
Role of Autophagy and AMPK in Cancer Stem Cells: Therapeutic Opportunities and Obstacles in Cancer


Abstract
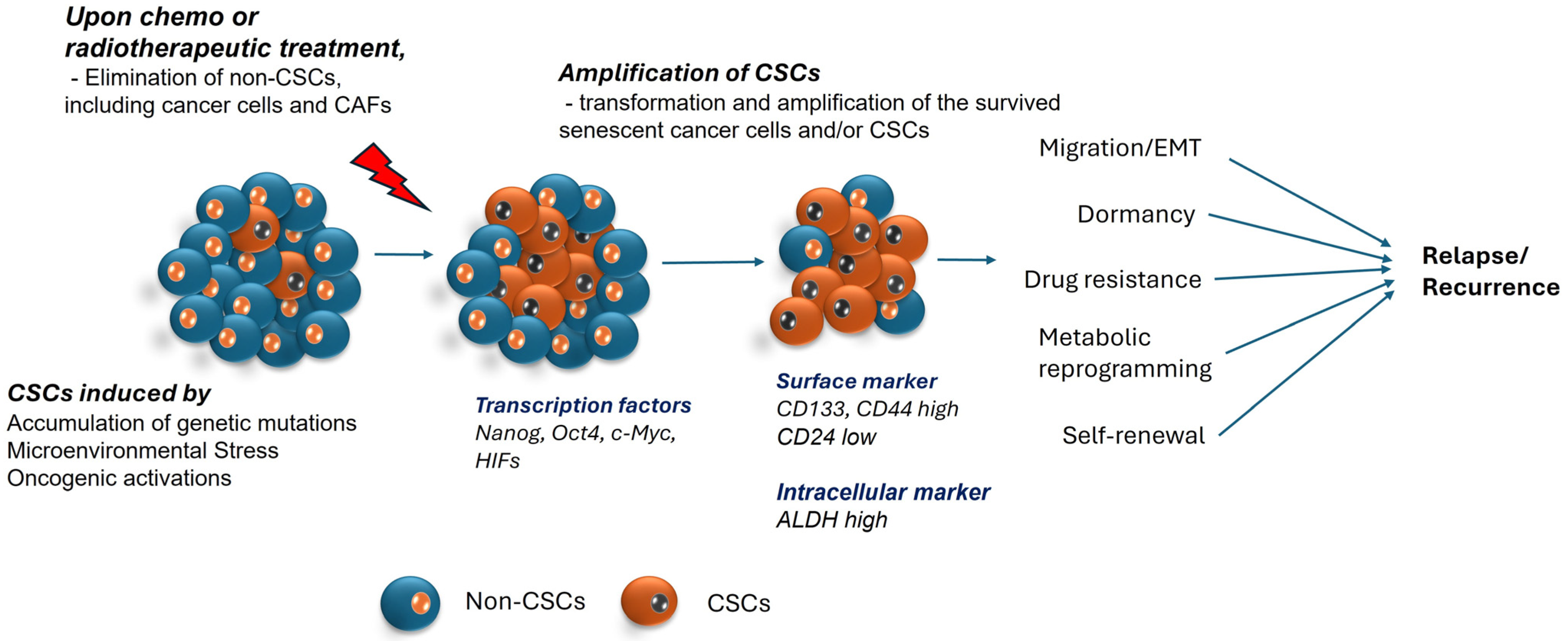
:1. Introduction
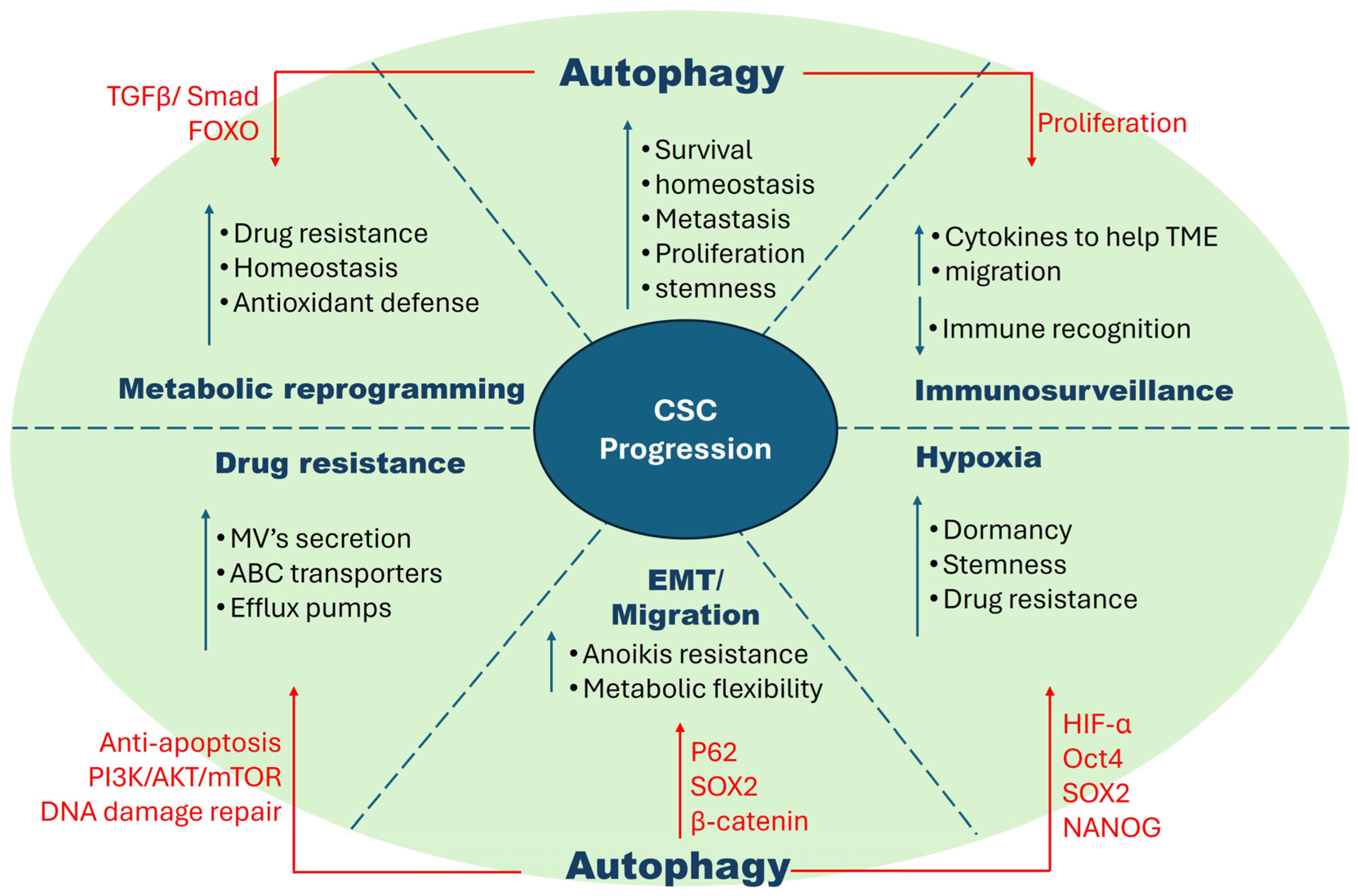
2. Cancer Stem Cells
2.1. Dormancy
2.2. Hypoxia
2.3. Drug Resistance
2.4. EMT (Epithelial–Mesenchymal Transition)
2.5. Immunosurveillance
2.6. Metabolic Flexibility
2.7. Regulatory Mechanisms in CSCs vs. Non-CSCs vs. Normal Cells
3. Autophagy
3.1. Autophagy in Normal Cells vs. Cancer Cells
3.2. Tumor-Suppressive Effects of Autophagy in Early Cancers
3.3. Tumor Protective Properties of Autophagy in Established Cancers
3.3.1. Relationship between Tumorigenicity and Autophagy in CSCs
3.3.2. Autophagy in Hypoxic Environments
3.3.3. Influence of Autophagy in EMT
3.3.4. Signaling Pathways Involved in Autophagy Dependent CSCs
3.3.5. Autophagy in Drug Resistance
3.4. Mitophagy in CSCs
3.4.1. Mitophagy and Tumorigenesis
3.4.2. The Role of Mitophagy in CSCs
3.5. Importance of Context Specificity in Cancer
4. Regulators of Autophagy in Cancer
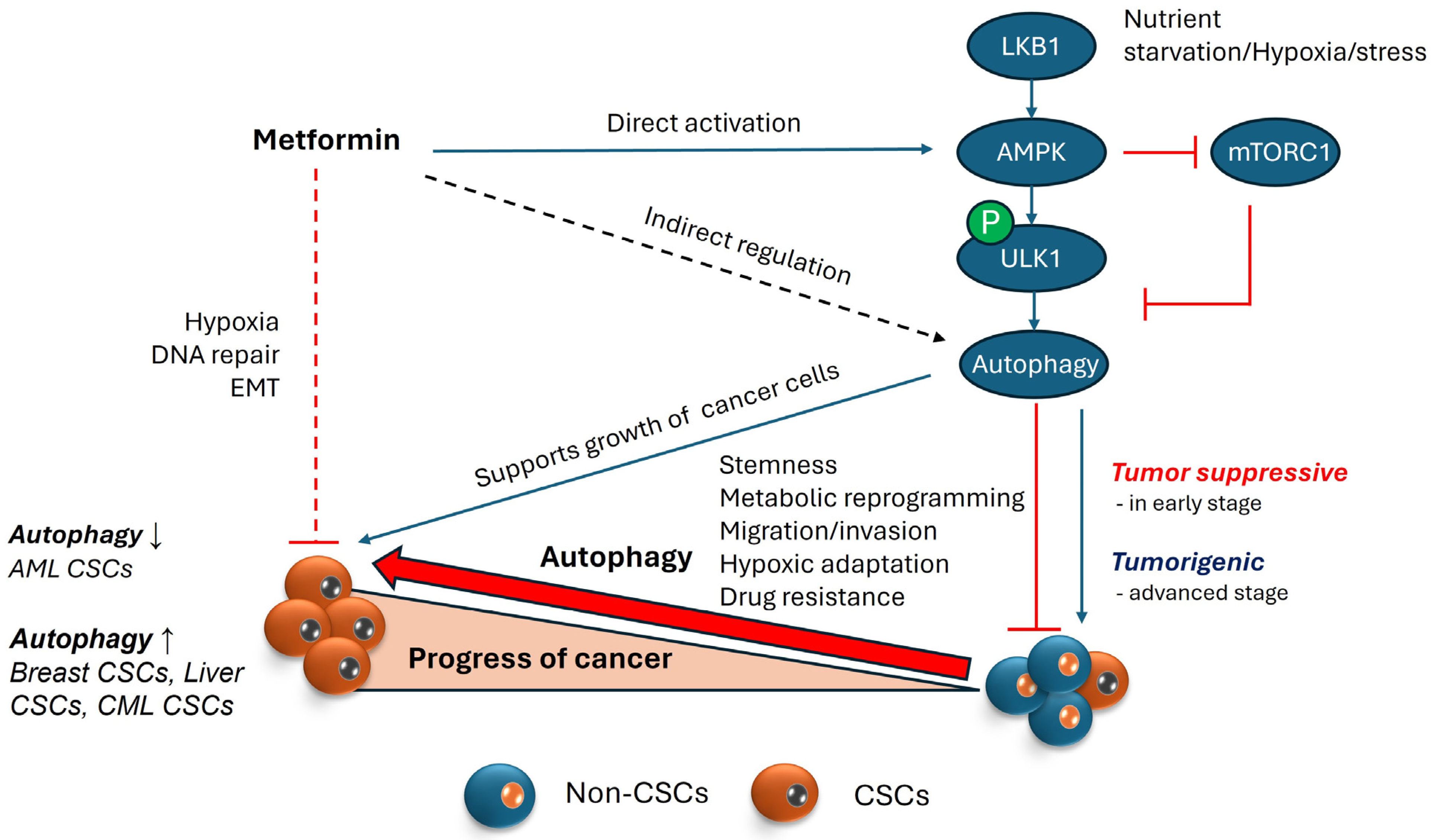
4.1. AMPK and Its Role in Autophagy
4.2. Involvement of AMPK in Regulating Autophagy within the Context of Cancer
5. Therapeutic Approaches
5.1. Metformin
5.2. Autophagy Inhibitors
5.3. In Silico/Computational Models
5.4. Peptide/Small Molecules
5.5. Natural Compounds
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Soerjomataram, I.; Bray, F. Planning for tomorrow: Global cancer incidence and the role of prevention 2020–2070. Nat. Rev. Clin. Oncol. 2021, 18, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.C. Cancer stem cells: Role in tumor growth, recurrence, metastasis, and treatment resistance. Medicine 2016, 95, S20–S25. [Google Scholar] [CrossRef] [PubMed]
- Nazio, F.; Bordi, M.; Cianfanelli, V.; Locatelli, F.; Cecconi, F. Autophagy and cancer stem cells: Molecular mechanisms and therapeutic applications. Cell Death Differ. 2019, 26, 690–702. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Alarcon, T. Metabostemness: A new cancer hallmark. Front. Oncol. 2014, 4, 262. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Vellon, L.; Oliveras-Ferraros, C.; Cufi, S.; Vazquez-Martin, A. mTOR-regulated senescence and autophagy during reprogramming of somatic cells to pluripotency: A roadmap from energy metabolism to stem cell renewal and aging. Cell Cycle 2011, 10, 3658–3677. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.G.; Macleod, K.F. Autophagy, cancer stem cells and drug resistance. J. Pathol. 2019, 247, 708–718. [Google Scholar] [CrossRef] [PubMed]
- Takakura, N. Formation and regulation of the cancer stem cell niche. Cancer Sci. 2012, 103, 1177–1181. [Google Scholar] [CrossRef] [PubMed]
- Paul, R.; Dorsey, J.F.; Fan, Y. Cell plasticity, senescence, and quiescence in cancer stem cells: Biological and therapeutic implications. Pharmacol. Ther. 2022, 231, 107985. [Google Scholar] [CrossRef] [PubMed]
- Enderling, H.; Hahnfeldt, P.; Hlatky, L.; Almog, N. Systems biology of tumor dormancy: Linking biology and mathematics on multiple scales to improve cancer therapy. Cancer Res. 2012, 72, 2172–2175. [Google Scholar] [CrossRef]
- Talukdar, S.; Bhoopathi, P.; Emdad, L.; Das, S.; Sarkar, D.; Fisher, P.B. Dormancy and cancer stem cells: An enigma for cancer therapeutic targeting. Adv. Cancer Res. 2019, 141, 43–84. [Google Scholar] [CrossRef]
- Cabarcas, S.M.; Mathews, L.A.; Farrar, W.L. The cancer stem cell niche—There goes the neighborhood? Int. J. Cancer 2011, 129, 2315–2327. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Chen, W.B.; Zhang, X.Y.; Kang, X.N.; Jin, L.J.; Zhang, H.; Wang, Z.Y. HIF-2alpha regulates CD44 to promote cancer stem cell activation in triple-negative breast cancer via PI3K/AKT/mTOR signaling. World J. Stem Cells 2020, 12, 87–99. [Google Scholar] [CrossRef] [PubMed]
- McCord, A.M.; Jamal, M.; Shankavaram, U.T.; Lang, F.F.; Camphausen, K.; Tofilon, P.J. Physiologic oxygen concentration enhances the stem-like properties of CD133+ human glioblastoma cells in vitro. Mol. Cancer Res. 2009, 7, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Das, B.; Tsuchida, R.; Malkin, D.; Koren, G.; Baruchel, S.; Yeger, H. Hypoxia enhances tumor stemness by increasing the invasive and tumorigenic side population fraction. Stem Cells 2008, 26, 1818–1830. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Z.; Ajani, J.A.; Song, S. Drug resistance and Cancer stem cells. Cell Commun. Signal 2021, 19, 19. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.P.; Yang, C.J.; Huang, M.S.; Yeh, C.T.; Wu, A.T.; Lee, Y.C.; Lai, T.C.; Lee, C.H.; Hsiao, Y.W.; Lu, J.; et al. Cisplatin selects for multidrug-resistant CD133+ cells in lung adenocarcinoma by activating Notch signaling. Cancer Res. 2013, 73, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Oskarsson, T.; Batlle, E.; Massague, J. Metastatic stem cells: Sources, niches, and vital pathways. Cell Stem Cell 2014, 14, 306–321. [Google Scholar] [CrossRef] [PubMed]
- Babaei, G.; Aziz, S.G.; Jaghi, N.Z.Z. EMT, cancer stem cells and autophagy; The three main axes of metastasis. Biomed. Pharmacother. 2021, 133, 110909. [Google Scholar] [CrossRef] [PubMed]
- Sahlgren, C.; Gustafsson, M.V.; Jin, S.; Poellinger, L.; Lendahl, U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc. Natl. Acad. Sci. USA 2008, 105, 6392–6397. [Google Scholar] [CrossRef]
- Yang, M.H.; Wu, M.Z.; Chiou, S.H.; Chen, P.M.; Chang, S.Y.; Liu, C.J.; Teng, S.C.; Wu, K.J. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat. Cell Biol. 2008, 10, 295–305. [Google Scholar] [CrossRef]
- Aptsiauri, N.; Cabrera, T.; Mendez, R.; Garcia-Lora, A.; Ruiz-Cabello, F.; Garrido, F. Role of altered expression of HLA class I molecules in cancer progression. Adv. Exp. Med. Biol. 2007, 601, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Sun, Y.; Ma, L. ZEB1: At the crossroads of epithelial-mesenchymal transition, metastasis and therapy resistance. Cell Cycle 2015, 14, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.C.; Kim, J.H. Cancer stem cell metabolism: Target for cancer therapy. BMB Rep. 2018, 51, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Wang, J.H.; Fan, W.J.; Meng, Y.T.; Li, M.M.; Li, T.T.; Cui, B.; Wang, H.F.; Zhao, Y.; An, F.; et al. Glycolysis gatekeeper PDK1 reprograms breast cancer stem cells under hypoxia. Oncogene 2018, 37, 1062–1074. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.; Wikman, H.; Pantel, K.; Haigis, M.C.; de Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014, 16, 992–1003, 1001–1015. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.M.; Giltnane, J.M.; Balko, J.M.; Schwarz, L.J.; Guerrero-Zotano, A.L.; Hutchinson, K.E.; Nixon, M.J.; Estrada, M.V.; Sanchez, V.; Sanders, M.E.; et al. MYC and MCL1 Cooperatively Promote Chemotherapy-Resistant Breast Cancer Stem Cells via Regulation of Mitochondrial Oxidative Phosphorylation. Cell Metab. 2017, 26, 633–647 e637. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Vidal, C.; Dey, S.; Zhang, L. Mitochondria Targeting as an Effective Strategy for Cancer Therapy. Int. J. Mol. Sci. 2020, 21, 3363. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, A.; Sahara, H. The Metabolic Heterogeneity and Flexibility of Cancer Stem Cells. Cancers 2020, 12, 278. [Google Scholar] [CrossRef]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef]
