
Eosinophilic Esophagitis and Inflammatory Bowel Disease: What Are the Differences?


Abstract
:1. Introduction
2. Increasing EoE and IBD Incidence and Prevalence
2.1. EoE
2.2. IBD
3. Genetics Polymorphisms
3.1. EoE
3.2. IBD
3.3. Overlapping Genetic Features
4. The Exposome in EoE and IBD
5. Diagnosis
6. Treatments
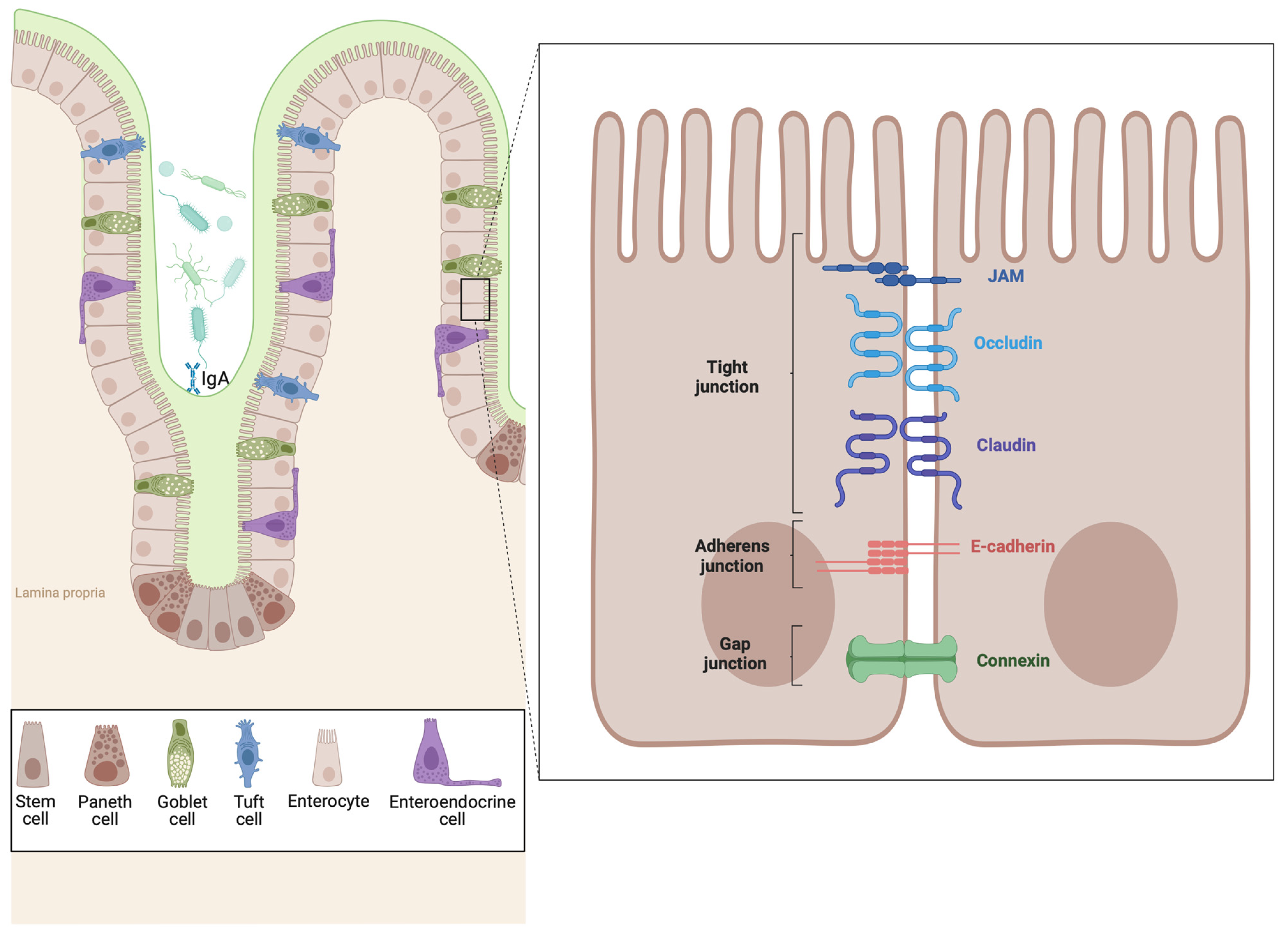
7. The Epithelial Barrier in EoE and IBD
7.1. Epithelial Barrier Dysfunction in EoE
7.2. Epithelial Barrier Dysfunction in IBD
7.3. Immune and Epithelial Interfaces: Soluble Immune Effectors
7.3.1. EoE
7.3.2. IBD
8. Comparative Immunopathogenesis of Eosinophilic Esophagitis and Inflammatory Bowel Disease
8.1. Eosinophils
8.1.1. EoE
8.1.2. IBD
8.2. T Cells
8.2.1. EoE
8.2.2. IBD
8.3. Mast Cells
8.3.1. EoE
8.3.2. IBD
8.4. B Cells
8.4.1. EoE
8.4.2. IBD
8.5. Dendritic Cells and Macrophages
8.5.1. EoE
8.5.2. IBD
9. Summary and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Brown, L.F.; Goldman, H.; Antonioli, D.A. Intraepithelial eosinophils in endoscopic biopsies of adults with reflux esophagitis. Am. J. Surg. Pathol. 1984, 8, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Winter, H.S.; Madara, J.L.; Stafford, R.J.; Grand, R.J.; Quinlan, J.-E.; Goldman, H. Intraepithelial Eosinophils: A New Diagnostic Criterion for Reflux Esophagitis. Gastroenterology 1982, 83, 818–823. [Google Scholar] [CrossRef] [PubMed]
- Furuta, G.T.; Liacouras, C.A.; Collins, M.H.; Gupta, S.K.; Justinich, C.; Putnam, P.E.; Bonis, P.; Hassall, E.; Straumann, A.; Rothenberg, M.E. Eosinophilic Esophagitis in Children and Adults: A Systematic Review and Consensus Recommendations for Diagnosis and Treatment. Gastroenterology 2007, 133, 1342–1363. [Google Scholar] [CrossRef] [PubMed]
- Liacouras, C.A.; Furuta, G.T.; Hirano, I.; Atkins, D.; Attwood, S.E.; Bonis, P.A.; Burks, A.W.; Chehade, M.; Collins, M.H.; Dellon, E.S.; et al. Eosinophilic esophagitis: Updated consensus recommendations for children and adults. J. Allergy Clin. Immunol. 2011, 128, 3–20.e6. [Google Scholar] [CrossRef] [PubMed]
- Biedermann, L.; Straumann, A. Mechanisms and clinical management of eosinophilic oesophagitis: An overview. Nat. Rev. Gastroenterol. Hepatol. 2022, 20, 101–119. [Google Scholar] [CrossRef] [PubMed]
- Rothenberg, M.E. Biology and Treatment of Eosinophilic Esophagitis. Gastroenterology 2009, 137, 1238–1249. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Z.; Li, Y.Y. Inflammatory bowel disease: Pathogenesis. World J. Gastroenterol. 2014, 20, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Rochman, M.; Azouz, N.P.; Rothenberg, M.E. Epithelial origin of eosinophilic esophagitis. J. Allergy Clin. Immunol. 2018, 142, 10–23. [Google Scholar] [CrossRef] [PubMed]
- Underwood, B.; Troutman, T.D.; Schwartz, J.T. Breaking down the complex pathophysiology of eosinophilic esophagitis. Ann. Allergy Asthma Immunol. 2022, 130, 28–39. [Google Scholar] [CrossRef]
- Mulder, D.J.; Justinich, C.J. Understanding eosinophilic esophagitis: The cellular and molecular mechanisms of an emerging disease. Mucosal Immunol. 2011, 4, 139–147. [Google Scholar] [CrossRef]
- Raheem, M.; Leach, S.T.; Day, A.S.; Lemberg, D.A. The Pathophysiology of Eosinophilic Esophagitis. Front. Pediatr. 2014, 2, 41. [Google Scholar] [CrossRef] [PubMed]
- Coskun, M. Intestinal Epithelium in Inflammatory Bowel Disease. Front. Med. 2014, 1, 24. [Google Scholar] [CrossRef] [PubMed]
- Martini, E.; Krug, S.M.; Siegmund, B.; Neurath, M.F.; Becker, C. Mend Your Fences: The Epithelial Barrier and its Relationship with Mucosal Immunity in Inflammatory Bowel Disease. Cell Mol. Gastroenterol. Hepatol. 2017, 4, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, C.; Stucke, E.M.; Rodriguez-Jimenez, B.; Burwinkel, K.; Collins, M.H.; Ahrens, A.; Alexander, E.S.; Butz, B.K.B.; Jameson, S.C.; Kaul, A.; et al. A striking local esophageal cytokine expression profile in eosinophilic esophagitis. J. Allergy Clin. Immunol. 2011, 127, 208–217.e7. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.T. Pathophysiology of Inflammatory Bowel Diseases. N. Engl. J. Med. 2020, 383, 2652–2664. [Google Scholar] [CrossRef]
- Racca, F.; Pellegatta, G.; Cataldo, G.; Vespa, E.; Carlani, E.; Pelaia, C.; Paoletti, G.; Messina, M.R.; Nappi, E.; Canonica, G.W.; et al. Type 2 Inflammation in Eosinophilic Esophagitis: From Pathophysiology to Therapeutic Targets. Front. Physiol. 2022, 12, 815842. [Google Scholar] [CrossRef]
- Meyer, F.; Wendling, D.; Demougeot, C.; Prati, C.; Verhoeven, F. Cytokines and intestinal epithelial permeability: A systematic review. Autoimmun. Rev. 2023, 22, 103331. [Google Scholar] [CrossRef]
- Rosenblum, D.; Naik, S. Epithelial–immune crosstalk in health and disease. Curr. Opin. Genet. Dev. 2022, 74, 101910. [Google Scholar] [CrossRef]
- Prasad, G.A.; Alexander, J.A.; Schleck, C.D.; Zinsmeister, A.R.; Smyrk, T.C.; Elias, R.M.; Locke, G.R.; Talley, N.J. Epidemiology of Eosinophilic Esophagitis Over Three Decades in Olmsted County, Minnesota. Clin. Gastroenterol. Hepatol. 2009, 7, 1055–1061. [Google Scholar] [CrossRef]
- Hruz, P.; Straumann, A.; Bussmann, C.; Heer, P.; Simon, H.-U.; Zwahlen, M.; Beglinger, C.; Schoepfer, A.M.; Swiss EoE Study Group. Escalating incidence of eosinophilic esophagitis: A 20-year prospective, population-based study in Olten County, Switzerland. J. Allergy Clin. Immunol. 2011, 128, 1349–1350.e5. [Google Scholar] [CrossRef]
- van Rhijn, B.D.; Verheij, J.; Smout, A.J.P.M.; Bredenoord, A.J. Rapidly increasing incidence of eosinophilic esophagitis in a large cohort. Neurogastroenterol. Motil. 2012, 25, 47-e5. [Google Scholar] [CrossRef]
- Soon, I.S.; Butzner, J.D.; Kaplan, G.G.; de Bruyn, J.C. Incidence and Prevalence of Eosinophilic Esophagitis in Children. J. Pediatr. Gastroenterol. Nutr. 2013, 57, 72–80. [Google Scholar] [CrossRef]
- Hurtado, C.W.; Furuta, G.T.; E Kramer, R. Etiology of Esophageal Food Impactions in Children. J. Pediatr. Gastroenterol. Nutr. 2011, 52, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, S.H.; Go, M.; Chadwick, B.; Thomas, K.; Fang, J.; Kuwada, S.; Lamphier, S.; Hilden, K.; Peterson, K. Eosinophilic oesophagitis in patients presenting with dysphagia—A prospective analysis. Aliment. Pharmacol. Ther. 2008, 28, 1140–1146. [Google Scholar] [CrossRef]
