
CRKL Enhances YAP Signaling through Binding and JNK/JUN Pathway Activation in Liver Cancer
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Establishment of Biotin-Ligase Tagged YAP and TAZ
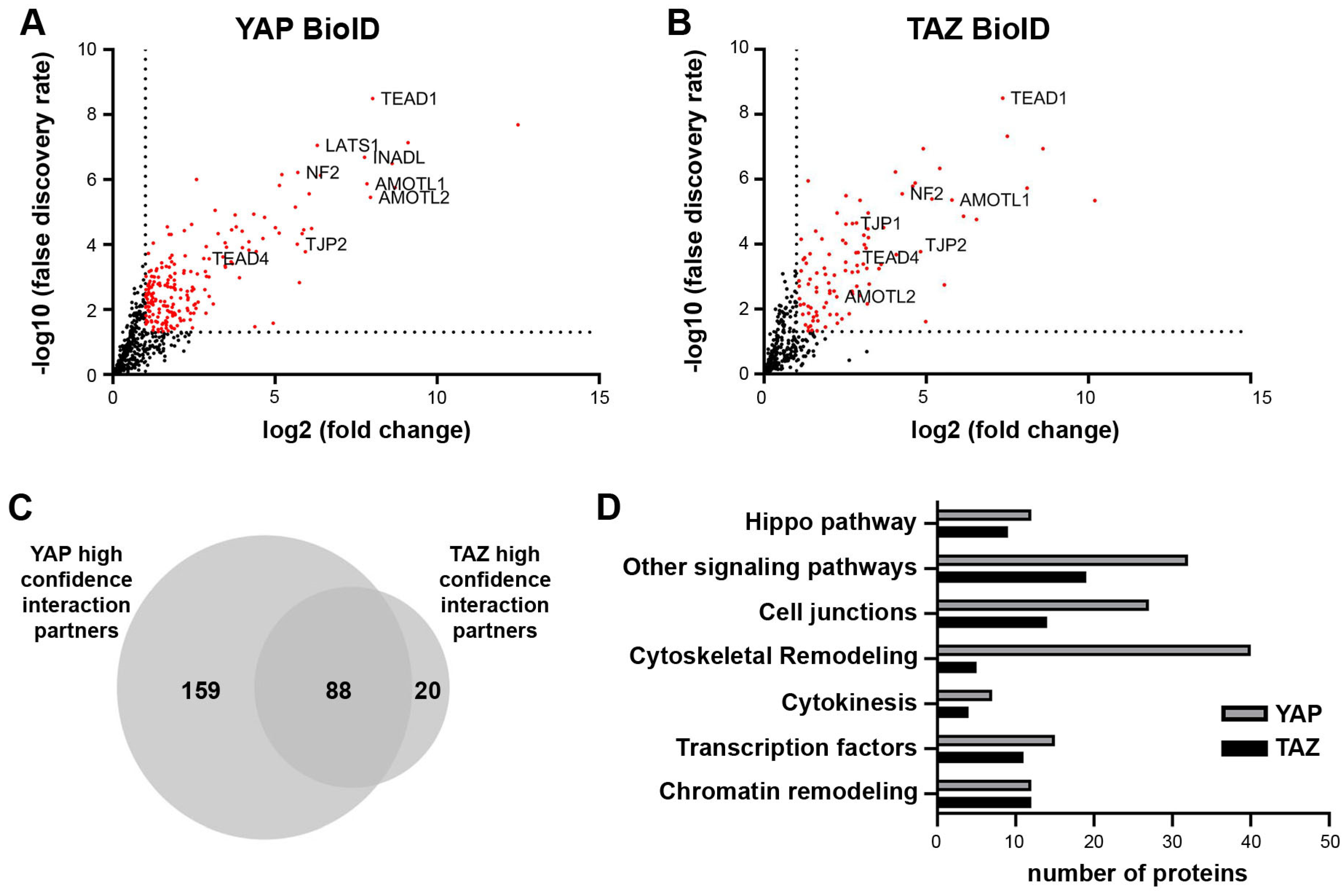
2.2. BioID Identifies Functionally Relevant Interaction Partners of YAP/TAZ in Cancer Cells
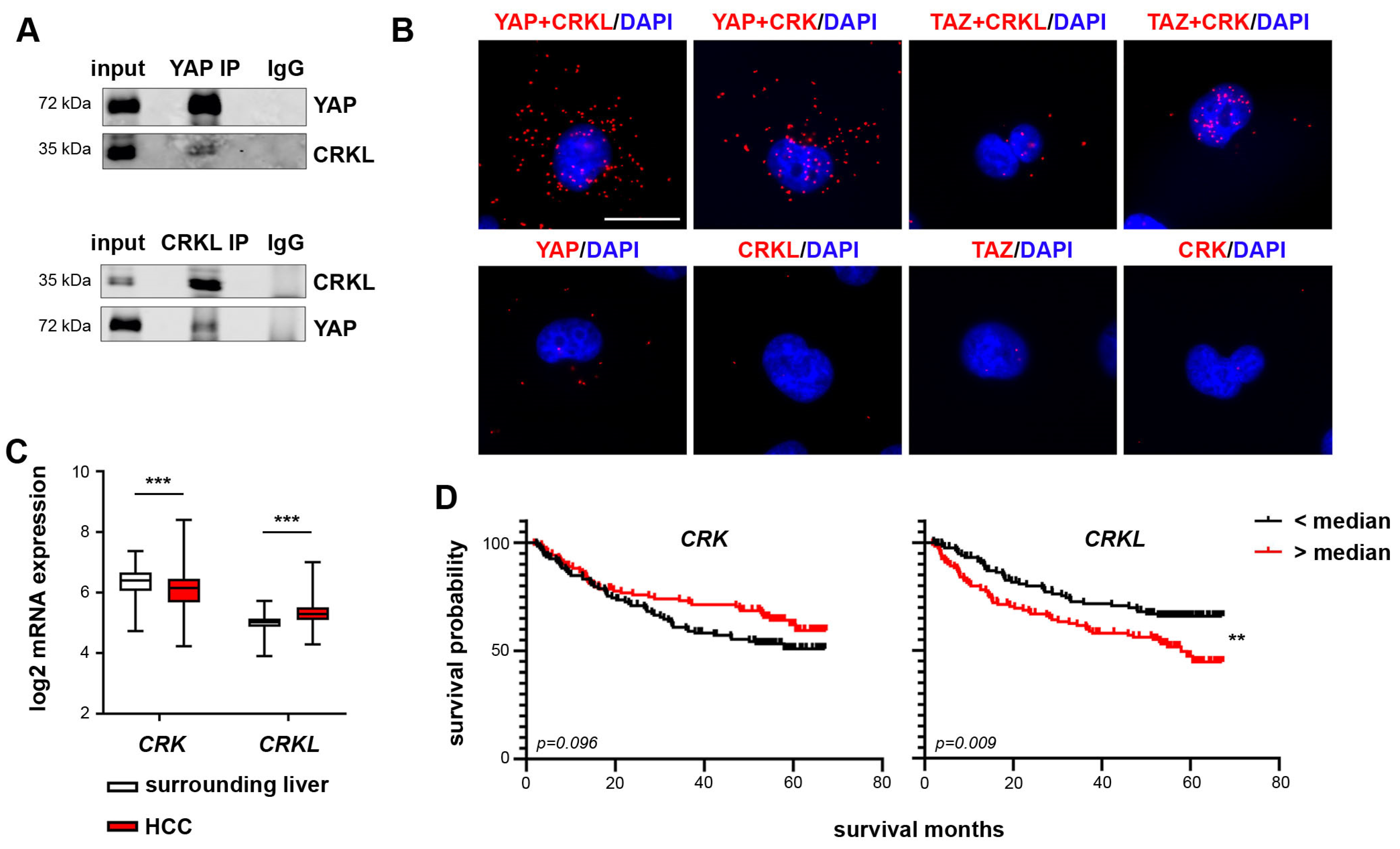
2.3. CRKL Is a YAP Specific Interaction Partner
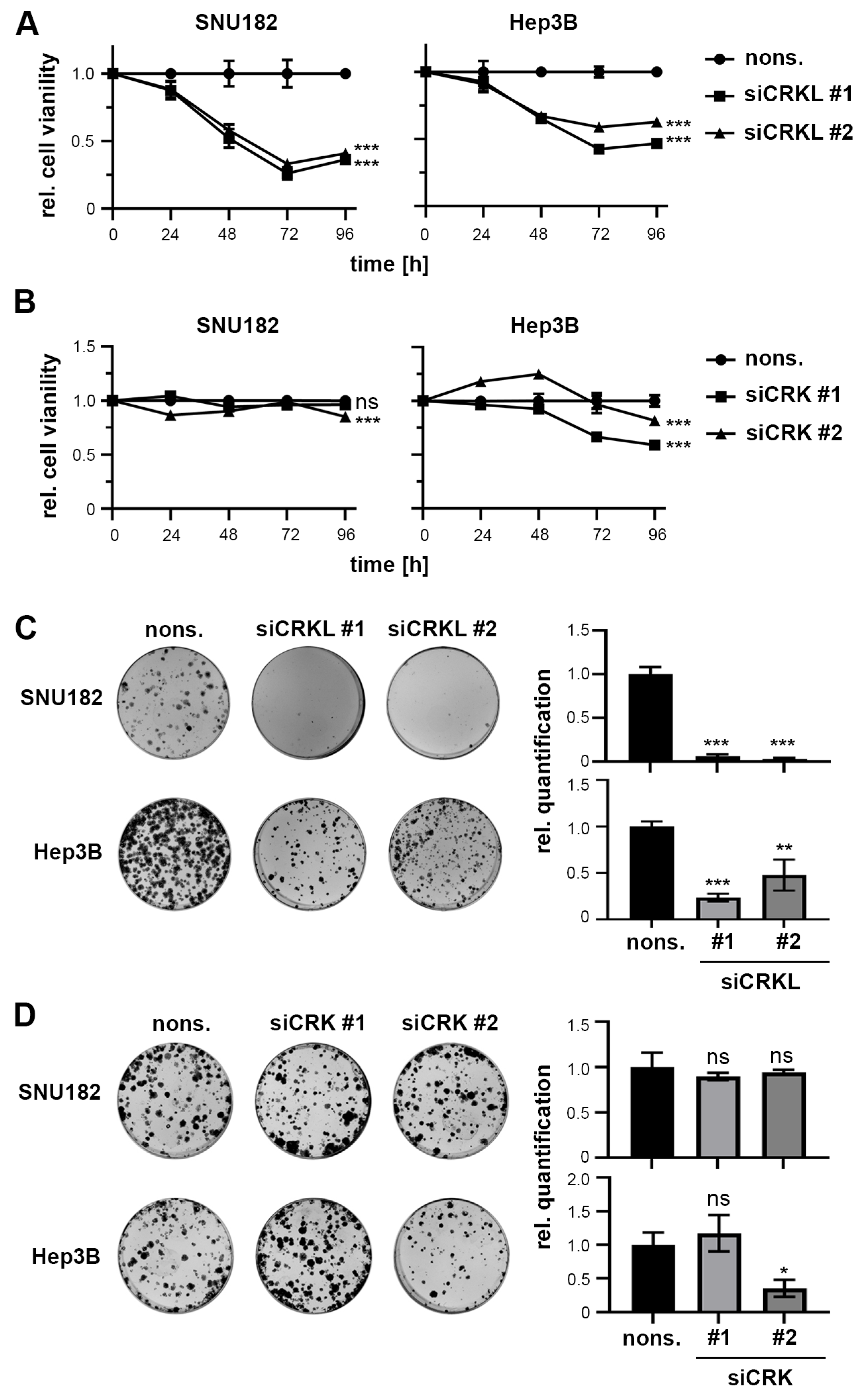
2.4. CRKL but Not CRK Influences HCC Cell Viability and Colony Formation
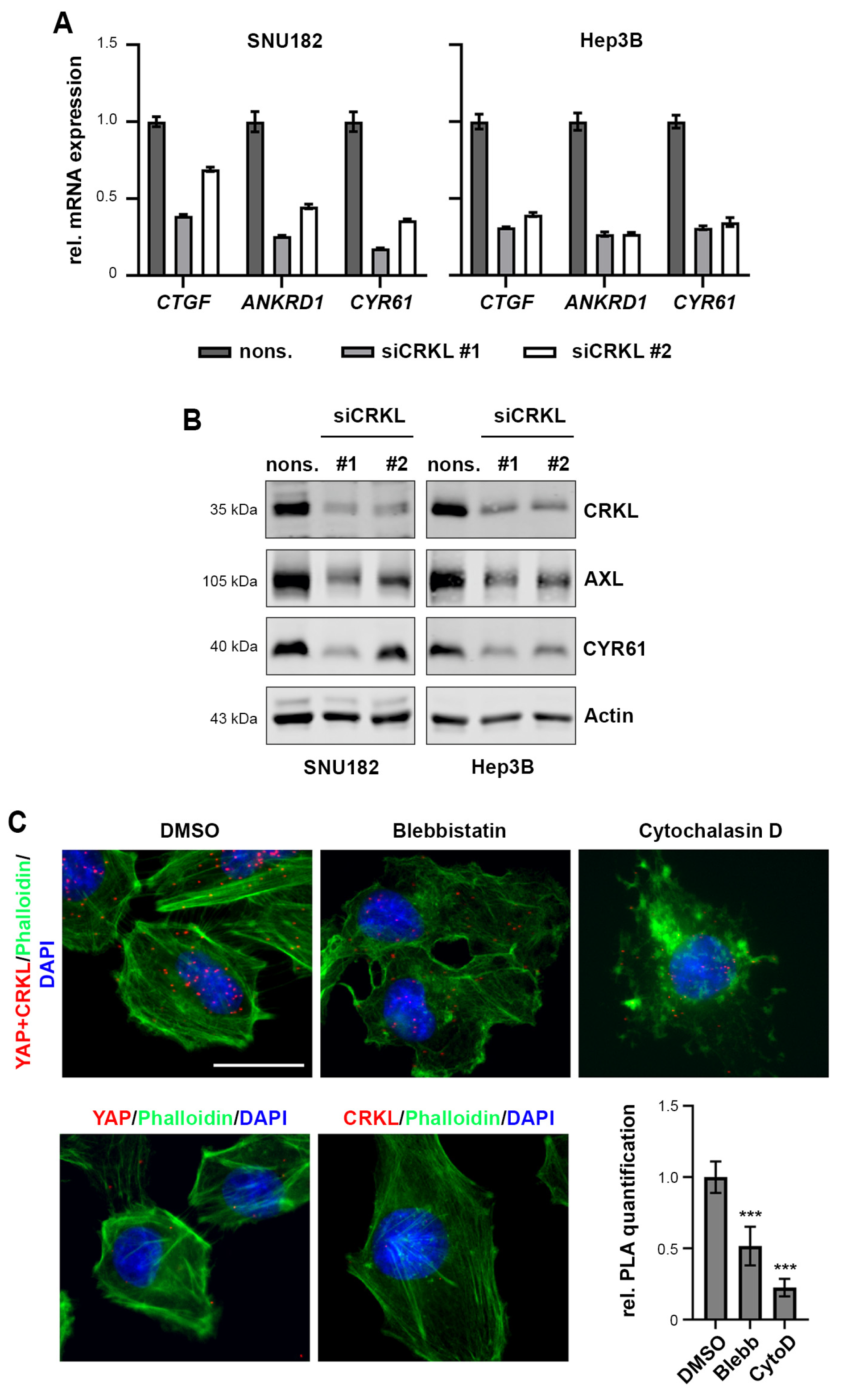
2.5. CRKL Activates YAP Signaling
2.6. CRKL Activates JNK/JUN Signaling to Induce YAP mRNA Expression
2.7. CRKL Expression Correlates with Active YAP in HCC Patients
3. Discussion
4. Materials and Methods
4.1. Cultivation of Cells, siRNA Transfections and Inhibitor Treatments
4.2. Gateway Cloning
4.3. Lentiviral Transduction
4.4. RNA Isolation, cDNA Synthesis and Real-Time PCR
4.5. Protein Isolation, Nuclear/Cytoplasmic Fractionation and Western Immunoblotting
4.6. Biotin Treatment and BioID Pulldown
4.7. Tryptic In-Gel Digest and LC-MS Analysis
4.8. Co-Immunoprecipitation (CoIP)
4.9. Proximity Ligation Assay (PLA)
4.10. Cell Viability and Cytotoxicity Assays, Colony Formation Assays
4.11. Immunohistochemistry and Tissue Microarray Analysis
4.12. Chromatin Immunoprecipitation (ChIP)
4.13. Statistical, Correlation, Expression and Survival Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef]