- Ryoo, I.G.; Lee, S.H.; Kwak, M.K. Redox Modulating NRF2: A Potential Mediator of Cancer Stem Cell Resistance. Oxid. Med. Cell Longev. 2016, 2016, 2428153. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhang, Z.; Song, L.; Gao, J.; Liu, Y. Lipid metabolism of cancer stem cells. Oncol. Lett. 2022, 23, 119. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Tuy, K.; Rickenbacker, L.; Hjelmeland, A.B. Reactive oxygen species produced by altered tumor metabolism impacts cancer stem cell maintenance. Redox Biol. 2021, 44, 101953. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Chen, J.; Zhu, H.; Jia, Z.H.; Cui, M.H. Aberrantly elevated redox sensing factor Nrf2 promotes cancer stem cell survival via enhanced transcriptional regulation of ABCG2 and Bcl-2/Bmi-1 genes. Oncol. Rep. 2015, 34, 2296–2304. [Google Scholar] [CrossRef] [PubMed]
- Achuthan, S.; Santhoshkumar, T.R.; Prabhakar, J.; Nair, S.A.; Pillai, M.R. Drug-induced senescence generates chemoresistant stemlike cells with low reactive oxygen species. J. Biol. Chem. 2011, 286, 37813–37829. [Google Scholar] [CrossRef] [PubMed]
- Emmink, B.L.; Verheem, A.; Van Houdt, W.J.; Steller, E.J.; Govaert, K.M.; Pham, T.V.; Piersma, S.R.; Borel Rinkes, I.H.; Jimenez, C.R.; Kranenburg, O. The secretome of colon cancer stem cells contains drug-metabolizing enzymes. J. Proteom. 2013, 91, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Dokic, I.; Hartmann, C.; Herold-Mende, C.; Regnier-Vigouroux, A. Glutathione peroxidase 1 activity dictates the sensitivity of glioblastoma cells to oxidative stress. Glia 2012, 60, 1785–1800. [Google Scholar] [CrossRef]
- Yu, W.K.; Wang, Z.; Fong, C.C.; Liu, D.; Yip, T.C.; Au, S.K.; Zhu, G.; Yang, M. Chemoresistant lung cancer stem cells display high DNA repair capability to remove cisplatin-induced DNA damage. Br. J. Pharmacol. 2017, 174, 302–313. [Google Scholar] [CrossRef]
- Yan, J.; Tang, D. Prostate cancer stem-like cells proliferate slowly and resist etoposide-induced cytotoxicity via enhancing DNA damage response. Exp. Cell Res. 2014, 328, 132–142. [Google Scholar] [CrossRef]
- Ichimiya, T.; Yamakawa, T.; Hirano, T.; Yokoyama, Y.; Hayashi, Y.; Hirayama, D.; Wagatsuma, K.; Itoi, T.; Nakase, H. Autophagy and Autophagy-Related Diseases: A Review. Int. J. Mol. Sci. 2020, 21, 8974. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Lee, J.; Ha, J.; Kang, I.; Choe, W. Endoplasmic Reticulum Stress and Its Impact on Adipogenesis: Molecular Mechanisms Implicated. Nutrients 2023, 15, 5082. [Google Scholar] [CrossRef] [PubMed]
- King, J.S.; Veltman, D.M.; Insall, R.H. The induction of autophagy by mechanical stress. Autophagy 2011, 7, 1490–1499. [Google Scholar] [CrossRef] [PubMed]
- Lamb, C.A.; Yoshimori, T.; Tooze, S.A. The autophagosome: Origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 759–774. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Li, J.; Yang, K.; Cao, D. An overview of autophagy: Mechanism, regulation and research progress. Bull. Du Cancer 2021, 108, 304–322. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; He, S.; Ma, B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 2020, 19, 12. [Google Scholar] [CrossRef] [PubMed]
- Vitale, I.; Manic, G.; Dandrea, V.; De Maria, R. Role of autophagy in the maintenance and function of cancer stem cells. Int. J. Dev. Biol. 2015, 59, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3446. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Galluzzi, L.; Zitvogel, L.; Kroemer, G. Autophagy and cellular immune responses. Immunity 2013, 39, 211–227. [Google Scholar] [CrossRef] [PubMed]
- Phadwal, K.; Watson, A.S.; Simon, A.K. Tightrope act: Autophagy in stem cell renewal, differentiation, proliferation, and aging. Cell Mol. Life Sci. 2013, 70, 89–103. [Google Scholar] [CrossRef]
- Mathew, R.; Karp, C.M.; Beaudoin, B.; Vuong, N.; Chen, G.; Chen, H.Y.; Bray, K.; Reddy, A.; Bhanot, G.; Gelinas, C.; et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009, 137, 1062–1075. [Google Scholar] [CrossRef] [PubMed]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Li, D.D.; Wang, L.L.; Deng, R.; Zhu, X.F. Decreased expression of autophagy-related proteins in malignant epithelial ovarian cancer. Autophagy 2008, 4, 1067–1068. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Coppola, D.; Matsushita, N.; Cualing, H.D.; Sun, M.; Sato, Y.; Liang, C.; Jung, J.U.; Cheng, J.Q.; Mule, J.J.; et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat. Cell Biol. 2007, 9, 1142–1151. [Google Scholar] [CrossRef] [PubMed]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes. Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Mainz, L.; Rosenfeldt, M.T. Autophagy and cancer—Insights from mouse models. FEBS J. 2018, 285, 792–808. [Google Scholar] [CrossRef] [PubMed]
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006, 126, 121–134. [Google Scholar] [CrossRef]
- Feng, Z.; Hu, W.; de Stanchina, E.; Teresky, A.K.; Jin, S.; Lowe, S.; Levine, A.J. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: Stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007, 67, 3043–3053. [Google Scholar] [CrossRef]
- Kenzelmann Broz, D.; Spano Mello, S.; Bieging, K.T.; Jiang, D.; Dusek, R.L.; Brady, C.A.; Sidow, A.; Attardi, L.D. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes. Dev. 2013, 27, 1016–1031. [Google Scholar] [CrossRef] [PubMed]
- Chavez-Dominguez, R.; Perez-Medina, M.; Lopez-Gonzalez, J.S.; Galicia-Velasco, M.; Aguilar-Cazares, D. The Double-Edge Sword of Autophagy in Cancer: From Tumor Suppression to Pro-tumor Activity. Front. Oncol. 2020, 10, 578418. [Google Scholar] [CrossRef] [PubMed]
- Luo, T.; Fu, J.; Xu, A.; Su, B.; Ren, Y.; Li, N.; Zhu, J.; Zhao, X.; Dai, R.; Cao, J.; et al. PSMD10/gankyrin induces autophagy to promote tumor progression through cytoplasmic interaction with ATG7 and nuclear transactivation of ATG7 expression. Autophagy 2016, 12, 1355–1371. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Bauvy, C.; Tonelli, G.; Yue, W.; Delomenie, C.; Nicolas, V.; Zhu, Y.; Domergue, V.; Marin-Esteban, V.; Tharinger, H.; et al. Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem-like/progenitor cells. Oncogene 2013, 32, 2261–2272. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Fan, S.; Qin, T.; Yang, J.; Sun, Y.; Lu, Y.; Mao, J.; Li, L. Role of autophagy in breast cancer and breast cancer stem cells (Review). Int. J. Oncol. 2018, 52, 1057–1070. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Hartleben, B.; Kretz, O.; Wiech, T.; Igarashi, P.; Mizushima, N.; Walz, G.; Huber, T.B. Autophagy plays a critical role in kidney tubule maintenance, aging and ischemia-reperfusion injury. Autophagy 2012, 8, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Wolf, J.; Dewi, D.L.; Fredebohm, J.; Muller-Decker, K.; Flechtenmacher, C.; Hoheisel, J.D.; Boettcher, M. A mammosphere formation RNAi screen reveals that ATG4A promotes a breast cancer stem-like phenotype. Breast Cancer Res. 2013, 15, R109. [Google Scholar] [CrossRef] [PubMed]