- Navarro, P.; Arias, Á.; Arias-González, L.; Laserna-Mendieta, E.J.; Ruiz-Ponce, M.; Lucendo, A.J. Systematic review with meta-analysis: The growing incidence and prevalence of eosinophilic oesophagitis in children and adults in population-based studies. Aliment. Pharmacol. Ther. 2019, 49, 1116–1125. [Google Scholar] [CrossRef]
- Arias, Á.; Perezmartinez, I.; Tenias, J.M.; Lucendo, A.J. Systematic review with meta-analysis: The incidence and prevalence of eosinophilic oesophagitis in children and adults in population-based studies. Aliment. Pharmacol. Ther. 2016, 43, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Arias, Á.; Lucendo, A.J. Prevalence of eosinophilic oesophagitis in adult patients in a central region of Spain. Eur. J. Gastroenterol. Hepatol. 2013, 25, 208–212. [Google Scholar] [CrossRef]
- Cherian, S.; Smith, N.M.; A Forbes, D. Rapidly increasing prevalence of eosinophilic oesophagitis in Western Australia. Arch. Dis. Child. 2006, 91, 1000–1004. [Google Scholar] [CrossRef] [PubMed]
- Fouad, M.; Fouad, Y.M.; Mokareb, H.A.; Mohamed, E.A.; Abdel-Rehim, D.M. Prevalence of Eosinophilic Esophagitis in Adult Patients with Upper Gastrointestinal Symptoms in a Locality in Upper Egypt. Clin. Endosc. 2018, 51, 357–361. [Google Scholar] [CrossRef]
- Dellon, E.S. Epidemiology of Eosinophilic Esophagitis. Gastroenterol. Clin. N. Am. 2014, 43, 201–218. [Google Scholar] [CrossRef]
- Giriens, B.; Yan, P.; Safroneeva, E.; Zwahlen, M.; Reinhard, A.; Nydegger, A.; Vavricka, S.; Sempoux, C.; Straumann, A.; Schoepfer, A.M. Escalating incidence of eosinophilic esophagitis in Canton of Vaud, Switzerland, 1993–2013: A population-based study. Allergy 2015, 70, 1633–1639. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet 2017, 390, 2769–2778. [Google Scholar] [CrossRef] [PubMed]
- Burisch, J.; Jess, T.; Martinato, M.; Lakatos, P.L. The burden of inflammatory bowel disease in Europe. J. Crohn’s Colitis 2013, 7, 322–337. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Li, Z.; Liu, S.; Zhang, D. Global, regional and national burden of inflammatory bowel disease in 204 countries and territories from 1990 to 2019: A systematic analysis based on the Global Burden of Disease Study 2019. BMJ Open 2023, 13, e065186. [Google Scholar] [CrossRef]
- Coward, S.; Clement, F.; Benchimol, E.I.; Bernstein, C.N.; Avina-Zubieta, J.A.; Bitton, A.; Carroll, M.W.; Hazlewood, G.; Jacobson, K.; Jelinski, S.; et al. Past and Future Burden of Inflammatory Bowel Diseases Based on Modeling of Population-Based Data. Gastroenterology 2019, 156, 1345–1353.e4. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.-C.; Weng, M.-T.; Tung, C.-C.; Chang, Y.-T.; Leong, Y.-L.; Wang, Y.-T.; Wang, H.-Y.; Wong, J.-M.; Wei, S.-C. Trends and risk factors of mortality analysis in patients with inflammatory bowel disease: A Taiwanese nationwide population-based study. J. Transl. Med. 2019, 17, 414. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-K.; Hong, W.-S.; Min, Y.I.; Kim, H.Y.; Rhee, P.-L.; Chang, D.K.; Song, I.S.; Jung, S.A.; Park, E.-B.; Yoo, H.M.; et al. Incidence and prevalence of ulcerative colitis in the Songpa-Kangdong district, Seoul, Korea, 1986–1997. Gastroenterology 2000, 118, A1377. [Google Scholar] [CrossRef]
- Yang, S.K.; Yun, S.; Kim, J.-H.; Park J., Y.; Kim, H.Y.; Kim, Y.-H.; Chang, D.K.; Kim, J.S.; Song, I.S.; Park, J.B.; et al. Epidemiology of inflammatory bowel disease in the Songpa-Kangdong district, Seoul, Korea, 1986–2005: A KASID study. Inflamm. Bowel Dis. 2008, 14, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Uchida, A.M.; Garber, J.J.; Pyne, A.; Peterson, K.; Roelstraete, B.; Olén, O.; Halfvarson, J.; Ludvigsson, J.F. Eosinophilic esophagitis is associated with increased risk of later inflammatory bowel disease in a nationwide Swedish population cohort. United Eur. Gastroenterol. J. 2023, 12, 34–43. [Google Scholar] [CrossRef]
- Moore, H.; Wechsler, J.; Frost, C.; Whiteside, E.; Baldassano, R.; Markowitz, J.; Muir, A.B. Comorbid Diagnosis of Eosinophilic Esophagitis and Inflammatory Bowel Disease in the Pediatric Population. J. Pediatr. Gastroenterol. Nutr. 2020, 72, 398–403. [Google Scholar] [CrossRef]
- Limketkai, B.N.; Shah, S.C.; Hirano, I.; Bellaguarda, E.; Colombel, J.-F. Epidemiology and implications of concurrent diagnosis of eosinophilic oesophagitis and IBD based on a prospective population-based analysis. Gut 2019, 68, 2152–2160. [Google Scholar] [CrossRef] [PubMed]
- Sleiman, P.M.A.; Wang, M.-L.; Cianferoni, A.; Aceves, S.; Gonsalves, N.; Nadeau, K.; Bredenoord, A.J.; Furuta, G.T.; Spergel, J.M.; Hakonarson, H. GWAS identifies four novel eosinophilic esophagitis loci. Nat. Commun. 2014, 5, 5593. [Google Scholar] [CrossRef] [PubMed]
- E Rothenberg, M.; Spergel, J.M.; Sherrill, J.D.; Annaiah, K.; Martin, L.J.; Cianferoni, A.; Gober, L.; Kim, C.; Glessner, J.; Frackelton, E.; et al. Common variants at 5q22 associate with pediatric eosinophilic esophagitis. Nat. Genet. 2010, 42, 289–291. [Google Scholar] [CrossRef] [PubMed]
- Kottyan, L.C.; Davis, B.P.; Sherrill, J.D.; Liu, K.; Rochman, M.; Kaufman, K.; Weirauch, M.T.; Vaughn, S.; Lazaro, S.; Rupert, A.M.; et al. Genome-wide association analysis of eosinophilic esophagitis provides insight into the tissue specificity of this allergic disease. Nat. Genet. 2014, 46, 895–900. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, C.; Wang, N.; Stringer, K.F.; Mishra, A.; Fulkerson, P.C.; Abonia, J.P.; Jameson, S.C.; Kirby, C.; Konikoff, M.R.; Collins, M.H.; et al. Eotaxin-3 and a uniquely conserved gene-expression profile in eosinophilic esophagitis. J. Clin. Investig. 2006, 116, 536–547. [Google Scholar] [CrossRef] [PubMed]
- Ávila-Castellano, R.; García-Lozano, J.R.; Cimbollek, S.; Lucendo, A.J.; Bozada, J.M.; Quiralte, J. Genetic variations in the TLR3 locus are associated with eosinophilic esophagitis. United Eur. Gastroenterol. J. 2018, 6, 349–357. [Google Scholar] [CrossRef]
- Mirkov, M.U.; Verstockt, B.; Cleynen, I. Genetics of inflammatory bowel disease: Beyond NOD2. Lancet Gastroenterol. Hepatol. 2017, 2, 224–234. [Google Scholar] [CrossRef]