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Weiler, S.M.E.; Pinna, F.; Wolf, T.; Lutz, T.; Geldiyev, A.; Sticht, C.; Knaub, M.; Thomann, S.; Bissinger, M.; Wan, S.; et al. Induction of Chromosome Instability by Activation of Yes-Associated Protein and Forkhead Box M1 in Liver Cancer. Gastroenterology 2017, 152, 2037–2051.e22. [Google Scholar] [CrossRef]
- Wei, T.; Weiler, S.M.E.; Tóth, M.; Sticht, C.; Lutz, T.; Thomann, S.; de La Torre, C.; Straub, B.; Merker, S.; Ruppert, T.; et al. YAP-dependent induction of UHMK1 supports nuclear enrichment of the oncogene MYBL2 and proliferation in liver cancer cells. Oncogene 2019, 38, 5541–5550. [Google Scholar] [CrossRef] [PubMed]
- Weiler, S.M.E.; Lutz, T.; Bissinger, M.; Sticht, C.; Knaub, M.; Gretz, N.; Schirmacher, P.; Breuhahn, K. TAZ target gene ITGAV regulates invasion and feeds back positively on YAP and TAZ in liver cancer cells. Cancer Lett. 2020, 473, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Feldmann, G.; Huang, J.; Wu, S.; Zhang, N.; Comerford, S.A.; Gayyed, M.F.; Anders, R.A.; Maitra, A.; Pan, D. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 2007, 130, 1120–1133. [Google Scholar] [CrossRef]
- Camargo, F.D.; Gokhale, S.; Johnnidis, J.B.; Fu, D.; Bell, G.W.; Jaenisch, R.; Brummelkamp, T.R. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr. Biol. 2007, 17, 2054–2060. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zheng, Z.; Caviglia, J.M.; Corey, K.E.; Herfel, T.M.; Cai, B.; Masia, R.; Chung, R.T.; Lefkowitch, J.H.; Schwabe, R.F.; et al. Hepatocyte TAZ/WWTR1 Promotes Inflammation and Fibrosis in Nonalcoholic Steatohepatitis. Cell Metab. 2016, 24, 848–862. [Google Scholar] [CrossRef]