- Troumpoukis, D.; Papadimitropoulou, A.; Charalampous, C.; Kogionou, P.; Palamaris, K.; Sarantis, P.; Serafimidis, I. Targeting autophagy in pancreatic cancer: The cancer stem cell perspective. Front. Oncol. 2022, 12, 1049436. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.J.; Zhang, S.S.; Guo, X.L.; Sun, K.; Han, Z.P.; Li, R.; Zhao, Q.D.; Deng, W.J.; Xie, X.Q.; Zhang, J.W.; et al. Autophagy contributes to the survival of CD133+ liver cancer stem cells in the hypoxic and nutrient-deprived tumor microenvironment. Cancer Lett. 2013, 339, 70–81. [Google Scholar] [CrossRef]
- Zhang, D.; Zhao, Q.; Sun, H.; Yin, L.; Wu, J.; Xu, J.; He, T.; Yang, C.; Liang, C. Defective autophagy leads to the suppression of stem-like features of CD271(+) osteosarcoma cells. J. Biomed. Sci. 2016, 23, 82. [Google Scholar] [CrossRef]
- Peng, Q.; Qin, J.; Zhang, Y.; Cheng, X.; Wang, X.; Lu, W.; Xie, X.; Zhang, S. Autophagy maintains the stemness of ovarian cancer stem cells by FOXA2. J. Exp. Clin. Cancer Res. 2017, 36, 171. [Google Scholar] [CrossRef] [PubMed]
- Buccarelli, M.; Marconi, M.; Pacioni, S.; De Pascalis, I.; D’Alessandris, Q.G.; Martini, M.; Ascione, B.; Malorni, W.; Larocca, L.M.; Pallini, R.; et al. Inhibition of autophagy increases susceptibility of glioblastoma stem cells to temozolomide by igniting ferroptosis. Cell Death Dis. 2018, 9, 841. [Google Scholar] [CrossRef] [PubMed]
- Seo, W.; Silwal, P.; Song, I.C.; Jo, E.K. The dual role of autophagy in acute myeloid leukemia. J. Hematol. Oncol. 2022, 15, 51. [Google Scholar] [CrossRef] [PubMed]
- Torgersen, M.L.; Simonsen, A. Autophagy: Friend or foe in the treatment of fusion protein-associated leukemias? Autophagy 2013, 9, 2175–2177. [Google Scholar] [CrossRef] [PubMed]
- Altman, J.K.; Szilard, A.; Goussetis, D.J.; Sassano, A.; Colamonici, M.; Gounaris, E.; Frankfurt, O.; Giles, F.J.; Eklund, E.A.; Beauchamp, E.M.; et al. Autophagy is a survival mechanism of acute myelogenous leukemia precursors during dual mTORC2/mTORC1 targeting. Clin. Cancer Res. 2014, 20, 2400–2409. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, M.; Watson, A.S.; Simon, A.K. Lack of autophagy in the hematopoietic system leads to loss of hematopoietic stem cell function and dysregulated myeloid proliferation. Autophagy 2011, 7, 1069–1070. [Google Scholar] [CrossRef] [PubMed]
- Elzinga, B.M.; Nyhan, M.J.; Crowley, L.C.; O’Donovan, T.R.; Cahill, M.R.; McKenna, S.L. Induction of autophagy by Imatinib sequesters Bcr-Abl in autophagosomes and down-regulates Bcr-Abl protein. Am. J. Hematol. 2013, 88, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Isakson, P.; Bjoras, M.; Boe, S.O.; Simonsen, A. Autophagy contributes to therapy-induced degradation of the PML/RARA oncoprotein. Blood 2010, 116, 2324–2331. [Google Scholar] [CrossRef] [PubMed]
- Helgason, G.V.; Mukhopadhyay, A.; Karvela, M.; Salomoni, P.; Calabretta, B.; Holyoake, T.L. Autophagy in chronic myeloid leukaemia: Stem cell survival and implication in therapy. Curr. Cancer Drug Targets 2013, 13, 724–734. [Google Scholar] [CrossRef]
- Rothe, K.; Lin, H.; Lin, K.B.; Leung, A.; Wang, H.M.; Malekesmaeili, M.; Brinkman, R.R.; Forrest, D.L.; Gorski, S.M.; Jiang, X. The core autophagy protein ATG4B is a potential biomarker and therapeutic target in CML stem/progenitor cells. Blood 2014, 123, 3622–3634. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yamamoto, A.; Matsui, M.; Yoshimori, T.; Ohsumi, Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell 2004, 15, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gelinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Mabjeesh, N.J.; Amir, S. Hypoxia-inducible factor (HIF) in human tumorigenesis. Histol. Histopathol. 2007, 22, 559–572. [Google Scholar] [CrossRef]
- Pouyssegur, J.; Dayan, F.; Mazure, N.M. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 2006, 441, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, J.; Zhang, Z.; Zhou, W.; Wang, A.J.; Heddleston, J.M.; Pinna, C.M.; Hubaud, A.; Stadler, B.; Choi, M.; Bar, M.; et al. HIF induces human embryonic stem cell markers in cancer cells. Cancer Res. 2011, 71, 4640–4652. [Google Scholar] [CrossRef]
- Zhu, H.; Wang, D.; Liu, Y.; Su, Z.; Zhang, L.; Chen, F.; Zhou, Y.; Wu, Y.; Yu, M.; Zhang, Z.; et al. Role of the Hypoxia-inducible factor-1 alpha induced autophagy in the conversion of non-stem pancreatic cancer cells into CD133+ pancreatic cancer stem-like cells. Cancer Cell Int. 2013, 13, 119. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K.; Weinberg, R.A. Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat. Rev. Cancer 2009, 9, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Morel, A.P.; Lievre, M.; Thomas, C.; Hinkal, G.; Ansieau, S.; Puisieux, A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS ONE 2008, 3, e2888. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.S.; Jiang, J.; Liang, X.H.; Tang, Y.L. Links between cancer stem cells and epithelial-mesenchymal transition. Onco Targets Ther. 2015, 8, 2973–2980. [Google Scholar] [CrossRef]
- Galavotti, S.; Bartesaghi, S.; Faccenda, D.; Shaked-Rabi, M.; Sanzone, S.; McEvoy, A.; Dinsdale, D.; Condorelli, F.; Brandner, S.; Campanella, M.; et al. The autophagy-associated factors DRAM1 and p62 regulate cell migration and invasion in glioblastoma stem cells. Oncogene 2013, 32, 699–712. [Google Scholar] [CrossRef]
- Zhu, Y.; Huang, S.; Chen, S.; Chen, J.; Wang, Z.; Wang, Y.; Zheng, H. SOX2 promotes chemoresistance, cancer stem cells properties, and epithelial-mesenchymal transition by beta-catenin and Beclin1/autophagy signaling in colorectal cancer. Cell Death Dis. 2021, 12, 449. [Google Scholar] [CrossRef] [PubMed]
- Yeo, S.K.; Wen, J.; Chen, S.; Guan, J.L. Autophagy Differentially Regulates Distinct Breast Cancer Stem-like Cells in Murine Models via EGFR/Stat3 and Tgfbeta/Smad Signaling. Cancer Res. 2016, 76, 3397–3410. [Google Scholar] [CrossRef] [PubMed]
- Maycotte, P.; Jones, K.L.; Goodall, M.L.; Thorburn, J.; Thorburn, A. Autophagy Supports Breast Cancer Stem Cell Maintenance by Regulating IL6 Secretion. Mol. Cancer Res. 2015, 13, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Liang, R.; Ghaffari, S. Stem Cells Seen Through the FOXO Lens: An Evolving Paradigm. Curr. Top. Dev. Biol. 2018, 127, 23–47. [Google Scholar] [CrossRef] [PubMed]
- Dubrovska, A.; Kim, S.; Salamone, R.J.; Walker, J.R.; Maira, S.M.; Garcia-Echeverria, C.; Schultz, P.G.; Reddy, V.A. The role of PTEN/Akt/PI3K signaling in the maintenance and viability of prostate cancer stem-like cell populations. Proc. Natl. Acad. Sci. USA 2009, 106, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Sunayama, J.; Sato, A.; Matsuda, K.; Tachibana, K.; Watanabe, E.; Seino, S.; Suzuki, K.; Narita, Y.; Shibui, S.; Sakurada, K.; et al. FoxO3a functions as a key integrator of cellular signals that control glioblastoma stem-like cell differentiation and tumorigenicity. Stem Cells 2011, 29, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Ning, Y.; Luo, C.; Ren, K.; Quan, M.; Cao, J. FOXO3a-mediated suppression of the self-renewal capacity of sphere-forming cells derived from the ovarian cancer SKOV3 cell line by 7-difluoromethoxyl-5,4’-di-n-octyl genistein. Mol. Med. Rep. 2014, 9, 1982–1988. [Google Scholar] [CrossRef] [PubMed]