- Ellinghaus, D.; Ellinghaus, E.; Nair, R.P.; Stuart, P.E.; Esko, T.; Metspalu, A.; Debrus, S.; Raelson, J.V.; Tejasvi, T.; Belouchi, M.; et al. Combined Analysis of Genome-wide Association Studies for Crohn Disease and Psoriasis Identifies Seven Shared Susceptibility Loci. Am. J. Hum. Genet. 2012, 90, 636–647. [Google Scholar] [CrossRef] [PubMed]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Schumm, L.P.; Sharma, Y.; Anderson, C.A.; et al. Host–microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef]
- Abraham, C.; Medzhitov, R. Interactions Between the Host Innate Immune System and Microbes in Inflammatory Bowel Disease. Gastroenterology 2011, 140, 1729–1737. [Google Scholar] [CrossRef]
- Adler, J.; Rangwalla, S.C.; A Dwamena, B.; Higgins, P.D. The Prognostic Power of the NOD2 Genotype for Complicated Crohn’s Disease: A Meta-Analysis. Am. J. Gastroenterol. 2011, 106, 699–712. [Google Scholar] [CrossRef]
- Cho, J.H.; Brant, S.R. Recent Insights Into the Genetics of Inflammatory Bowel Disease. Gastroenterology 2011, 140, 1704–1712.e2. [Google Scholar] [CrossRef]
- Lauro, R.; Mannino, F.; Irrera, N.; Squadrito, F.; Altavilla, D.; Squadrito, G.; Pallio, G.; Bitto, A. Pharmacogenetics of Biological Agents Used in Inflammatory Bowel Disease: A Systematic Review. Biomedicines 2021, 9, 1748. [Google Scholar] [CrossRef]
- Klein, W.; Tromm, A.; Folwaczny, C.; Hagedorn, M.; Duerig, N.; Epplen, J.; Schmiegel, W.; Griga, T. The G2964A polymorphism of the STAT6 gene in inflammatory bowel disease. Dig. Liver Dis. 2005, 37, 159–161. [Google Scholar] [CrossRef] [PubMed]
- Namjou, B.; Marsolo, K.; Caroll, R.J.; Denny, J.C.; Ritchie, M.D.; Verma, S.S.; Lingren, T.; Porollo, A.; Cobb, B.L.; Perry, C.; et al. Phenome-wide association study (PheWAS) in EMR-linked pediatric cohorts, genetically links PLCL1 to speech language development and IL5-IL13 to Eosinophilic Esophagitis. Front. Genet. 2014, 5, 401. [Google Scholar] [CrossRef] [PubMed]
- Walczak, A.; Przybylowska, K.; Dziki, L.; Sygut, A.; Chojnacki, C.; Chojnacki, J.; Dziki, A.; Majsterek, I. The lL-8 and IL-13 gene polymorphisms in inflammatory bowel disease and colorectal cancer. DNA Cell Biol. 2012, 31, 1431–1438. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, C.; Stucke, E.M.; Burwinkel, K.; Caldwell, J.M.; Collins, M.H.; Ahrens, A.; Buckmeier, B.K.; Jameson, S.C.; Greenberg, A.; Kaul, A.; et al. Coordinate Interaction between IL-13 and Epithelial Differentiation Cluster Genes in Eosinophilic Esophagitis. J. Immunol. 2010, 184, 4033–4041. [Google Scholar] [CrossRef] [PubMed]
- Van Limbergen, J.; Russell, R.K.; Nimmo, E.R.; Zhao, Y.; Liao, H.; Drummond, H.E.; Davies, G.; Gillett, P.M.; McGrogan, P.; Bisset, W.M.; et al. Filaggrin loss-of-function variants are associated with atopic comorbidity in pediatric inflammatory bowel disease. Inflamm. Bowel Dis. 2009, 15, 1492–1498. [Google Scholar] [CrossRef]
- Kagalwalla, A.F.; Shah, A.; Li, B.U.; Sentongo, T.A.; Ritz, S.; Manuel-Rubio, M.; Jacques, K.; Wang, D.; Melin-Aldana, H.; Nelson, S.P. Identification of Specific Foods Responsible for Inflammation in Children With Eosinophilic Esophagitis Successfully Treated With Empiric Elimination Diet. J. Pediatr. Gastroenterol. Nutr. 2011, 53, 145–149. [Google Scholar] [CrossRef]
- Spergel, J.M.; Brown-Whitehorn, T.F.; Cianferoni, A.; Shuker, M.; Wang, M.-L.; Verma, R.; Liacouras, C.A. Identification of causative foods in children with eosinophilic esophagitis treated with an elimination diet. J. Allergy Clin. Immunol. 2012, 130, 461–467.e5. [Google Scholar] [CrossRef]
- Simon, D.; Cianferoni, A.; Spergel, J.M.; Aceves, S.; Holbreich, M.; Venter, C.; Rothenberg, M.E.; Terreehorst, I.; Muraro, A.; Lucendo, A.J.; et al. Eosinophilic esophagitis is characterized by a non-IgE-mediated food hypersensitivity. Allergy 2016, 71, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Hogan, S.P.; Brandt, E.B.; Rothenberg, M.E. An etiological role for aeroallergens and eosinophils in experimental esophagitis. J. Clin. Investig. 2001, 107, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Jensen, E.T.; Dellon, E.S. Environmental factors and eosinophilic esophagitis. J. Allergy Clin. Immunol. 2018, 142, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Muir, A.B.; Benitez, A.J.; Dods, K.; Spergel, J.M.; Fillon, S.A. Microbiome and its impact on gastrointestinal atopy. Allergy 2016, 71, 1256–1263. [Google Scholar] [CrossRef] [PubMed]
- Doyle, A.D.; Masuda, M.Y.; Pyon, G.C.; Luo, H.; Putikova, A.; LeSuer, W.E.; Flashner, S.; Rank, M.A.; Nakagawa, H.; Kita, H.; et al. Detergent exposure induces epithelial barrier dysfunction and eosinophilic inflammation in the esophagus. Allergy 2022, 78, 192–201. [Google Scholar] [CrossRef]
- Abegunde, A.T.; Muhammad, B.H.; Bhatti, O.; Ali, T. Environmental risk factors for inflammatory bowel diseases: Evidence based literature review. World J. Gastroenterol. 2016, 22, 6296–6317. [Google Scholar] [CrossRef]
- Chapman-Kiddell, C.A.; Davies, P.S.; Gillen, L.; Radford-Smith, G.L. Role of diet in the development of inflammatory bowel disease. Inflamm. Bowel Dis. 2010, 16, 137–151. [Google Scholar] [CrossRef]
- Higuchi, L.M.; Khalili, H.; Chan, A.T.; Richter, J.M.; Bousvaros, A.; Fuchs, C.S. A Prospective Study of Cigarette Smoking and the Risk of Inflammatory Bowel Disease in Women. Am. J. Gastroenterol. 2012, 107, 1399–1406. [Google Scholar] [CrossRef]
- Thomas, T.; Chandan, J.S.; Li, V.S.W.; Lai, C.Y.; Tang, W.; Bhala, N.; Kaplan, G.G.; Ng, S.C.; Ghosh, S. Global smoking trends in inflammatory bowel disease: A systematic review of inception cohorts. PLoS ONE 2019, 14, e0221961. [Google Scholar] [CrossRef]
- Devkota, S.; Wang, Y.; Musch, M.W.; Leone, V.; Fehlner-Peach, H.; Nadimpalli, A.; Antonopoulos, D.A.; Jabri, B.; Chang, E.B. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10−/− mice. Nature 2012, 487, 104–108. [Google Scholar] [CrossRef]
- Hildebrand, H.; Malmborg, P.; Askling, J.; Ekbom, A.; Montgomery, S.M. Early-life exposures associated with antibiotic use and risk of subsequent Crohn’s disease. Scand. J. Gastroenterol. 2008, 43, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Ungaro, R.; Bernstein, C.N.; Gearry, R.; Hviid, A.; Kolho, K.-L.; Kronman, M.P.; Shaw, S.; Van Kruiningen, H.; Colombel, J.-F.; Atreja, A. Antibiotics Associated With Increased Risk of New-Onset Crohn’s Disease But Not Ulcerative Colitis: A Meta-Analysis. Am. J. Gastroenterol. 2014, 109, 1728–1738. [Google Scholar] [CrossRef] [PubMed]