- Tschaharganeh, D.F.; Chen, X.; Latzko, P.; Malz, M.; Gaida, M.M.; Felix, K.; Ladu, S.; Singer, S.; Pinna, F.; Gretz, N.; et al. Yes-associated protein up-regulates Jagged-1 and activates the Notch pathway in human hepatocellular carcinoma. Gastroenterology 2013, 144, 1530–1542.e12. [Google Scholar] [CrossRef]
- Lin, C.; Hu, Z.; Lei, B.; Tang, B.; Yu, H.; Qiu, X.; He, S. Overexpression of Yes-associated protein and its association with clinicopathological features of hepatocellular carcinoma: A meta-analysis: A meta-analysis. Liver Int. 2017, 37, 1675–1681. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Jiang, N.; Zhou, B.; Liu, Q.; Du, C. TAZ regulates cell proliferation and epithelial-mesenchymal transition of human hepatocellular carcinoma. Cancer Sci. 2015, 106, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.T.; Konradi, A.W.; Feng, Y.; Peng, X.; Ma, M.; Li, J.; Yu, F.-X.; Guan, K.-L.; Post, L. Small Molecule Inhibitors of TEAD Auto-palmitoylation Selectively Inhibit Proliferation and Tumor Growth of NF2-deficient Mesothelioma. Mol. Cancer Ther. 2021, 20, 986–998. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, T.; Peterson, C.; Schneider, R.; Garg, S.; Schwarz, D.; Gunera, J.; Seshire, A.; Kötzner, L.; Schlesiger, S.; Musil, D.; et al. Optimization of TEAD P-Site Binding Fragment Hit into In Vivo Active Lead MSC-4106. J. Med. Chem. 2022, 65, 9206–9229. [Google Scholar] [CrossRef] [PubMed]
- Pocaterra, A.; Romani, P.; Dupont, S. YAP/TAZ functions and their regulation at a glance. J. Cell Sci. 2020, 133, jcs230425. [Google Scholar] [CrossRef] [PubMed]