- Smit, L.; Berns, K.; Spence, K.; Ryder, W.D.; Zeps, N.; Madiredjo, M.; Beijersbergen, R.; Bernards, R.; Clarke, R.B. An integrated genomic approach identifies that the PI3K/AKT/FOXO pathway is involved in breast cancer tumor initiation. Oncotarget 2016, 7, 2596–2610. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, V.V.; Allen, J.E.; Dicker, D.T.; El-Deiry, W.S. Small-Molecule ONC201/TIC10 Targets Chemotherapy-Resistant Colorectal Cancer Stem-like Cells in an Akt/Foxo3a/TRAIL-Dependent Manner. Cancer Res. 2015, 75, 1423–1432. [Google Scholar] [CrossRef]
- Naka, K.; Hoshii, T.; Muraguchi, T.; Tadokoro, Y.; Ooshio, T.; Kondo, Y.; Nakao, S.; Motoyama, N.; Hirao, A. TGF-beta-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature 2010, 463, 676–680. [Google Scholar] [CrossRef]
- Van der Vos, K.E.; Coffer, P.J. FOXO-binding partners: It takes two to tango. Oncogene 2008, 27, 2289–2299. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, L.N.; Chow, E.K. Mechanisms of chemoresistance in cancer stem cells. Clin. Transl. Med. 2013, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Kleffel, S.; Schatton, T. Tumor dormancy and cancer stem cells: Two sides of the same coin? Adv. Exp. Med. Biol. 2013, 734, 145–179. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Zhao, G.; Yang, Y.; Jiang, Z.; Cai, J.; Hu, H. Inhibition of CD133 Overcomes Cisplatin Resistance Through Inhibiting PI3K/AKT/mTOR Signaling Pathway and Autophagy in CD133-Positive Gastric Cancer Cells. Technol. Cancer Res. Treat. 2019, 18, 1533033819864311. [Google Scholar] [CrossRef] [PubMed]
- Wendler, F.; Favicchio, R.; Simon, T.; Alifrangis, C.; Stebbing, J.; Giamas, G. Extracellular vesicles swarm the cancer microenvironment: From tumor-stroma communication to drug intervention. Oncogene 2017, 36, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Attar-Schneider, O.; Dabbah, M.; Drucker, L.; Gottfried, M. Niche origin of mesenchymal stem cells derived microvesicles determines opposing effects on NSCLC: Primary versus metastatic. Cell Signal 2020, 65, 109456. [Google Scholar] [CrossRef] [PubMed]
- Ibraheem, A.; Attar-Schneider, O.; Dabbah, M.; Dolberg Jarchowsky, O.; Tartakover Matalon, S.; Lishner, M.; Drucker, L. BM-MSCs-derived ECM modifies multiple myeloma phenotype and drug response in a source-dependent manner. Transl. Res. 2019, 207, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Ojha, R.; Jha, V.; Singh, S.K.; Bhattacharyya, S. Autophagy inhibition suppresses the tumorigenic potential of cancer stem cell enriched side population in bladder cancer. Biochim. Biophys. Acta 2014, 1842, 2073–2086. [Google Scholar] [CrossRef]
- Yang, H.Z.; Ma, Y.; Zhou, Y.; Xu, L.M.; Chen, X.J.; Ding, W.B.; Zou, H.B. Autophagy contributes to the enrichment and survival of colorectal cancer stem cells under oxaliplatin treatment. Cancer Lett. 2015, 361, 128–136. [Google Scholar] [CrossRef]
- Ma, K.; Chen, G.; Li, W.; Kepp, O.; Zhu, Y.; Chen, Q. Mitophagy, Mitochondrial Homeostasis, and Cell Fate. Front. Cell Dev. Biol. 2020, 8, 467. [Google Scholar] [CrossRef] [PubMed]
- Poole, L.P.; Macleod, K.F. Mitophagy in tumorigenesis and metastasis. Cell Mol. Life Sci. 2021, 78, 3817–3851. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Dorn, G.W., 2nd. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Geisler, S.; Holmstrom, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Sarraf, S.A.; Raman, M.; Guarani-Pereira, V.; Sowa, M.E.; Huttlin, E.L.; Gygi, S.P.; Harper, J.W. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 2013, 496, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Ney, P.A. Mitochondrial autophagy: Origins, significance, and role of BNIP3 and NIX. Biochim. Biophys. Acta 2015, 1853, 2775–2783. [Google Scholar] [CrossRef] [PubMed]
- Rogov, V.V.; Suzuki, H.; Marinkovic, M.; Lang, V.; Kato, R.; Kawasaki, M.; Buljubasic, M.; Sprung, M.; Rogova, N.; Wakatsuki, S.; et al. Phosphorylation of the mitochondrial autophagy receptor Nix enhances its interaction with LC3 proteins. Sci. Rep. 2017, 7, 1131. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Massen, S.; Terenzio, M.; Lang, V.; Chen-Lindner, S.; Eils, R.; Novak, I.; Dikic, I.; Hamacher-Brady, A.; Brady, N.R. Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis. J. Biol. Chem. 2013, 288, 1099–1113. [Google Scholar] [CrossRef]
- Ding, W.X.; Ni, H.M.; Li, M.; Liao, Y.; Chen, X.; Stolz, D.B.; Dorn, G.W., 2nd; Yin, X.M. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J. Biol. Chem. 2010, 285, 27879–27890. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Chen, D.; Si, J.; Hu, Q.; Qin, Z.; Fang, M.; Wang, G. The mitochondrial protein BNIP3L is the substrate of PARK2 and mediates mitophagy in PINK1/PARK2 pathway. Hum. Mol. Genet. 2015, 24, 2528–2538. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Xue, L.; Li, L.; Tang, C.; Wan, Z.; Wang, R.; Tan, J.; Tan, Y.; Han, H.; Tian, R.; et al. BNIP3 Protein Suppresses PINK1 Kinase Proteolytic Cleavage to Promote Mitophagy. J. Biol. Chem. 2016, 291, 21616–21629. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Y.; Kim, E.; Beemiller, P.; Wang, C.Y.; Swanson, J.; You, M.; Guan, K.L. Bnip3 mediates the hypoxia-induced inhibition on mammalian target of rapamycin by interacting with Rheb. J. Biol. Chem. 2007, 282, 35803–35813. [Google Scholar] [CrossRef] [PubMed]
- Melser, S.; Chatelain, E.H.; Lavie, J.; Mahfouf, W.; Jose, C.; Obre, E.; Goorden, S.; Priault, M.; Elgersma, Y.; Rezvani, H.R.; et al. Rheb regulates mitophagy induced by mitochondrial energetic status. Cell Metab. 2013, 17, 719–730. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Tian, W.; Hu, Z.; Chen, G.; Huang, L.; Li, W.; Zhang, X.; Xue, P.; Zhou, C.; Liu, L.; et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 2014, 15, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Han, Z.; Feng, D.; Chen, Y.; Chen, L.; Wu, H.; Huang, L.; Zhou, C.; Cai, X.; Fu, C.; et al. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol. Cell 2014, 54, 362–377. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Karuppagounder, S.S.; Springer, D.A.; Allen, M.D.; Zheng, L.; Chao, B.; Zhang, Y.; Dawson, V.L.; Dawson, T.M.; Lenardo, M. Genetic deficiency of the mitochondrial protein PGAM5 causes a Parkinson’s-like movement disorder. Nat. Commun. 2014, 5, 4930. [Google Scholar] [CrossRef]
- Agnihotri, S.; Golbourn, B.; Huang, X.; Remke, M.; Younger, S.; Cairns, R.A.; Chalil, A.; Smith, C.A.; Krumholtz, S.L.; Mackenzie, D.; et al. PINK1 Is a Negative Regulator of Growth and the Warburg Effect in Glioblastoma. Cancer Res. 2016, 76, 4708–4719. [Google Scholar] [CrossRef]
- Cesari, R.; Martin, E.S.; Calin, G.A.; Pentimalli, F.; Bichi, R.; McAdams, H.; Trapasso, F.; Drusco, A.; Shimizu, M.; Masciullo, V.; et al. Parkin, a gene implicated in autosomal recessive juvenile parkinsonism, is a candidate tumor suppressor gene on chromosome 6q25-q27. Proc. Natl. Acad. Sci. USA 2003, 100, 5956–5961. [Google Scholar] [CrossRef] [PubMed]