- Leslie, W.D.; Miller, N.; Rogala, L.; Bernstein, C.N. Vitamin D Status and Bone Density in Recently Diagnosed Inflammatory Bowel Disease: The Manitoba IBD Cohort Study. Am. J. Gastroenterol. 2008, 103, 1451–1459. [Google Scholar] [CrossRef] [PubMed]
- Cantorna, M.T.; Munsick, C.; Bemiss, C.; Mahon, B.D. 1,25-Dihydroxycholecalciferol Prevents and Ameliorates Symptoms of Experimental Murine Inflammatory Bowel Disease. J. Nutr. 2000, 130, 2648–2652. [Google Scholar] [CrossRef]
- Brusilovsky, M.; Rochman, M.; Shoda, T.; Kotliar, M.; Caldwell, J.M.; E Mack, L.; A Besse, J.; Chen, X.; Weirauch, M.T.; Barski, A.; et al. Vitamin D receptor and STAT6 interactome governs oesophageal epithelial barrier responses to IL-13 signalling. Gut 2023, 72, 834–845. [Google Scholar] [CrossRef] [PubMed]
- Dellon, E.S.; Liacouras, C.A.; Molina-Infante, J.; Furuta, G.T.; Spergel, J.M.; Zevit, N.; Spechler, S.J.; Attwood, S.E.; Straumann, A.; Aceves, S.S.; et al. Updated International Consensus Diagnostic Criteria for Eosinophilic Esophagitis: Proceedings of the AGREE Conference. Gastroenterology 2018, 155, 1022–1033.e10. [Google Scholar] [CrossRef]
- Dellon, E.S.; Gonsalves, N.; Hirano, I.; Furuta, G.T.; A Liacouras, C.; A Katzka, D. ACG Clinical Guideline: Evidenced Based Approach to the Diagnosis and Management of Esophageal Eosinophilia and Eosinophilic Esophagitis (EoE). Am. J. Gastroenterol. 2013, 108, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Ashton, J.J.; Beattie, R.M. Inflammatory bowel disease: Recent developments. Arch. Dis. Child. 2023, 109, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Falvey, J.D.; Hoskin, T.; Meijer, B.; Ashcroft, A.; Walmsley, R.; Day, A.S.; Gearry, R.B. Disease activity assessment in IBD: Clinical indices and biomarkers fail to predict endoscopic remission. Inflamm. Bowel Dis. 2015, 21, 824–831. [Google Scholar] [CrossRef]
- Liacouras, C.A.; Spergel, J.M.; Ruchelli, E.; Verma, R.; Mascarenhas, M.; Semeao, E.; Flick, J.; Kelly, J.; Brown–Whitehorn, T.; Mamula, P.; et al. Eosinophilic Esophagitis: A 10-Year Experience in 381 Children. Clin. Gastroenterol. Hepatol. 2005, 3, 1198–1206. [Google Scholar] [CrossRef]
- Rank, M.A.; Sharaf, R.N.; Furuta, G.T.; Aceves, S.S.; Greenhawt, M.; Spergel, J.M.; Falck-Ytter, Y.T.; Dellon, E.S.; Chachu, K.A.; Day, L.; et al. Technical Review on the Management of Eosinophilic Esophagitis: A Report From the AGA Institute and the Joint Task Force on Allergy-Immunology Practice Parameters. Gastroenterology 2020, 158, 1789–1810.e15. [Google Scholar] [CrossRef] [PubMed]
- Lucendo, A.J.; Arias, Á.; Molina-Infante, J. Efficacy of Proton Pump Inhibitor Drugs for Inducing Clinical and Histologic Remission in Patients With Symptomatic Esophageal Eosinophilia: A Systematic Review and Meta-Analysis. Clin. Gastroenterol. Hepatol. 2016, 14, 13–22.e1. [Google Scholar] [CrossRef] [PubMed]
- Molina-Infante, J.; Bredenoord, A.J.; Cheng, E.; Dellon, E.S.; Furuta, G.T.; Gupta, S.K.; Hirano, I.; A Katzka, D.; Moawad, F.J.; E Rothenberg, M.; et al. Proton pump inhibitor-responsive oesophageal eosinophilia: An entity challenging current diagnostic criteria for eosinophilic oesophagitis. Gut 2015, 65, 524–531. [Google Scholar] [CrossRef]
- Dellon, E.S.; Woosley, J.T.; Arrington, A.; McGee, S.J.; Covington, J.; Moist, S.E.; Gebhart, J.H.; Tylicki, A.E.; Shoyoye, S.O.; Martin, C.F.; et al. Efficacy of Budesonide vs Fluticasone for Initial Treatment of Eosinophilic Esophagitis in a Randomized Controlled Trial. Gastroenterology 2019, 157, 65–73.e5. [Google Scholar] [CrossRef] [PubMed]
- Lucendo, A.J.; Miehlke, S.; Schlag, C.; Vieth, M.; von Arnim, U.; Molina-Infante, J.; Hartmann, D.; Bredenoord, A.J.; de Los Rios, C.C.; Schubert, S.; et al. Efficacy of Budesonide Orodispersible Tablets as Induction Therapy for Eosinophilic Esophagitis in a Randomized Placebo-Controlled Trial. Gastroenterology 2019, 157, 74–86.e15. [Google Scholar] [CrossRef] [PubMed]
- Lucendo, A.J.; De Rezende, L.C.; Jiménez-Contreras, S.; Yagüe-Compadre, J.L.; González-Cervera, J.; Mota-Huertas, T.; Guagnozzi, D.; Angueira, T.; González-Castillo, S.; Arias, A. Montelukast Was Inefficient in Maintaining Steroid-Induced Remission in Adult Eosinophilic Esophagitis. Dig. Dis. Sci. 2011, 56, 3551–3558. [Google Scholar] [CrossRef]
- Rothenberg, M.E.; Wen, T.; Greenberg, A.; Alpan, O.; Enav, B.; Hirano, I.; Nadeau, K.; Kaiser, S.; Peters, T.; Perez, A.; et al. Intravenous anti–IL-13 mAb QAX576 for the treatment of eosinophilic esophagitis. J. Allergy Clin. Immunol. 2014, 135, 500–507. [Google Scholar] [CrossRef]
- Dellon, E.S.; Rothenberg, M.E.; Collins, M.H.; Hirano, I.; Chehade, M.; Bredenoord, A.J.; Lucendo, A.J.; Spergel, J.M.; Aceves, S.; Sun, X.; et al. Dupilumab in Adults and Adolescents with Eosinophilic Esophagitis. N. Engl. J. Med. 2022, 387, 2317–2330. [Google Scholar] [CrossRef]
- Higashiyama, M.; Hokari, R. New and Emerging Treatments for Inflammatory Bowel Disease. Digestion 2023, 104, 74–81. [Google Scholar] [CrossRef]
- Aditi, K.; Philip, J.S. Horizon scanning: New and future therapies in the management of inflammatory bowel disease. eGastroenterology 2023, 1, e100012. [Google Scholar]
- Oka, A.; Sartor, R.B. Microbial-Based and Microbial-Targeted Therapies for Inflammatory Bowel Diseases. Dig. Dis. Sci. 2020, 65, 757–788. [Google Scholar] [CrossRef]
- Moayyedi, P.; Surette, M.G.; Kim, P.T.; Libertucci, J.; Wolfe, M.; Onischi, C.; Armstrong, D.; Marshall, J.K.; Kassam, Z.; Reinisch, W.; et al. Fecal Microbiota Transplantation Induces Remission in Patients With Active Ulcerative Colitis in a Randomized Controlled Trial. Gastroenterology 2015, 149, 102–109.e6. [Google Scholar] [CrossRef] [PubMed]
- Paramsothy, S.; Kamm, M.A.; Kaakoush, N.O.; Walsh, A.J.; van den Bogaerde, J.; Samuel, D.; Leong, R.W.L.; Connor, S.; Ng, W.; Paramsothy, R.; et al. Multidonor intensive faecal microbiota transplantation for active ulcerative colitis: A randomised placebo-controlled trial. Lancet 2017, 389, 1218–1228. [Google Scholar] [CrossRef] [PubMed]