- Weiler, S.M.E.; Bissinger, M.; Rose, F.; von Bubnoff, F.; Lutz, T.; Ori, A.; Schirmacher, P.; Breuhahn, K. SEPTIN10-mediated crosstalk between cytoskeletal networks controls mechanotransduction and oncogenic YAP/TAZ signaling. Cancer Lett. 2024, 584, 216637. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.H.; Camargo, F.D.; Yimlamai, D. Hippo Signaling in the Liver Regulates Organ Size, Cell Fate, and Carcinogenesis. Gastroenterology 2017, 152, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Fischer, R.S.; Pan, D.; Waterman, C.M. YAP Nuclear Localization in the Absence of Cell-Cell Contact Is Mediated by a Filamentous Actin-dependent, Myosin II- and Phospho-YAP-independent Pathway during Extracellular Matrix Mechanosensing. J. Biol. Chem. 2016, 291, 6096–6110. [Google Scholar] [CrossRef]
- Seo, J.; Kim, J. Regulation of Hippo signaling by actin remodeling. BMB Rep. 2018, 51, 151–156. [Google Scholar] [CrossRef]
- Aragona, M.; Panciera, T.; Manfrin, A.; Giulitti, S.; Michielin, F.; Elvassore, N.; Dupont, S.; Piccolo, S. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell 2013, 154, 1047–1059. [Google Scholar] [CrossRef] [PubMed]
- Pocaterra, A.; Santinon, G.; Romani, P.; Brian, I.; Dimitracopoulos, A.; Ghisleni, A.; Carnicer-Lombarte, A.; Forcato, M.; Braghetta, P.; Montagner, M.; et al. F-actin dynamics regulates mammalian organ growth and cell fate maintenance. J. Hepatol. 2019, 71, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.-W.; Kim, M.-K.; Lee, Y.-S.; Lee, J.-W.; Kim, D.-M.; Song, S.-H.; Lee, J.