- Drake, L.E.; Springer, M.Z.; Poole, L.P.; Kim, C.J.; Macleod, K.F. Expanding perspectives on the significance of mitophagy in cancer. Semin. Cancer Biol. 2017, 47, 110–124. [Google Scholar] [CrossRef] [PubMed]
- Veeriah, S.; Taylor, B.S.; Meng, S.; Fang, F.; Yilmaz, E.; Vivanco, I.; Janakiraman, M.; Schultz, N.; Hanrahan, A.J.; Pao, W.; et al. Somatic mutations of the Parkinson’s disease-associated gene PARK2 in glioblastoma and other human malignancies. Nat. Genet. 2010, 42, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, M.; Marusawa, H.; Wang, H.Q.; Iwai, A.; Ikeuchi, K.; Imai, Y.; Kataoka, A.; Nukina, N.; Takahashi, R.; Chiba, T. Parkin as a tumor suppressor gene for hepatocellular carcinoma. Oncogene 2008, 27, 6002–6011. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Lin, M.; Wu, R.; Wang, X.; Yang, B.; Levine, A.J.; Hu, W.; Feng, Z. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc. Natl. Acad. Sci. USA 2011, 108, 16259–16264. [Google Scholar] [CrossRef] [PubMed]
- Bernardini, J.P.; Lazarou, M.; Dewson, G. Parkin and mitophagy in cancer. Oncogene 2017, 36, 1315–1327. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhang, Y.; Cheng, X.; Yuan, H.; Zhu, S.; Liu, J.; Wen, Q.; Xie, Y.; Liu, J.; Kroemer, G.; et al. PINK1 and PARK2 Suppress Pancreatic Tumorigenesis through Control of Mitochondrial Iron-Mediated Immunometabolism. Dev. Cell 2018, 46, 441–455 e448. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Zhao, Y.; Yue, X.; Wu, H.; Huang, S.; Chen, J.; Tomsky, K.; Xie, H.; Khella, C.A.; et al. Parkin targets HIF-1alpha for ubiquitination and degradation to inhibit breast tumor progression. Nat. Commun. 2017, 8, 1823. [Google Scholar] [CrossRef] [PubMed]
- Sarraf, S.A.; Sideris, D.P.; Giagtzoglou, N.; Ni, L.; Kankel, M.W.; Sen, A.; Bochicchio, L.E.; Huang, C.H.; Nussenzweig, S.C.; Worley, S.H.; et al. PINK1/Parkin Influences Cell Cycle by Sequestering TBK1 at Damaged Mitochondria, Inhibiting Mitosis. Cell Rep. 2019, 29, 225–235 e225. [Google Scholar] [CrossRef]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef]
- Erkan, M.; Kleeff, J.; Esposito, I.; Giese, T.; Ketterer, K.; Buchler, M.W.; Giese, N.A.; Friess, H. Loss of BNIP3 expression is a late event in pancreatic cancer contributing to chemoresistance and worsened prognosis. Oncogene 2005, 24, 4421–4432. [Google Scholar] [CrossRef] [PubMed]
- Murai, M.; Toyota, M.; Suzuki, H.; Satoh, A.; Sasaki, Y.; Akino, K.; Ueno, M.; Takahashi, F.; Kusano, M.; Mita, H.; et al. Aberrant methylation and silencing of the BNIP3 gene in colorectal and gastric cancer. Clin. Cancer Res. 2005, 11, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Okami, J.; Simeone, D.M.; Logsdon, C.D. Silencing of the hypoxia-inducible cell death protein BNIP3 in pancreatic cancer. Cancer Res. 2004, 64, 5338–5346. [Google Scholar] [CrossRef] [PubMed]
- Sowter, H.M.; Ferguson, M.; Pym, C.; Watson, P.; Fox, S.B.; Han, C.; Harris, A.L. Expression of the cell death genes BNip3 and NIX in ductal carcinoma in situ of the breast; correlation of BNip3 levels with necrosis and grade. J. Pathol. 2003, 201, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Akada, M.; Crnogorac-Jurcevic, T.; Lattimore, S.; Mahon, P.; Lopes, R.; Sunamura, M.; Matsuno, S.; Lemoine, N.R. Intrinsic chemoresistance to gemcitabine is associated with decreased expression of BNIP3 in pancreatic cancer. Clin. Cancer Res. 2005, 11, 3094–3101. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Agarwal, E.; Bertolini, I.; Seo, J.H.; Caino, M.C.; Ghosh, J.C.; Kossenkov, A.V.; Liu, Q.; Tang, H.Y.; Goldman, A.R.; et al. The mitophagy effector FUNDC1 controls mitochondrial reprogramming and cellular plasticity in cancer cells. Sci. Signal 2020, 13. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zhang, D.; Zhou, L.; Pei, Y.; Zhuang, Y.; Cui, W.; Chen, J. FUN14 domain-containing 1 promotes breast cancer proliferation and migration by activating calcium-NFATC1-BMI1 axis. EBioMedicine 2019, 41, 384–394. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.; Foster, R.E.; Horgan, K.; Mounsey, K.; Nixon, H.; Smalle, N.; Hughes, T.A.; Carter, C.R. Lymphocyte depletion and repopulation after chemotherapy for primary breast cancer. Breast Cancer Res. 2016, 18, 10. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Joven, J.; Cufi, S.; Corominas-Faja, B.; Oliveras-Ferraros, C.; Cuyas, E.; Martin-Castillo, B.; Lopez-Bonet, E.; Alarcon, T.; Vazquez-Martin, A. The Warburg effect version 2.0: Metabolic reprogramming of cancer stem cells. Cell Cycle 2013, 12, 1166–1179. [Google Scholar] [CrossRef] [PubMed]
- Pacini, N.; Borziani, F. Cancer stem cell theory and the warburg effect, two sides of the same coin? Int. J. Mol. Sci. 2014, 15, 8893–8930. [Google Scholar] [CrossRef]
- Zhou, K.; Yao, Y.L.; He, Z.C.; Chen, C.; Zhang, X.N.; Yang, K.D.; Liu, Y.Q.; Liu, Q.; Fu, W.J.; Chen, Y.P.; et al. VDAC2 interacts with PFKP to regulate glucose metabolism and phenotypic reprogramming of glioma stem cells. Cell Death Dis. 2018, 9, 988. [Google Scholar] [CrossRef] [PubMed]
- Poltavets, V.; Kochetkova, M.; Pitson, S.M.; Samuel, M.S. The Role of the Extracellular Matrix and Its Molecular and Cellular Regulators in Cancer Cell Plasticity. Front. Oncol. 2018, 8, 431. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, A.; Scorrano, L. Mitochondria: From cell death executioners to regulators of cell differentiation. Trends Cell Biol. 2014, 24, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Shirihai, O.S. The interplay between mitochondrial dynamics and mitophagy. Antioxid. Redox Signal 2011, 14, 1939–1951. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chan, D.C. Mitochondrial Dynamics in Regulating the Unique Phenotypes of Cancer and Stem Cells. Cell Metab. 2017, 26, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Wu, Q.; Horbinski, C.M.; Flavahan, W.A.; Yang, K.; Zhou, W.; Dombrowski, S.M.; Huang, Z.; Fang, X.; Shi, Y.; et al. Mitochondrial control by DRP1 in brain tumor initiating cells. Nat. Neurosci. 2015, 18, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Boya, P.; Codogno, P.; Rodriguez-Muela, N. Autophagy in stem cells: Repair, remodelling and metabolic reprogramming. Development 2018, 145. [Google Scholar] [CrossRef] [PubMed]
- Kashatus, J.A.; Nascimento, A.; Myers, L.J.; Sher, A.; Byrne, F.L.; Hoehn, K.L.; Counter, C.M.; Kashatus, D.F. Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol. Cell 2015, 57, 537–551. [Google Scholar] [CrossRef]
- Serasinghe, M.N.; Wieder, S.Y.; Renault, T.T.; Elkholi, R.; Asciolla, J.J.; Yao, J.L.; Jabado, O.; Hoehn, K.; Kageyama, Y.; Sesaki, H.; et al. Mitochondrial division is requisite to RAS-induced transformation and targeted by oncogenic MAPK pathway inhibitors. Mol. Cell 2015, 57, 521–536. [Google Scholar] [CrossRef] [PubMed]
- Wieder, S.Y.; Serasinghe, M.N.; Sung, J.C.; Choi, D.C.; Birge, M.B.; Yao, J.L.; Bernstein, E.; Celebi, J.T.; Chipuk, J.E. Activation of the Mitochondrial Fragmentation Protein DRP1 Correlates with BRAF(V600E) Melanoma. J. Investig. Dermatol. 2015, 135, 2544–2547. [Google Scholar] [CrossRef]