- Squier, C.A.; Kremer, M.J. Biology of Oral Mucosa and Esophagus. JNCI Monogr. 2001, 2001, 7–15. [Google Scholar] [CrossRef]
- Kottyan, L.C.; Trimarchi, M.P.; Lu, X.; Caldwell, J.M.; Maddox, A.; Parameswaran, S.; Lape, M.; D’mello, R.J.; Bonfield, M.; Ballaban, A.; et al. Replication and meta-analyses nominate numerous eosinophilic esophagitis risk genes. J. Allergy Clin. Immunol. 2020, 147, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Sherrill, J.D.; Rothenberg, M.E. Genetic dissection of eosinophilic esophagitis provides insight into disease pathogenesis and treatment strategies. J. Allergy Clin. Immunol. 2011, 128, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Rochman, M.; Travers, J.; Miracle, C.E.; Bedard, M.C.; Wen, T.; Azouz, N.P.; Caldwell, J.M.; Kc, K.; Sherrill, J.D.; Davis, B.P.; et al. Profound loss of esophageal tissue differentiation in patients with eosinophilic esophagitis. J. Allergy Clin. Immunol. 2017, 140, 738–749.e3. [Google Scholar] [CrossRef]
- Azouz, N.P.; Ynga-Durand, M.A.; Caldwell, J.M.; Jain, A.; Rochman, M.; Fischesser, D.M.; Ray, L.M.; Bedard, M.C.; Mingler, M.K.; Forney, C.; et al. The antiprotease SPINK7 serves as an inhibitory checkpoint for esophageal epithelial inflammatory responses. Sci. Transl. Med. 2018, 10, eaap9736. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, T.; Oka, T.; Demehri, S. Alarmin Cytokines as Central Regulators of Cutaneous Immunity. Front. Immunol. 2022, 13, 876515. [Google Scholar] [CrossRef]
- Brusilovsky, M.; Rochman, M.; Rochman, Y.; Caldwell, J.M.; Mack, L.E.; Felton, J.M.; Habel, J.E.; Porollo, A.; Pasare, C.; Rothenberg, M.E. Environmental allergens trigger type 2 inflammation through ripoptosome activation. Nat. Immunol. 2021, 22, 1316–1326. [Google Scholar] [CrossRef]
- Steiner, S.J.; Kernek, K.M.; Fitzgerald, J.F. Severity of Basal Cell Hyperplasia Differs in Reflux Versus Eosinophilic Esophagitis. J. Pediatr. Gastroenterol. Nutr. 2006, 42, 506–509. [Google Scholar] [CrossRef] [PubMed]
- Noel, R.J.; E Putnam, P.; Collins, M.H.; Assa’ad, A.H.; Guajardo, J.R.; Jameson, S.C.; E Rothenberg, M. Clinical and immunopathologic effects of swallowed fluticasone for eosinophilic esophagitis. Clin. Gastroenterol. Hepatol. 2004, 2, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Wang, M.; Pemmaraju, V.R.; Collins, M.H.; Fulkerson, P.C.; Abonia, J.P.; Blanchard, C.; Putnam, P.E.; Rothenberg, M.E. Esophageal Remodeling Develops as a Consequence of Tissue Specific IL-5-Induced Eosinophilia. Gastroenterology 2008, 134, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Rochman, M.; Wen, T.; Kotliar, M.; Dexheimer, P.J.; Morgenstern, N.B.-B.; Caldwell, J.M.; Lim, H.-W.; Rothenberg, M.E. Single-cell RNA-Seq of human esophageal epithelium in homeostasis and allergic inflammation. J. Clin. Investig. 2022, 7, e159093. [Google Scholar] [CrossRef] [PubMed]
- Sherrill, J.D.; Kc, K.; Wu, D.; Djukic, Z.; Caldwell, J.M.; Stucke, E.M.; A Kemme, K.; Costello, M.S.; Mingler, M.K.; Blanchard, C.; et al. Desmoglein-1 regulates esophageal epithelial barrier function and immune responses in eosinophilic esophagitis. Mucosal Immunol. 2013, 7, 718–729. [Google Scholar] [CrossRef] [PubMed]
- Shoda, T.; Kaufman, K.M.; Wen, T.; Caldwell, J.M.; Osswald, G.A.; Purnima, P.; Zimmermann, N.; Collins, M.H.; Rehn, K.; Foote, H.; et al. Desmoplakin and periplakin genetically and functionally contribute to eosinophilic esophagitis. Nat. Commun. 2021, 12, 6795. [Google Scholar] [CrossRef] [PubMed]
- Abdulnour-Nakhoul, S.M.; Al-Tawil, Y.; Gyftopoulos, A.A.; Brown, K.L.; Hansen, M.; Butcher, K.F.; Eidelwein, A.P.; Noel, R.A.; Rabon, E.; Posta, A.; et al. Alterations in junctional proteins, inflammatory mediators and extracellular matrix molecules in eosinophilic esophagitis. Clin. Immunol. 2013, 148, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Kurashima, Y.; Kiyono, H. Mucosal Ecological Network of Epithelium and Immune Cells for Gut Homeostasis and Tissue Healing. Annu. Rev. Immunol. 2017, 35, 119–147. [Google Scholar] [CrossRef] [PubMed]
- van der Flier, L.G.; Clevers, H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu. Rev. Physiol. 2009, 71, 241–260. [Google Scholar] [CrossRef]
- Noah, T.K.; Donahue, B.; Shroyer, N.F. Intestinal development and differentiation. Exp. Cell Res. 2011, 317, 2702–2710. [Google Scholar] [CrossRef]
- Vivinus-Nébot, M.; Frin-Mathy, G.; Bzioueche, H.; Dainese, R.; Bernard, G.; Anty, R.; Filippi, J.; Saint-Paul, M.C.; Tulic, M.K.; Verhasselt, V.; et al. Functional bowel symptoms in quiescent inflammatory bowel diseases: Role of epithelial barrier disruption and low-grade inflammation. Gut 2013, 63, 744–752. [Google Scholar] [CrossRef] [PubMed]
- Pizarro, T.T.; Pastorelli, L.; Bamias, G.; Garg, R.R.; Reuter, B.K.; Mercado, J.R.; Chieppa, M.; Arseneau, K.O.; Ley, K.; Cominelli, F. SAMP1/YitFc mouse strain: A spontaneous model of Crohn’s disease-like ileitis. Inflamm. Bowel Dis. 2011, 17, 2566–2584. [Google Scholar] [CrossRef]
- Resta-Lenert, S.; Smitham, J.; Barrett, K.E. Epithelial dysfunction associated with the development of colitis in conventionally housed mdr1a-/- mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G153–G162. [Google Scholar] [CrossRef]
- Pullan, R.D.; A Thomas, G.; Rhodes, M.; Newcombe, R.G.; Williams, G.T.; Allen, A.; Rhodes, J. Thickness of adherent mucus gel on colonic mucosa in humans and its relevance to colitis. Gut 1994, 35, 353–359. [Google Scholar] [CrossRef]
- Van der Sluis, M.; De Koning, B.A.; De Bruijn, A.C.; Velcich, A.; Meijerink, J.P.; Van Goudoever, J.B.; Büller, H.A.; Dekker, J.; Van Seuningen, I.; Renes, I.B.; et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology 2006, 131, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.C.; Lee, J.C.; Lees, C.W.; Prescott, N.J.; Anderson, C.A.; Phillips, A.; Wesley, E.; Parnell, K.; Zhang, H.; Drummond, H.; et al. Genome-wide association study of ulcerative colitis identifies three new susceptibility loci, including the HNF4A region. Nat. Genet. 2009, 41, 1330–1334. [Google Scholar] [PubMed]