-Y.; Choi, B.-Y.; Min, B.; Chi, X.-Z.; et al. RAC-LATS1/2 signaling regulates YAP activity by switching between the YAP-binding partners TEAD4 and RUNX3. Oncogene 2017, 36, 999–1011. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, X.; Liu, L.; Yang, X.; Qian, Q.; Du, B. c-Src promotes the growth and tumorigenesis of hepatocellular carcinoma via the Hippo signaling pathway. Life Sci. 2021, 264, 118711. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Wehling, L.; Wan, S.; Weiler, S.M.E.; Tóth, M.; Ibberson, D.; Marhenke, S.; Ali, A.; Lam, M.; Guo, T.; et al. Dynamic YAP expression in the non-parenchymal liver cell compartment controls heterologous cell communication. Cell. Mol. Life Sci. 2024, 81, 115. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, J.; Zhang, S.; Jia, J.; Liu, X.; Zhang, J.; Wang, P.; Song, X.; Che, L.; Liu, K.; et al. Distinct and Overlapping Roles of Hippo Effectors YAP and TAZ During Human and Mouse Hepatocarcinogenesis. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 1095–1117. [Google Scholar] [CrossRef] [PubMed]
- Roux, K.J.; Kim, D.I.; Raida, M.; Burke, B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 2012, 196, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucl. Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef] [PubMed]
- Roessler, S.; Jia, H.-L.; Budhu, A.; Forgues, M.; Ye, Q.-H.; Lee, J.-S.; Thorgeirsson, S.S.; Sun, Z.; Tang, Z.-Y.; Qin, L.-X.; et al. A unique metastasis gene signature enables prediction of tumor relapse in early-stage hepatocellular carcinoma patients. Cancer Res. 2010, 70, 10202–10212. [Google Scholar] [CrossRef]