- Trotta, A.P.; Gelles, J.D.; Serasinghe, M.N.; Loi, P.; Arbiser, J.L.; Chipuk, J.E. Disruption of mitochondrial electron transport chain function potentiates the pro-apoptotic effects of MAPK inhibition. J. Biol. Chem. 2017, 292, 11727–11739. [Google Scholar] [CrossRef] [PubMed]
- Nagdas, S.; Kashatus, J.A.; Nascimento, A.; Hussain, S.S.; Trainor, R.E.; Pollock, S.R.; Adair, S.J.; Michaels, A.D.; Sesaki, H.; Stelow, E.B.; et al. Drp1 Promotes KRas-Driven Metabolic Changes to Drive Pancreatic Tumor Growth. Cell Rep. 2019, 28, 1845–1859 e1845. [Google Scholar] [CrossRef] [PubMed]
- Bryant, K.L.; Stalnecker, C.A.; Zeitouni, D.; Klomp, J.E.; Peng, S.; Tikunov, A.P.; Gunda, V.; Pierobon, M.; Waters, A.M.; George, S.D.; et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med. 2019, 25, 628–640. [Google Scholar] [CrossRef]
- Kinsey, C.G.; Camolotto, S.A.; Boespflug, A.M.; Guillen, K.P.; Foth, M.; Truong, A.; Schuman, S.S.; Shea, J.E.; Seipp, M.T.; Yap, J.T.; et al. Protective autophagy elicited by RAF-->MEK-->ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med. 2019, 25, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Martin, A.; Van den Haute, C.; Cufi, S.; Corominas-Faja, B.; Cuyas, E.; Lopez-Bonet, E.; Rodriguez-Gallego, E.; Fernandez-Arroyo, S.; Joven, J.; Baekelandt, V.; et al. Mitophagy-driven mitochondrial rejuvenation regulates stem cell fate. Aging 2016, 8, 1330–1352. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.T.; Warr, M.R.; Adelman, E.R.; Lansinger, O.M.; Flach, J.; Verovskaya, E.V.; Figueroa, M.E.; Passegue, E. Autophagy maintains the metabolism and function of young and old stem cells. Nature 2017, 543, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Medarde, A.; Santos, E. Ras in cancer and developmental diseases. Genes. Cancer 2011, 2, 344–358. [Google Scholar] [CrossRef] [PubMed]
- Chippalkatti, R.; Abankwa, D. Promotion of cancer cell stemness by Ras. Biochem. Soc. Trans. 2021, 49, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Nyfeler, B.; Eng, C.H. Revisiting autophagy addiction of tumor cells. Autophagy 2016, 12, 1206–1207. [Google Scholar] [CrossRef]
- Guo, J.Y.; Chen, H.Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.; Karantza, V.; et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes. Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef]
- Piffoux, M.; Eriau, E.; Cassier, P.A. Autophagy as a therapeutic target in pancreatic cancer. Br. J. Cancer 2021, 124, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes. Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Rausch, V.; Liu, L.; Apel, A.; Rettig, T.; Gladkich, J.; Labsch, S.; Kallifatidis, G.; Kaczorowski, A.; Groth, A.; Gross, W.; et al. Autophagy mediates survival of pancreatic tumour-initiating cells in a hypoxic microenvironment. J. Pathol. 2012, 227, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeldt, M.T.; O’Prey, J.; Morton, J.P.; Nixon, C.; MacKay, G.; Mrowinska, A.; Au, A.; Rai, T.S.; Zheng, L.; Ridgway, R.; et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature 2013, 504, 296–300. [Google Scholar] [CrossRef]
- Guo, J.Y.; Karsli-Uzunbas, G.; Mathew, R.; Aisner, S.C.; Kamphorst, J.J.; Strohecker, A.M.; Chen, G.; Price, S.; Lu, W.; Teng, X.; et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes. Dev. 2013, 27, 1447–1461. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, H.; Yuan, M.; Fan, H.; Cai, Z. Role of AMPK in autophagy. Front. Physiol. 2022, 13, 1015500. [Google Scholar] [CrossRef]
- Zhang, R.; Kang, R.; Klionsky, D.J.; Tang, D. Ion channels and transporters in autophagy. Autophagy 2022, 18, 4–23. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Sadria, M.; Seo, D.; Layton, A.T. The mixed blessing of AMPK signaling in Cancer treatments. BMC Cancer 2022, 22, 105. [Google Scholar] [CrossRef]
- Chen, C.; Gao, H.; Su, X. Autophagy-related signaling pathways are involved in cancer. Exp. Ther. Med. 2021, 22, 1–13. [Google Scholar] [CrossRef]
- Zou, L.; Liao, M.; Zhen, Y.; Zhu, S.; Chen, X.; Zhang, J.; Hao, Y.; Liu, B. Autophagy and beyond: Unraveling the complexity of UNC-51-like kinase 1 (ULK1) from biological functions to therapeutic implications. Acta Pharm. Sin. B 2022, 12, 3743–3782. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.J.; Gubas, A.; Tooze, S.A. A molecular perspective of mammalian autophagosome biogenesis. J. Biol. Chem. 2018, 293, 5386–5395. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhang, L.; Ma, A.; Liu, L.; Li, J.; Gu, J.; Liu, Y. Transient mTOR inhibition facilitates continuous growth of liver tumors by modulating the maintenance of CD133+ cell populations. PLoS ONE 2011, 6, e28405. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.B.; Liu, Y.; Wang, G.H.; Xu, X.; Cai, Y.; Wang, H.Y.; Li, Y.Q.; Meng, H.F.; Dai, F.; Jin, J.D. Mesenchymal stem cells promote colorectal cancer progression through AMPK/mTOR-mediated NF-kappaB activation. Sci. Rep. 2016, 6, 21420. [Google Scholar] [CrossRef]
- Matsubara, S.; Ding, Q.; Miyazaki, Y.; Kuwahata, T.; Tsukasa, K.; Takao, S. mTOR plays critical roles in pancreatic cancer stem cells through specific and stemness-related functions. Sci. Rep. 2013, 3, 3230. [Google Scholar] [CrossRef] [PubMed]
- Hibdon, E.S.; Razumilava, N.; Keeley, T.M.; Wong, G.; Solanki, S.; Shah, Y.M.; Samuelson, L.C. Notch and mTOR Signaling Pathways Promote Human Gastric Cancer Cell Proliferation. Neoplasia 2019, 21, 702–712. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Matsui, A.; Akabane, S.; Tamura, Y.; Hatano, A.; Miyano, Y.; Omote, H.; Kajikawa, M.; Maenaka, K.; Moriyama, Y. The mitochondrial inner membrane protein LETM1 modulates cristae organization through its LETM domain. Commun. Biol. 2020, 3, 99. [Google Scholar] [CrossRef]
- Che, N.; Yang, Z.; Liu, X.; Li, M.; Feng, Y.; Zhang, C.; Li, C.; Cui, Y.; Xuan, Y. Suppression of LETM1 inhibits the proliferation and stemness of colorectal cancer cells through reactive oxygen species–induced autophagy. J. Cell. Mol. Med. 2021, 25, 2110–2120. [Google Scholar] [CrossRef]
- Xu, D.Q.; Wang, Z.; Wang, C.Y.; Zhang, D.Y.; Wan, H.D.; Zhao, Z.L.; Gu, J.; Zhang, Y.X.; Li, Z.G.; Man, K.Y. PAQR 3 controls autophagy by integrating AMPK signaling to enhance ATG 14L-associated PI 3K activity. EMBO J. 2016, 35, 496–514. [Google Scholar] [CrossRef]
- Weerasekara, V.K.; Panek, D.J.; Broadbent, D.G.; Mortenson, J.B.; Mathis, A.D.; Logan, G.N.; Prince, J.T.; Thomson, D.M.; Thompson, J.W.; Andersen, J.L. Metabolic-stress-induced rearrangement of the 14-3-3ζ interactome promotes autophagy via a ULK1-and AMPK-regulated 14-3-3ζ interaction with phosphorylated Atg9. Mol. Cell. Biol. 2014, 34, 4379–4388. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wang, W.; Sun, X.; Xu, D.; Wang, C.; Zhang, Q.; Wang, H.; Luo, W.; Chen, Y.; Chen, H. AMPK regulates autophagy by phosphorylating BECN1 at threonine 388. Autophagy 2016, 12, 1447–1459. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, Q.; Qiu, G.; Zhou, S.; Jing, Z.; Wang, J.; Wang, W.; Cao, J.; Han, K.; Cheng, Q. RACK1 promotes autophagy by enhancing the Atg14L-Beclin 1-Vps34-Vps15 complex formation upon phosphorylation by AMPK. Cell Rep. 2015, 13, 1407–1417. [Google Scholar] [CrossRef] [PubMed]