- Khounlotham, M.; Kim, W.; Peatman, E.; Nava, P.; Medina-Contreras, O.; Addis, C.; Koch, S.; Fournier, B.; Nusrat, A.; Denning, T.L.; et al. Compromised Intestinal Epithelial Barrier Induces Adaptive Immune Compensation that Protects from Colitis. Immunity 2012, 37, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Chaturvedi, R.; Olivares-Villagómez, D.; Habib, T.; Asim, M.; Shivesh, P.; Polk, D.B.; Wilson, K.T.; Washington, M.K.; Van Kaer, L.; et al. Targeted colonic claudin-2 expression renders resistance to epithelial injury, induces immune suppression, and protects from colitis. Mucosal Immunol. 2014, 7, 1340–1353. [Google Scholar] [CrossRef]
- Zeissig, S.; Bürgel, N.; Günzel, D.; Richter, J.; Mankertz, J.; Wahnschaffe, U.; Kroesen, A.J.; Zeitz, M.; Fromm, M.; Schulzke, J.D. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut 2007, 56, 61–72. [Google Scholar] [CrossRef]
- Oshima, T.; Miwa, H.; Joh, T. Changes in the expression of claudins in active ulcerative colitis. J. Gastroenterol. Hepatol. 2008, 23 (Suppl. S2), S146–S150. [Google Scholar] [CrossRef]
- Van Itallie, C.M.; Fanning, A.S.; Holmes, J.; Anderson, J.M. Occludin is required for cytokine-induced regulation of tight junction barriers. J. Cell Sci. 2010, 123 Pt 16, 2844–2852. [Google Scholar] [CrossRef] [PubMed]
- Kagalwalla, A.F.; Akhtar, N.; Woodruff, S.A.; Rea, B.A.; Masterson, J.C.; Mukkada, V.; Parashette, K.R.; Du, J.; Fillon, S.; Protheroe, C.A.; et al. Eosinophilic esophagitis: Epithelial mesenchymal transition contributes to esophageal remodeling and reverses with treatment. J. Allergy Clin. Immunol. 2012, 129, 1387–1396.e7. [Google Scholar] [CrossRef]
- Muir, A.B.; Lim, D.M.; Benitez, A.J.; Chandramouleeswaran, P.M.; Lee, A.J.; Ruchelli, E.D.; Spergel, J.M.; Wang, M.-L. Esophageal epithelial and mesenchymal cross-talk leads to features of epithelial to mesenchymal transition in vitro. Exp. Cell Res. 2012, 319, 850–859. [Google Scholar] [CrossRef] [PubMed]
- Aceves, S.S.; Chen, D.; Newbury, R.O.; Dohil, R.; Bastian, J.F.; Broide, D.H. Mast cells infiltrate the esophageal smooth muscle in patients with eosinophilic esophagitis, express TGF-β1, and increase esophageal smooth muscle contraction. J. Allergy Clin. Immunol. 2010, 126, 1198–1204.e4. [Google Scholar] [CrossRef] [PubMed]
- Aceves, S.S.; Ackerman, S.J. Relationships Between Eosinophilic Inflammation, Tissue Remodeling, and Fibrosis in Eosinophilic Esophagitis. Immunol. Allergy Clin. N. Am. 2009, 29, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.; Fernando, S.D.; A Biette, K.; A Hammer, J.; E Capocelli, K.; A Kitzenberg, D.; E Glover, L.; Colgan, S.P.; Furuta, G.T.; Masterson, J.C. TGF-β1 alters esophageal epithelial barrier function by attenuation of claudin-7 in eosinophilic esophagitis. Mucosal Immunol. 2018, 11, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Laky, K.; Kinard, J.L.; Li, J.M.; Moore, I.N.; Lack, J.; Fischer, E.R.; Kabat, J.; Latanich, R.; Zachos, N.C.; Limkar, A.R.; et al. Epithelial-intrinsic defects in TGFβR signaling drive local allergic inflammation manifesting as eosinophilic esophagitis. Sci. Immunol. 2023, 8, eabp9940. [Google Scholar] [CrossRef]
- Shoda, T.; Wen, T.; Caldwell, J.M.; Morgenstern, N.B.-B.; Osswald, G.A.; Rochman, M.; Mack, L.E.; Felton, J.M.; Abonia, J.P.; Arva, N.C.; et al. Loss of Endothelial TSPAN12 Promotes Fibrostenotic Eosinophilic Esophagitis via Endothelial Cell–Fibroblast Crosstalk. Gastroenterology 2021, 162, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, L.Y.; Chiang, A.W.; Duong, L.D.; Kuo, C.-C.; Dong, S.X.; Dohil, R.; Kurten, R.; Lewis, N.E.; Aceves, S.S. A unique esophageal extracellular matrix proteome alters normal fibroblast function in severe eosinophilic esophagitis. J. Allergy Clin. Immunol. 2021, 148, 486–494. [Google Scholar] [CrossRef]
- Caldwell, J.; Collins, M.; Kemme, K.; Sherrill, J.; Wen, T.; Rochman, M.; Stucke, E.; Amin, L.; Tai, H.; Putnam, P.; et al. Cadherin 26 is an alpha integrin-binding epithelial receptor regulated during allergic inflammation. Mucosal Immunol. 2017, 10, 1190–1201. [Google Scholar] [CrossRef]
- Blanchard, C.; Mingler, M.K.; Vicario, M.; Abonia, J.P.; Wu, Y.Y.; Lu, T.X.; Collins, M.H.; Putnam, P.E.; Wells, S.I.; Rothenberg, M.E. IL-13 involvement in eosinophilic esophagitis: Transcriptome analysis and reversibility with glucocorticoids. J. Allergy Clin. Immunol. 2007, 120, 1292–1300. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, J.M.; Blanchard, C.; Collins, M.H.; Putnam, P.E.; Kaul, A.; Aceves, S.S.; Bouska, C.A.; Rothenberg, M.E. Glucocorticoid-regulated genes in eosinophilic esophagitis: A role for FKBP51. J. Allergy Clin. Immunol. 2010, 125, 879–888.e8. [Google Scholar] [CrossRef] [PubMed]
- Sa, S.M.; Valdez, P.A.; Wu, J.; Jung, K.; Zhong, F.; Hall, L.; Kasman, I.; Winer, J.; Modrusan, Z.; Danilenko, D.M.; et al. The Effects of IL-20 Subfamily Cytokines on Reconstituted Human Epidermis Suggest Potential Roles in Cutaneous Innate Defense and Pathogenic Adaptive Immunity in Psoriasis. J. Immunol. 2007, 178, 2229–2240. [Google Scholar] [CrossRef] [PubMed]
- Kaymak, T.; Kaya, B.; Wuggenig, P.; Nuciforo, S.; Göldi, A.; Oswald, F.; Roux, J.; Noti, M.; Melhem, H.; Hruz, P.; et al. IL-20 subfamily cytokines impair the oesophageal epithelial barrier by diminishing filaggrin in eosinophilic oesophagitis. Gut 2022, 72, 821–833. [Google Scholar] [CrossRef] [PubMed]
- Madsen, K.; Lewis, S.; Tavernini, M.; Hibbard, J.; Fedorak, R.N. Interleukin 10 prevents cytokine-induced disruption of T84 monolayer barrier integrity and limits chloride secretion. Gastroenterology 1997, 113, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Kominsky, D.J.; Campbell, E.L.; Ehrentraut, S.F.; Wilson, K.E.; Kelly, C.J.; Glover, L.E.; Collins, C.B.; Bayless, A.J.; Saeedi, B.; Dobrinskikh, E.; et al. IFN-γ–Mediated Induction of an Apical IL-10 Receptor on Polarized Intestinal Epithelia. J. Immunol. 2014, 192, 1267–1276. [Google Scholar] [CrossRef] [PubMed]
- Lorén, V.; Cabré, E.; Ojanguren, I.; Domènech, E.; Pedrosa, E.; García-Jaraquemada, A.; Mañosa, M.; Manyé, J. Interleukin-10 Enhances the Intestinal Epithelial Barrier in the Presence of Corticosteroids through p38 MAPK Activity in Caco-2 Monolayers: A Possible Mechanism for Steroid Responsiveness in Ulcerative Colitis. PLoS ONE 2015, 10, e0130921. [Google Scholar] [CrossRef] [PubMed]