- Cheung, H.W.; Du, J.; Boehm, J.S.; He, F.; Weir, B.A.; Wang, X.; Butaney, M.; Sequist, L.V.; Luo, B.; Engelman, J.A.; et al. Amplification of CRKL induces transformation and epidermal growth factor receptor inhibitor resistance in human non-small cell lung cancers. Cancer Discov. 2011, 1, 608–625. [Google Scholar] [CrossRef]
- Zhang, J.; Gao, X.; Schmit, F.; Adelmant, G.; Eck, M.J.; Marto, J.A.; Zhao, J.J.; Roberts, T.M. CRKL Mediates p110β-Dependent PI3K Signaling in PTEN-Deficient Cancer Cells. Cell Rep. 2017, 20, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Girardin, S.E.; Yaniv, M. A direct interaction between JNK1 and CrkII is critical for Rac1-induced JNK activation. EMBO J. 2001, 20, 3437–3446. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Forcato, M.; Battilana, G.; Azzolin, L.; Quaranta, E.; Bodega, B.; Rosato, A.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol. 2015, 17, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Stein, C.; Bardet, A.F.; Roma, G.; Bergling, S.; Clay, I.; Ruchti, A.; Agarinis, C.; Schmelzle, T.; Bouwmeester, T.; Schübeler, D.; et al. YAP1 Exerts Its Transcriptional Control via TEAD-Mediated Activation of Enhancers. PLoS Genet 2015, 11, e1005465. [Google Scholar] [CrossRef] [PubMed]
- The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Mathelier, A.; Zhao, X.; Zhang, A.W.; Parcy, F.; Worsley-Hunt, R.; Arenillas, D.J.; Buchman, S.; Chen, C.; Chou, A.; Ienasescu, H.; et al. JASPAR 2014: An extensively expanded and updated open-access database of transcription factor binding profiles. Nucl. Acids Res. 2013, 42, D142–D147. [Google Scholar] [CrossRef] [PubMed]
