, C.E. Nuclear receptors and other nuclear transcription factors in mitochondria: Regulatory molecules in a new environment. Biochim. Biophys. Acta 2008, 1783, 1–11. [Google Scholar] [CrossRef]
- Leigh-Brown, S.; Enriquez, J.A.; Odom, D.T. Nuclear transcription factors in mammalian mitochondria. Genome Biol. 2010, 11, 215. [Google Scholar] [CrossRef]
- Wong, D.; Teixeira, A.; Oikonomopoulos, S.; Humburg, P.; Lone, I.N.; Saliba, D.; Siggers, T.; Bulyk, M.; Angelov, D.; Dimitrov, S.; et al. Extensive characterization of NF-kappaB binding uncovers non-canonical motifs and advances the interpretation of genetic functional traits. Genome Biol. 2011, 12, R70. [Google Scholar] [CrossRef]
- Tang, H.L.; Yuen, K.L.; Tang, H.M.; Fung, M.C. Reversibility of apoptosis in cancer cells. Br. J. Cancer 2009, 100, 118–122. [Google Scholar] [CrossRef]
- Xu, Y.; Fang, F.; Clair, D.K.S.; Sompol, P.; Josson, S.; Clair, W.H.S. SN52, a novel nuclear factor-kappaB inhibitor, blocks nuclear import of RelB:p52 dimer and sensitizes prostate cancer cells to ionizing radiation. Mol. Cancer Ther. 2008, 7, 2367–2376. [Google Scholar] [CrossRef]
- Zhang, X.; Dang, C.V. Time to hit pause on mitochondria-targeting cancer therapies. Nat. Med. 2023, 29, 29–30. [Google Scholar] [CrossRef] [PubMed]
- Kalyanaraman, B.; Cheng, G.; Hardy, M.; You, M. OXPHOS-targeting drugs in oncology: New perspectives. Expert Opin. Ther. Targets 2023, 27, 939–952. [Google Scholar] [CrossRef]
- Lin, J.; Handschin, C.; Spiegelman, B.M. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005, 1, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, S.E.; Chandel, N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2015, 11, 9–15. [Google Scholar] [CrossRef]
- Salminen, A.; Huuskonen, J.; Ojala, J.; Kauppinen, A.; Kaarniranta, K.; Suuronen, T. Activation of innate immunity system during aging: NF-kB signaling is the molecular culprit of inflamm-aging. Ageing Res. Rev. 2008, 7, 83–105. [Google Scholar] [CrossRef] [PubMed]
- Albensi, B.C. What Is Nuclear Factor Kappa B (NF-kappaB) Doing in and to the Mitochondrion? Front. Cell Dev. Biol. 2019, 7, 154. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.F.; Witzel, I.I.; Perkins, N.D. p53-dependent regulation of mitochondrial energy production by the RelA subunit of NF-kappaB. Cancer Res. 2011, 71, 5588–5597. [Google Scholar] [CrossRef]
- Mauro, C.; Leow, S.C.; Anso, E.; Rocha, S.; Thotakura, A.K.; Tornatore, L.; Moretti, M.; De Smaele, E.; Beg, A.A.; Tergaonkar, V.; et al. NF-kappaB controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nat. Cell Biol. 2011, 13, 1272–1279. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Schon, K.R.; Elgar, G.; Orioli, A.; Tanguy, M.; Giess, A.; Tischkowitz, M.; Caulfield, M.J.; Chinnery, P.F. Nuclear-embedded mitochondrial DNA sequences in 66,083 human genomes. Nature 2022, 611, 105–114. [Google Scholar] [CrossRef]
- Waseem, M.; Wang, B.D. Promising Strategy of mPTP Modulation in Cancer Therapy: An Emerging Progress and Future Insight. Int. J. Mol. Sci. 2023, 24, 5564. [Google Scholar] [CrossRef]
- Halestrap, A.P. What is the mitochondrial permeability transition pore? J. Mol. Cell Cardiol. 2009, 46, 821–831. [Google Scholar] [CrossRef]
- Tsuboi, T.; Viana, M.P.; Xu, F.; Yu, J.; Chanchani, R.; Arceo, X.G.; Tutucci, E.; Choi, J.; Chen, Y.S.; Singer, R.H.; et al. Mitochondrial volume fraction and translation duration impact mitochondrial mRNA localization and protein synthesis. eLife 2020, 9, e57814. [Google Scholar] [CrossRef]
- Aibara, S.; Singh, V.; Modelska, A.; Amunts, A. Structural basis of mitochondrial translation. eLife 2020, 9, e58362. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Hino, C.; Baylink, D.J.; Xiao, J.; Reeves, M.E.; Zhong, J.F.; Mirshahidi, S.; Cao, H. Vitamin D activates FBP1 to block the Warburg effect and modulate blast metabolism in acute myeloid leukemia. Biomark. Res. 2022, 10, 16. [Google Scholar] [CrossRef]
- Thompson, C.B.; Vousden, K.H.; Johnson, R.S.; Koppenol, W.H.; Sies, H.; Lu, Z.; Finley, L.W.S.; Frezza, C.; Kim, J.; Hu, Z.; et al. A century of the Warburg effect. Nat. Metab. 2023, 5, 1840–1843. [Google Scholar] [CrossRef]
- Westermarck, J. Inhibition of adaptive therapy tolerance in cancer: Is triplet mitochondrial targeting the key? Mol. Oncol. 2023, 17, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Vasan, K.; Chandel, N.S. Molecular and cellular mechanisms underlying the failure of mitochondrial metabolism drugs in cancer clinical trials. J. Clin. Investig. 2024, 134, e176736. [Google Scholar] [CrossRef]