- Gan, B.; Hu, J.; Jiang, S.; Liu, Y.; Sahin, E.; Zhuang, L.; Fletcher-Sananikone, E.; Colla, S.; Wang, Y.A.; Chin, L.; et al. Lkb1 regulates quiescence and metabolic homeostasis of haematopoietic stem cells. Nature 2010, 468, 701–704. [Google Scholar] [CrossRef] [PubMed]
- Von Eyss, B.; Jaenicke, L.A.; Kortlever, R.M.; Royla, N.; Wiese, K.E.; Letschert, S.; McDuffus, L.A.; Sauer, M.; Rosenwald, A.; Evan, G.I.; et al. A MYC-Driven Change in Mitochondrial Dynamics Limits YAP/TAZ Function in Mammary Epithelial Cells and Breast Cancer. Cancer Cell 2015, 28, 743–757. [Google Scholar] [CrossRef] [PubMed]
- Loffler, A.S.; Alers, S.; Dieterle, A.M.; Keppeler, H.; Franz-Wachtel, M.; Kundu, M.; Campbell, D.G.; Wesselborg, S.; Alessi, D.R.; Stork, B. Ulk1-mediated phosphorylation of AMPK constitutes a negative regulatory feedback loop. Autophagy 2011, 7, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005, 330, 1304–1305. [Google Scholar] [CrossRef] [PubMed]
- Basen-Engquist, K.; Chang, M. Obesity and cancer risk: Recent review and evidence. Curr. Oncol. Rep. 2011, 13, 71–76. [Google Scholar] [CrossRef]
- Pierotti, M.A.; Berrino, F.; Gariboldi, M.; Melani, C.; Mogavero, A.; Negri, T.; Pasanisi, P.; Pilotti, S. Targeting metabolism for cancer treatment and prevention: Metformin, an old drug with multi-faceted effects. Oncogene 2013, 32, 1475–1487. [Google Scholar] [CrossRef]
- Saini, N.; Yang, X. Metformin as an anti-cancer agent: Actions and mechanisms targeting cancer stem cells. Acta Biochim. Biophys. Sin. 2018, 50, 133–143. [Google Scholar] [CrossRef]
- Choi, Y.K.; Park, K.G. Metabolic roles of AMPK and metformin in cancer cells. Mol. Cells 2013, 36, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Chen, J.; Yi, G.; Deng, M.; Liu, H.; Liang, M.; Shi, B.; Fu, X.; Chen, Y.; Chen, L.; et al. Metformin suppresses hypoxia-induced stabilization of HIF-1alpha through reprogramming of oxygen metabolism in hepatocellular carcinoma. Oncotarget 2016, 7, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Corominas-Faja, B.; Quirantes-Pine, R.; Oliveras-Ferraros, C.; Vazquez-Martin, A.; Cufi, S.; Martin-Castillo, B.; Micol, V.; Joven, J.; Segura-Carretero, A.; Menendez, J.A. Metabolomic fingerprint reveals that metformin impairs one-carbon metabolism in a manner similar to the antifolate class of chemotherapy drugs. Aging 2012, 4, 480–498. [Google Scholar] [CrossRef] [PubMed]
- Sinnett-Smith, J.; Kisfalvi, K.; Kui, R.; Rozengurt, E. Metformin inhibition of mTORC1 activation, DNA synthesis and proliferation in pancreatic cancer cells: Dependence on glucose concentration and role of AMPK. Biochem. Biophys. Res. Commun. 2013, 430, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Kim, D.; Kee, S.H. Metformin-activated AMPK regulates beta-catenin to reduce cell proliferation in colon carcinoma RKO cells. Oncol. Lett. 2019, 17, 2695–2702. [Google Scholar] [CrossRef] [PubMed]
- Kalender, A.; Selvaraj, A.; Kim, S.Y.; Gulati, P.; Brule, S.; Viollet, B.; Kemp, B.E.; Bardeesy, N.; Dennis, P.; Schlager, J.J.; et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. 2010, 11, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Kaufmann, A.M. The Significance of Cancer Stem Cells and Epithelial-Mesenchymal Transition in Metastasis and Anti-Cancer Therapy. Int. J. Mol. Sci. 2023, 24, 2555. [Google Scholar] [CrossRef] [PubMed]
- Koeck, S.; Amann, A.; Huber, J.M.; Gamerith, G.; Hilbe, W.; Zwierzina, H. The impact of metformin and salinomycin on transforming growth factor beta-induced epithelial-to-mesenchymal transition in non-small cell lung cancer cell lines. Oncol. Lett. 2016, 11, 2946–2952. [Google Scholar] [CrossRef]
- Kwan, H.T.; Chan, D.W.; Cai, P.C.; Mak, C.S.; Yung, M.M.; Leung, T.H.; Wong, O.G.; Cheung, A.N.; Ngan, H.Y. AMPK activators suppress cervical cancer cell growth through inhibition of DVL3 mediated Wnt/beta-catenin signaling activity. PLoS ONE 2013, 8, e53597. [Google Scholar] [CrossRef]
- Vazquez-Martin, A.; Oliveras-Ferraros, C.; Cufi, S.; Del Barco, S.; Martin-Castillo, B.; Menendez, J.A. Metformin regulates breast cancer stem cell ontogeny by transcriptional regulation of the epithelial-mesenchymal transition (EMT) status. Cell Cycle 2010, 9, 3807–3814. [Google Scholar] [CrossRef]
- Hoesel, B.; Schmid, J.A. The complexity of NF-kappaB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, H.A.; Iliopoulos, D.; Struhl, K. Metformin inhibits the inflammatory response associated with cellular transformation and cancer stem cell growth. Proc. Natl. Acad. Sci. USA 2013, 110, 972–977. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.; Kim, J.; Park, S.J.; Park, J.J.; Cheon, J.H.; Kim, W.H.; Kim, T.I. Metformin Suppresses Cancer Stem Cells through AMPK Activation and Inhibition of Protein Prenylation of the Mevalonate Pathway in Colorectal Cancer. Cancers 2020, 12, 2554. [Google Scholar] [CrossRef] [PubMed]
- Suissa, S.; Azoulay, L. Response to Bodmer et al. Metformin and the risk of cancer: Time-related biases in observational studies. Diabetes Care 2013, 36, e86. [Google Scholar] [CrossRef] [PubMed]
- Sharif, T.; Martell, E.; Dai, C.; Kennedy, B.E.; Murphy, P.; Clements, D.R.; Kim, Y.; Lee, P.W.; Gujar, S.A. Autophagic homeostasis is required for the pluripotency of cancer stem cells. Autophagy 2017, 13, 264–284. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Roy, S.; Lazar, A.J.; Wang, W.L.; McAuliffe, J.C.; Reynoso, D.; McMahon, J.; Taguchi, T.; Floris, G.; Debiec-Rychter, M.; et al. Autophagy inhibition and antimalarials promote cell death in gastrointestinal stromal tumor (GIST). Proc. Natl. Acad. Sci. USA 2010, 107, 14333–14338. [Google Scholar] [CrossRef] [PubMed]
- Pagotto, A.; Pilotto, G.; Mazzoldi, E.L.; Nicoletto, M.O.; Frezzini, S.; Pasto, A.; Amadori, A. Autophagy inhibition reduces chemoresistance and tumorigenic potential of human ovarian cancer stem cells. Cell Death Dis. 2017, 8, e2943. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hu, S.B.; Wang, L.Y.; Zhang, X.; Zhou, X.; Yang, B.; Li, J.H.; Xiong, J.; Liu, N.; Li, Y.; et al. Autophagy-dependent generation of Axin2+ cancer stem-like cells promotes hepatocarcinogenesis in liver cirrhosis. Oncogene 2017, 36, 6725–6737. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Song, J.; Liu, Z.; Pan, L.; Xu, G. Autophagy activation promotes bevacizumab resistance in glioblastoma by suppressing Akt/mTOR signaling pathway. Oncol. Lett. 2018, 15, 1487–1494. [Google Scholar] [CrossRef]
- De Francesco, E.M.; Bonuccelli, G.; Maggiolini, M.; Sotgia, F.; Lisanti, M.P. Vitamin C and Doxycycline: A synthetic lethal combination therapy targeting metabolic flexibility in cancer stem cells (CSCs). Oncotarget 2017, 8, 67269–67286. [Google Scholar] [CrossRef]