- Manmadhan, S.; Ehmer, U. Hippo Signaling in the Liver—A Long and Ever-Expanding Story. Front. Cell Dev. Biol. 2019, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Kobashigawa, Y.; Inagaki, F. Structural biology: CrkL is not Crk-like. Nat. Chem. Biol. 2012, 8, 504–505. [Google Scholar] [CrossRef] [PubMed]
- Senechal, K.; Halpern, J.; Sawyers, C.L. The CRKL adaptor protein transforms fibroblasts and functions in transformation by the BCR-ABL oncogene. J. Biol. Chem. 1996, 271, 23255–23261. [Google Scholar] [CrossRef]
- Matsuda, M.; Tanaka, S.; Nagata, S.; Kojima, A.; Kurata, T.; Shibuya, M. Two species of human CRK cDNA encode proteins with distinct biological activities. Mol. Cell. Biol. 1992, 12, 3482–3489. [Google Scholar] [CrossRef]
- Franke, F.C.; Slusarenko, B.O.; Engleitner, T.; Johannes, W.; Laschinger, M.; Rad, R.; Nitsche, U.; Janssen, K.-P. Novel role for CRK adaptor proteins as essential components of SRC/FAK signaling for epithelial-mesenchymal transition and colorectal cancer aggressiveness. Int. J. Cancer 2020, 147, 1715–1731. [Google Scholar] [CrossRef] [PubMed]
- Koudelková, L.; Brábek, J.; Rosel, D. Src kinase: Key effector in mechanosignalling. Int. J. Biochem. Cell Biol. 2021, 131, 105908. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, X.; Hou, Z.; Cai, S.; Guo, Y.; Sun, L.; Li, A.; Li, Q.; Wang, E.; Miao, Y. P130cas-FAK interaction is essential for YAP-mediated radioresistance of non-small cell lung cancer. Cell Death Dis. 2022, 13, 783. [Google Scholar] [CrossRef] [PubMed]