- Madsen, K.L.; Malfair, D.; Gray, D.; Doyle, J.S.; Jewell, L.D.; Fedorak, R.N. Interleukin-10 gene-deficient mice develop a primary intestinal permeability defect in response to enteric microflora. Inflamm. Bowel Dis. 1999, 5, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.R.; Raleigh, D.R.; Su, L.; Shen, L.; Sullivan, E.A.; Wang, Y.; Turner, J.R. Epithelial Myosin Light Chain Kinase Activation Induces Mucosal Interleukin-13 Expression to Alter Tight Junction Ion Selectivity. J. Biol. Chem. 2010, 285, 12037–12046. [Google Scholar] [CrossRef]
- Madden, K.B.; Whitman, L.; Sullivan, C.; Gause, W.C.; Urban, J.F.; Katona, I.M.; Finkelman, F.D.; Shea-Donohue, T. Role of STAT6 and Mast Cells in IL-4- and IL-13-Induced Alterations in Murine Intestinal Epithelial Cell Function. J. Immunol. 2002, 169, 4417–4422. [Google Scholar] [CrossRef]
- Rawat, M.; Nighot, M.; Al-Sadi, R.; Gupta, Y.; Viszwapriya, D.; Yochum, G.; Koltun, W.; Ma, T.Y. IL1B Increases Intestinal Tight Junction Permeability by Up-regulation of MIR200C-3p, Which Degrades Occludin mRNA. Gastroenterology 2020, 159, 1375–1389. [Google Scholar] [CrossRef] [PubMed]
- Clayburgh, D.R.; Musch, M.W.; Leitges, M.; Fu, Y.-X.; Turner, J.R. Coordinated epithelial NHE3 inhibition and barrier dysfunction are required for TNF-mediated diarrhea in vivo. J. Clin. Investig. 2006, 116, 2682–2694. [Google Scholar] [CrossRef] [PubMed]
- Colpaert, S.; Liu, Z.; De Greef, B.; Rutgeerts, P.; Ceuppens, J.L.; Geboes, K. Effects of anti-tumour necrosis factor, interleukin-10 and antibiotic therapy in the indometacin-induced bowel inflammation rat model. Aliment. Pharmacol. Ther. 2001, 15, 1827–1836. [Google Scholar] [CrossRef] [PubMed]
- Suenaert, P.; Bulteel, V.; Lemmens, L.; Noman, M.; Geypens, B.; Van Assche, G.; Geboes, K.; Ceuppens, J.L.; Rutgeerts, P. Anti-tumor necrosis factor treatment restores the gut barrier in Crohn’s disease. Am. J. Gastroenterol. 2002, 97, 2000–2004. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, V.; Ebel, J.-F.; Phuong, N.N.T.; Klopfleisch, R.; Vu, V.P.; Adamczyk, A.; Zöller, J.; Riedel, C.; Buer, J.; Krebs, P.; et al. Interleukin-33 signaling exacerbates experimental infectious colitis by enhancing gut permeability and inhibiting protective Th17 immunity. Mucosal Immunol. 2021, 14, 923–936. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Tato, C.M.; Joyce-Shaikh, B.; Gulen, M.F.; Cayatte, C.; Chen, Y.; Blumenschein, W.M.; Judo, M.; Ayanoglu, G.; McClanahan, T.K.; et al. Interleukin-23-Independent IL-17 Production Regulates Intestinal Epithelial Permeability. Immunity 2015, 43, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Donohoe, D.; Fredrich, K.; Gourlay, D.M.; Liedel, J.L.; Heinzerling, N.P. Interleukin-23 Increases Intestinal Epithelial Cell Permeability In Vitro. Eur. J. Pediatr. Surg. 2015, 26, 260–266. [Google Scholar] [CrossRef] [PubMed]
- O’shea, K.M.; Rochman, M.; Shoda, T.; Zimmermann, N.; Caldwell, J.; Rothenberg, M.E. Eosinophilic esophagitis with extremely high esophageal eosinophil counts. J. Allergy Clin. Immunol. 2021, 147, 409–412.e5. [Google Scholar] [CrossRef]
- Straumann, A.; Conus, S.; Grzonka, P.; Kita, H.; Kephart, G.; Bussmann, C.; Beglinger, C.; A Smith, D.; Patel, J.; Byrne, M.; et al. Anti-interleukin-5 antibody treatment (mepolizumab) in active eosinophilic oesophagitis: A randomised, placebo-controlled, double-blind trial. Gut 2009, 59, 21–30. [Google Scholar] [CrossRef]
- Spergel, J.M.; Rothenberg, M.E.; Collins, M.H.; Furuta, G.T.; Markowitz, J.E.; Fuchs, G.; O’gorman, M.A.; Abonia, J.P.; Young, J.; Henkel, T.; et al. Reslizumab in children and adolescents with eosinophilic esophagitis: Results of a double-blind, randomized, placebo-controlled trial. J. Allergy Clin. Immunol. 2011, 129, 456–463.e3. [Google Scholar] [CrossRef]
- Mueller, S.; Aigner, T.; Neureiter, D.; Stolte, M. Eosinophil infiltration and degranulation in oesophageal mucosa from adult patients with eosinophilic oesophagitis: A retrospective and comparative study on pathological biopsy. J. Clin. Pathol. 2006, 59, 1175–1180. [Google Scholar] [CrossRef] [PubMed]
- Aceves, S.S.; Newbury, R.O.; Dohil, R.; Bastian, J.F.; Broide, D.H. Esophageal remodeling in pediatric eosinophilic esophagitis. J. Allergy Clin. Immunol. 2007, 119, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Straumann, A.; Kristl, J.; Conus, S.; Vassina, E.; Spichtin, H.-P.; Beglinger, C.; Simon, H.-U. Cytokine Expression in Healthy and Inflamed Mucosa: Probing the Role of Eosinophils in the Digestive Tract. Inflamm. Bowel Dis. 2005, 11, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Coppi, L.C.; Thomazzi, S.M.; de Ayrizono, M.L.; Coy, C.S.; Fagundes, W.J.; Goes, J.R.; Franchi, G.C.; Nowill, A.E.; Montes, C.G.; Antunes, E.; et al. Comparative study of eosinophil chemotaxis, adhesion, and degranulation in vitro in ulcerative colitis and Crohn’s disease. Inflamm. Bowel Dis. 2007, 13, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Canavese, G.; Villanacci, V.; Antonelli, E.; Cadei, M.; Sapino, A.; Rocca, R.; Daperno, M.; Suriani, R.; Di Santo, M.G.; Cassoni, P.; et al. Eosinophilia—Associated basal plasmacytosis: An early and sensitive histologic feature of inflammatory bowel disease. APMIS 2017, 125, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, I.; Ceulemans, M.; Wauters, L.; Breynaert, C.; Vermeire, S.; Verstockt, B.; Vanuytsel, T. Role of Eosinophils in Intestinal Inflammation and Fibrosis in Inflammatory Bowel Disease: An Overlooked Villain? Front. Immunol. 2021, 12, 754413. [Google Scholar] [CrossRef]
- Lucendo, A.J.; Navarro, M.; Comas, C.; Pascual, J.M.; Burgos, E.; Santamaría, L.; Larrauri, J. Immunophenotypic characterization and quantification of the epithelial inflammatory infiltrate in eosinophilic esophagitis through stereology: An analysis of the cellular mechanisms of the disease and the immunologic capacity of the esophagus. Am. J. Surg. Pathol. 2007, 31, 598–606. [Google Scholar] [CrossRef]
- Mishra, A.; Schlotman, J.; Wang, M.; E Rothenberg, M. Critical role for adaptive T cell immunity in experimental eosinophilic esophagitis in mice. J. Leukoc. Biol. 2006, 81, 916–924. [Google Scholar] [CrossRef]