- Yue, W.; Hamai, A.; Tonelli, G.; Bauvy, C.; Nicolas, V.; Tharinger, H.; Codogno, P.; Mehrpour, M. Inhibition of the autophagic flux by salinomycin in breast cancer stem-like/progenitor cells interferes with their maintenance. Autophagy 2013, 9, 714–729. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Shen, S.; Zhang, Y.J.; Xu, C.F.; Cao, Z.T.; Wen, L.P.; Wang, J. Nanoparticle-facilitated autophagy inhibition promotes the efficacy of chemotherapeutics against breast cancer stem cells. Biomaterials 2016, 103, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.H.; Choi, D.S.; Ensor, J.E.; Kaipparettu, B.A.; Bass, B.L.; Chang, J.C. The autophagy inhibitor chloroquine targets cancer stem cells in triple negative breast cancer by inducing mitochondrial damage and impairing DNA break repair. Cancer Lett. 2016, 376, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Kim, C.K.; Alvarez, A.A.; Pangeni, R.P.; Wan, X.; Song, X.; Shi, T.; Yang, Y.; Sastry, N.; Horbinski, C.M.; et al. MST4 Phosphorylation of ATG4B Regulates Autophagic Activity, Tumorigenicity, and Radioresistance in Glioblastoma. Cancer Cell 2017, 32, 840–855 e848. [Google Scholar] [CrossRef]
- Wolf, R.; Wolf, D.; Ruocco, V. Antimalarials: Unapproved uses or indications. Clin. Dermatol. 2000, 18, 17–35. [Google Scholar] [CrossRef]
- Solomon, V.R.; Lee, H. Chloroquine and its analogs: A new promise of an old drug for effective and safe cancer therapies. Eur. J. Pharmacol. 2009, 625, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, D.; Mita, M.; Sarantopoulos, J.; Wood, L.; Amaravadi, R.K.; Davis, L.E.; Mita, A.C.; Curiel, T.J.; Espitia, C.M.; Nawrocki, S.T.; et al. Combined autophagy and HDAC inhibition: A phase I safety, tolerability, pharmacokinetic, and pharmacodynamic analysis of hydroxychloroquine in combination with the HDAC inhibitor vorinostat in patients with advanced solid tumors. Autophagy 2014, 10, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Rangwala, R.; Leone, R.; Chang, Y.C.; Fecher, L.A.; Schuchter, L.M.; Kramer, A.; Tan, K.S.; Heitjan, D.F.; Rodgers, G.; Gallagher, M.; et al. Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1369–1379. [Google Scholar] [CrossRef] [PubMed]
- Vogl, D.T.; Stadtmauer, E.A.; Tan, K.S.; Heitjan, D.F.; Davis, L.E.; Pontiggia, L.; Rangwala, R.; Piao, S.; Chang, Y.C.; Scott, E.C.; et al. Combined autophagy and proteasome inhibition: A phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy 2014, 10, 1380–1390. [Google Scholar] [CrossRef]
- Wolpin, B.M.; Rubinson, D.A.; Wang, X.; Chan, J.A.; Cleary, J.M.; Enzinger, P.C.; Fuchs, C.S.; McCleary, N.J.; Meyerhardt, J.A.; Ng, K.; et al. Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. Oncologist 2014, 19, 637–638. [Google Scholar] [CrossRef]
- Sadria, M.; Layton, A.T. Interactions among mTORC, AMPK and SIRT: A computational model for cell energy balance and metabolism. Cell Commun. Signal 2021, 19, 57. [Google Scholar] [CrossRef] [PubMed]
- Pineda, C.T.; Ramanathan, S.; Fon Tacer, K.; Weon, J.L.; Potts, M.B.; Ou, Y.H.; White, M.A.; Potts, P.R. Degradation of AMPK by a cancer-specific ubiquitin ligase. Cell 2015, 160, 715–728. [Google Scholar] [CrossRef] [PubMed]
- Pavlinov, I.; Salkovski, M.; Aldrich, L.N. Beclin 1-ATG14L Protein-Protein Interaction Inhibitor Selectively Inhibits Autophagy through Disruption of VPS34 Complex I. J. Am. Chem. Soc. 2020, 142, 8174–8182. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.P.; Uddin, M.N.; Chaudhari, R.; Sutton, M.N.; Yang, H.; Rask, P.; Locke, H.; Engel, B.J.; Batistatou, N.; Wang, J.; et al. Directed evolution of cyclic peptides for inhibition of autophagy. Chem. Sci. 2021, 12, 3526–3543. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chen, W.Q.; Zhang, S.Q.; Bai, J.X.; Lau, C.L.; Sze, S.C.; Yung, K.K.; Ko, J.K. The human cathelicidin peptide LL-37 inhibits pancreatic cancer growth by suppressing autophagy and reprogramming of the tumor immune microenvironment. Front. Pharmacol. 2022, 13, 906625. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Shi, K.; Sabet, Z.F.; Fu, W.; Zhou, H.; Xu, S.; Liu, T.; You, M.; Cao, M.; Xu, M.; et al. New power of self-assembling carbonic anhydrase inhibitor: Short peptide-constructed nanofibers inspire hypoxic cancer therapy. Sci. Adv. 2019, 5, eaax0937. [Google Scholar] [CrossRef]
- Mai, T.T.; Moon, J.; Song, Y.; Viet, P.Q.; Phuc, P.V.; Lee, J.M.; Yi, T.H.; Cho, M.; Cho, S.K. Ginsenoside F2 induces apoptosis accompanied by protective autophagy in breast cancer stem cells. Cancer Lett. 2012, 321, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Chang, H.; Peng, X.; Bai, Q.; Yi, L.; Zhou, Y.; Zhu, J.; Mi, M. Resveratrol inhibits breast cancer stem-like cells and induces autophagy via suppressing Wnt/beta-catenin signaling pathway. PLoS ONE 2014, 9, e102535. [Google Scholar] [CrossRef]
- Patel, V.B.; Misra, S.; Patel, B.B.; Majumdar, A.P. Colorectal cancer: Chemopreventive role of curcumin and resveratrol. Nutr. Cancer 2010, 62, 958–967. [Google Scholar] [CrossRef]
- Filippi-Chiela, E.C.; Villodre, E.S.; Zamin, L.L.; Lenz, G. Autophagy interplay with apoptosis and cell cycle regulation in the growth inhibiting effect of resveratrol in glioma cells. PLoS ONE 2011, 6, e20849. [Google Scholar] [CrossRef]
- Miki, H.; Uehara, N.; Kimura, A.; Sasaki, T.; Yuri, T.; Yoshizawa, K.; Tsubura, A. Resveratrol induces apoptosis via ROS-triggered autophagy in human colon cancer cells. Int. J. Oncol. 2012, 40, 1020–1028. [Google Scholar] [CrossRef] [PubMed]
- Puissant, A.; Auberger, P. AMPK- and p62/SQSTM1-dependent autophagy mediate resveratrol-induced cell death in chronic myelogenous leukemia. Autophagy 2010, 6, 655–657. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Shankar, S.; Srivastava, R.K. Rottlerin induces autophagy and apoptosis in prostate cancer stem cells via PI3K/Akt/mTOR signaling pathway. Cancer Lett. 2014, 343, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Maioli, E.; Daveri, E.; Maellaro, E.; Ietta, F.; Cresti, L.; Valacchi, G. Non-conventional rottlerin anticancer properties. Arch. Biochem. Biophys. 2018, 645, 50–53. [Google Scholar] [CrossRef]
- Tang, D.; Yao, R.; Zhao, D.; Zhou, L.; Wu, Y.; Yang, Y.; Sun, Y.; Lu, L.; Gao, W. Trichostatin A reverses the chemoresistance of lung cancer with high IGFBP2 expression through enhancing autophagy. Sci. Rep. 2018, 8, 3917. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Cancer | Role of Autophagy | Ref. |
|---|---|---|
| Pancreatic cancer |
| [68] |
| Liver cancer |
| [69] |
| Osteosarcoma |
| [70] |
| Ovarian cancer |
| [71] |
| Glioblastoma |
| [72,90] |
| Breast cancer |
| [64] |
| Bladder carcinoma |
| [109] |
| Colorectal cancer |
| [91,110] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovale, L.; Singh, M.K.; Kim, J.; Ha, J. Role of Autophagy and AMPK in Cancer Stem Cells: Therapeutic Opportunities and Obstacles in Cancer. Int. J. Mol. Sci. 2024, 25, 8647. https://doi.org/10.3390/ijms25168647
Kovale L, Singh MK, Kim J, Ha J. Role of Autophagy and AMPK in Cancer Stem Cells: Therapeutic Opportunities and Obstacles in Cancer. International Journal of Molecular Sciences. 2024; 25(16):8647. https://doi.org/10.3390/ijms25168647
Chicago/Turabian StyleKovale, Lochana, Manish Kumar Singh, Joungmok Kim, and Joohun Ha. 2024. "Role of Autophagy and AMPK in Cancer Stem Cells: Therapeutic Opportunities and Obstacles in Cancer" International Journal of Molecular Sciences 25, no. 16: 8647. https://doi.org/10.3390/ijms25168647