- Zhang, L.; Wei, Y.; Yuan, S.; Sun, L. Targeting Mitochondrial Metabolic Reprogramming as a Potential Approach for Cancer Therapy. Int. J. Mol. Sci. 2023, 24, 4954. [Google Scholar] [CrossRef]
- Nguyen, C.; Pandey, S. Exploiting Mitochondrial Vulnerabilities to Trigger Apoptosis Selectively in Cancer Cells. Cancers 2019, 11, 916. [Google Scholar] [CrossRef]
- Vringer, E.; Tait, S.W.G. Mitochondria and cell death-associated inflammation. Cell Death Differ. 2023, 30, 304–312. [Google Scholar] [CrossRef]
- Newman, L.E.; Novak, S.W.; Rojas, G.R.; Tadepalle, N.; Schiavon, C.R.; Grotjahn, D.A.; Towers, C.G.; Tremblay, M.; Donnelly, M.P.; Ghosh, S.; et al. Mitochondrial DNA replication stress triggers a pro-inflammatory endosomal pathway of nucleoid disposal. Nat. Cell Biol. 2024, 26, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. Nuclear factor-kappaB in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef]
- Ramadass, V.; Vaiyapuri, T.; Tergaonkar, V. Small Molecule NF-kappaB Pathway Inhibitors in Clinic. Int. J. Mol. Sci. 2020, 21, 5164. [Google Scholar] [CrossRef]
- Bushweller, J.H. Targeting transcription factors in cancer—From undruggable to reality. Nat. Rev. Cancer 2019, 19, 611–624. [Google Scholar] [CrossRef]
- Cann, R.L.; Stoneking, M.; Wilson, A.C. Mitochondrial DNA and human evolution. Nature 1987, 325, 31–36. [Google Scholar] [CrossRef]
- Gray, M.W.; Burger, G.; Lang, B.F. Mitochondrial evolution. Science 1999, 283, 1476–1481. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Xiao, J.; Reeves, M.E.; Payne, K.; Chen, C.S.; Baylink, D.J.; Marcucci, G.; Xu, Y. Discovery of proangiogenic CD44+mesenchymal cancer stem cells in an acute myeloid leukemia patient’s bone marrow. J. Hematol. Oncol. 2020, 13, 63. [Google Scholar] [CrossRef]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef]
- Frezza, C.; Cipolat, S.; Scorrano, L. Organelle isolation: Functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat. Protoc. 2007, 2, 287–295. [Google Scholar] [CrossRef]
- Xu, Y.; Payne, K.; Pham, L.H.G.; Eunwoo, P.; Xiao, J.; Chi, D.; Lyu, J.; Campion, R.; Wasnik, S.; Jeong, I.S.; et al. A novel vitamin D gene therapy for acute myeloid leukemia. Transl. Oncol. 2020, 13, 100869. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Diagnosis | Sex | Age | Gene Mutation |
|---|---|---|---|---|
| AML Patient 1 | AML (Newly diagnosed) | M | 76 | FLT3 (c.2503G > T; p.D835Y): Allele Frequency 41% |
| AML Patient 2 | AML (Newly diagnosed) | M | 62 | FLT3-ITD: Level = 0.98 |
| AML Patient 3 | AML (Newly diagnosed) | M | 75 | FLT3-ITD: Allele Frequency 0.27% |
| AML Patient 4 | AML (Refractory) | M | 24 | FLT3-ITD; 46,XY,t(6,11)(q27;q23) [11]/46,XY [9] |
| AML Patient 5 | AML (Refractory) | M | 69 | FLT3-ITD: Signal Ratio 0.89 (c.1789delins25; p.Y597delins9) (23% allele) |
| AML Patient 6 | AML (Newly diagnosed) | M | 52 | FLT3-ITD: FLT3 Internal Tandem Duplication (ITD) confirmed by PCR with Signal Ratio 0.12. |
| AML Patient 7 | AML (Refractory) | F | 65 | FLT 3-ITD: Signal Ratio 0.57 |
| Healthy Patient 8 | N/A | M | 50 | Normal |
| Healthy Patient 9 | N/A | M | 55 | Normal |
| Healthy Patient 10 | N/A | F | 40 | Normal |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Y.; Baylink, D.J.; Xiao, J.; Tran, L.; Nguyen, V.; Park, B.; Valladares, I.; Lee, S.; Codorniz, K.; Tan, L.; et al. Discovery of NFκB2-Coordinated Dual Regulation of Mitochondrial and Nuclear Genomes Leads to an Effective Therapy for Acute Myeloid Leukemia. Int. J. Mol. Sci. 2024, 25, 8532. https://doi.org/10.3390/ijms25158532
Xu Y, Baylink DJ, Xiao J, Tran L, Nguyen V, Park B, Valladares I, Lee S, Codorniz K, Tan L, et al. Discovery of NFκB2-Coordinated Dual Regulation of Mitochondrial and Nuclear Genomes Leads to an Effective Therapy for Acute Myeloid Leukemia. International Journal of Molecular Sciences. 2024; 25(15):8532. https://doi.org/10.3390/ijms25158532
Chicago/Turabian StyleXu, Yi, David J. Baylink, Jeffrey Xiao, Lily Tran, Vinh Nguyen, Brandon Park, Ismael Valladares, Scott Lee, Kevin Codorniz, Laren Tan, and et al. 2024. "Discovery of NFκB2-Coordinated Dual Regulation of Mitochondrial and Nuclear Genomes Leads to an Effective Therapy for Acute Myeloid Leukemia" International Journal of Molecular Sciences 25, no. 15: 8532. https://doi.org/10.3390/ijms25158532