- Mitson-Salazar, A.; Yin, Y.; Wansley, D.L.; Young, M.; Bolan, H.; Arceo, S.; Ho, N.; Koh, C.; Milner, J.D.; Stone, K.D.; et al. Hematopoietic prostaglandin D synthase defines a proeosinophilic pathogenic effector human T(H)2 cell subpopulation with enhanced function. J. Allergy Clin. Immunol. 2016, 137, 907–918.e9. [Google Scholar] [CrossRef]
- Cianferoni, A.; Ruffner, M.A.; Guzek, R.; Guan, S.; Brown-Whitehorn, T.; Muir, A.; Spergel, J.M. Elevated expression of activated T H 2 cells and milk-specific T H 2 cells in milk-induced eosinophilic esophagitis. Ann. Allergy Asthma Immunol. 2017, 120, 177–183.e2. [Google Scholar] [CrossRef]
- Stuck, M.C.; Straumann, A.; Simon, H.-U. Relative lack of T regulatory cells in adult eosinophilic esophagitis—No normalization after corticosteroid therapy. Allergy 2010, 66, 705–707. [Google Scholar] [CrossRef] [PubMed]
- Tantibhaedhyangkul, U.; Tatevian, N.; A Gilger, M.; Major, A.M.; Davis, C.M. Increased esophageal regulatory T cells and eosinophil characteristics in children with eosinophilic esophagitis and gastroesophageal reflux disease. Ann. Clin. Lab. Sci. 2009, 39, 99–107. [Google Scholar]
- Morgenstern, N.B.-B.; Ballaban, A.Y.; Wen, T.; Shoda, T.; Caldwell, J.M.; Kliewer, K.; Felton, J.M.; Abonia, J.P.; Mukkada, V.A.; Putnam, P.E.; et al. Single-cell RNA sequencing of mast cells in eosinophilic esophagitis reveals heterogeneity, local proliferation, and activation that persists in remission. J. Allergy Clin. Immunol. 2022, 149, 2062–2077. [Google Scholar] [CrossRef]
- Komi, D.E.A.; Wöhrl, S.; Bielory, L. Mast Cell Biology at Molecular Level: A Comprehensive Review. Clin. Rev. Allergy Immunol. 2019, 58, 342–365. [Google Scholar] [CrossRef]
- Chehade, M.; A Sampson, H.; A Morotti, R.; Magid, M.S. Esophageal Subepithelial Fibrosis in Children with Eosinophilic Esophagitis. J. Pediatr. Gastroenterol. Nutr. 2007, 45, 319–328. [Google Scholar] [CrossRef]
- Hamilton, M.J.; Frei, S.M.; Stevens, R.L. The Multifaceted Mast Cell in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2014, 20, 2364–2378. [Google Scholar] [CrossRef] [PubMed]
- Groschwitz, K.R.; Ahrens, R.; Osterfeld, H.; Gurish, M.F.; Han, X.; Åbrink, M.; Finkelman, F.D.; Pejler, G.; Hogan, S.P. Mast cells regulate homeostatic intestinal epithelial migration and barrier function by a chymase/Mcpt4-dependent mechanism. Proc. Natl. Acad. Sci. USA 2009, 106, 22381–22386. [Google Scholar] [CrossRef] [PubMed]
- Vicario, M.; Blanchard, C.; Stringer, K.F.; Collins, M.H.; Mingler, M.K.; Ahrens, A.; E Putnam, P.; Abonia, J.P.; Santos, J.; E Rothenberg, M. Local B cells and IgE production in the oesophageal mucosa in eosinophilic oesophagitis. Gut 2009, 59, 12–20. [Google Scholar] [CrossRef]
- Leiper, K.; Martin, K.; Ellis, A.; Subramanian, S.; Watson, A.J.; Christmas, S.E.; Howarth, D.; Campbell, F.; Rhodes, J.M. Randomised placebo-controlled trial of rituximab (anti-CD20) in active ulcerative colitis. Gut 2011, 60, 1520–1526. [Google Scholar] [CrossRef]
- Martin, J.C.; Chang, C.; Boschetti, G.; Ungaro, R.; Giri, M.; Grout, J.A.; Gettler, K.; Chuang, L.-S.; Nayar, S.; Greenstein, A.J.; et al. Single-Cell Analysis of Crohn’s Disease Lesions Identifies a Pathogenic Cellular Module Associated with Resistance to Anti-TNF Therapy. Cell 2019, 178, 1493–1508.e20. [Google Scholar] [CrossRef]
- Castro-Dopico, T.; Dennison, T.; Ferdinand, J.; Mathews, R.; Fleming, A.; Clift, D.; Stewart, B.J.; Jing, C.; Strongili, K.; I Labzin, L.; et al. Anti-commensal IgG Drives Intestinal Inflammation and Type 17 Immunity in Ulcerative Colitis. Immunity 2019, 50, 1099–1114.e10. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhao, Y.; Kong, P.; Liu, Y.; Huang, J.; Xu, E.; Wei, W.; Li, G.; Cheng, X.; Xue, L.; et al. Integrated multi-omics profiling yields a clinically relevant molecular classification for esophageal squamous cell carcinoma. Cancer Cell 2023, 41, 181–195.e9. [Google Scholar] [CrossRef] [PubMed]
- Stagg, A.J. Intestinal Dendritic Cells in Health and Gut Inflammation. Front. Immunol. 2018, 9, 2883. [Google Scholar] [CrossRef]
- Hegarty, L.M.; Jones, G.-R.; Bain, C.C. Macrophages in intestinal homeostasis and inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 538–553. [Google Scholar] [CrossRef] [PubMed]
- Na, Y.R.; Stakenborg, M.; Seok, S.H.; Matteoli, G. Macrophages in intestinal inflammation and resolution: A potential therapeutic target in IBD. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 531–543. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Aspect | EoE | IBD |
|---|---|---|
| Main Treatment Focus | Reduce esophageal inflammation to alleviate symptoms and prevent complications such as strictures | Induce and maintain remission to alleviate symptoms, enhance quality of life, and prevent complications |
| Dietary Modifications | One- or six food elimination diet (identifying and eliminating specific food triggers) | Not commonly used as primary treatment |
| Medications | PPIs to suppress gastric acid and reduce inflammation; swallowed topical corticosteroids for symptomatic relief and mucosal healing; the IL-4Ra antibody dupilumab targeting IL-13 and IL-4/IL-13 | 5-ASAs for mild-to-moderate UC; corticosteroids for acute flares; immunomodulators (azathioprine, 6-mercaptopurine, methotrexate) for maintenance therapy; biologics targeting TNF-α, interleukins, integrins (e.g., anti-TNF agents, anti-integrins, anti-IL-12/23 or anti-IL-23 agents); JAK inhibitors |
| FMT | Not applicable | Restoring microbiome diversity |
| Surgical Interventions | Reserved for complications, such as perforations | Bowel resection or ostomy for complications like strictures, perforations, or refractory disease unresponsive to medical therapy |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Melhem, H.; Niess, J.H. Eosinophilic Esophagitis and Inflammatory Bowel Disease: What Are the Differences? Int. J. Mol. Sci. 2024, 25, 8534. https://doi.org/10.3390/ijms25158534
Melhem H, Niess JH. Eosinophilic Esophagitis and Inflammatory Bowel Disease: What Are the Differences? International Journal of Molecular Sciences. 2024; 25(15):8534. https://doi.org/10.3390/ijms25158534
Chicago/Turabian StyleMelhem, Hassan, and Jan Hendrik Niess. 2024. "Eosinophilic Esophagitis and Inflammatory Bowel Disease: What Are the Differences?" International Journal of Molecular Sciences 25, no. 15: 8534. https://doi.org/10.3390/ijms25158534