- Sawada, Y.; Tamada, M.; Dubin-Thaler, B.J.; Cherniavskaya, O.; Sakai, R.; Tanaka, S.; Sheetz, M.P. Force sensing by mechanical extension of the Src family kinase substrate p130Cas. Cell 2006, 127, 1015–1026. [Google Scholar] [CrossRef]
- Koo, J.H.; Plouffe, S.W.; Meng, Z.; Lee, D.-H.; Yang, D.; Lim, D.-S.; Wang, C.-Y.; Guan, K.-L. Induction of AP-1 by YAP/TAZ contributes to cell proliferation and organ growth. Genes Dev. 2020, 34, 72–86. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wesener, M.C.; Weiler, S.M.E.; Bissinger, M.; Klessinger, T.F.; Rose, F.; Merker, S.; Luzarowski, M.; Ruppert, T.; Helm, B.; Klingmüller, U.; et al. CRKL Enhances YAP Signaling through Binding and JNK/JUN Pathway Activation in Liver Cancer. Int. J. Mol. Sci. 2024, 25, 8549. https://doi.org/10.3390/ijms25158549
Wesener MC, Weiler SME, Bissinger M, Klessinger TF, Rose F, Merker S, Luzarowski M, Ruppert T, Helm B, Klingmüller U, et al. CRKL Enhances YAP Signaling through Binding and JNK/JUN Pathway Activation in Liver Cancer. International Journal of Molecular Sciences. 2024; 25(15):8549. https://doi.org/10.3390/ijms25158549
Chicago/Turabian StyleWesener, Marie C., Sofia M. E. Weiler, Michaela Bissinger, Tobias F. Klessinger, Fabian Rose, Sabine Merker, Marcin Luzarowski, Thomas Ruppert, Barbara Helm, Ursula Klingmüller, and et al. 2024. "CRKL Enhances YAP Signaling through Binding and JNK/JUN Pathway Activation in Liver Cancer" International Journal of Molecular Sciences 25, no. 15: 8549. https://doi.org/10.3390/ijms25158549