
Transcription-Associated Metabolomic Analysis Reveals the Mechanism of Fruit Ripening during the Development of Chinese Bayberry
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
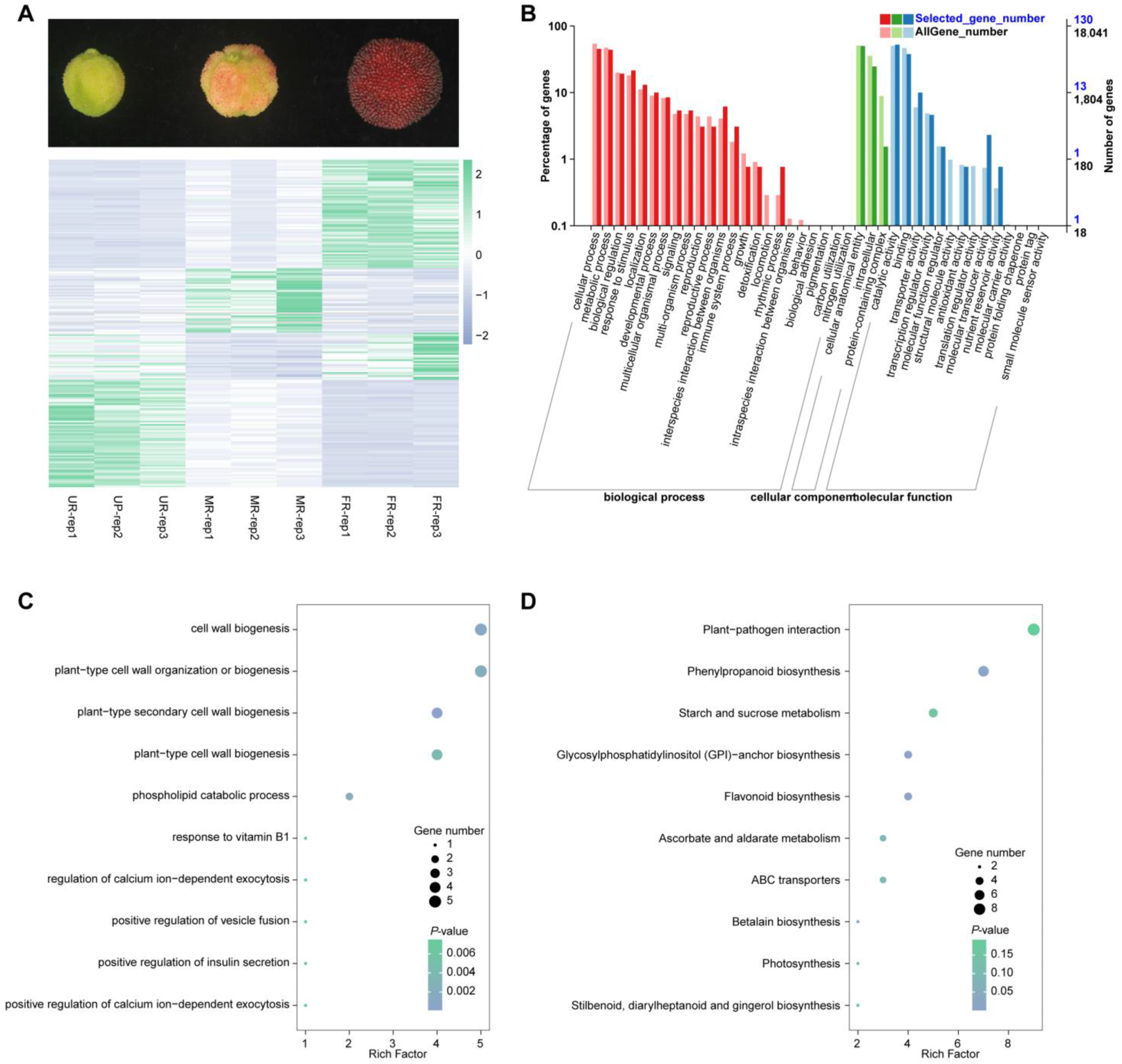
2.1. Transcriptomic Analysis and Differentially Expressed Genes
2.2. Metabolome Analysis and Differential Metabolites
2.3. Time Course of Metabolome
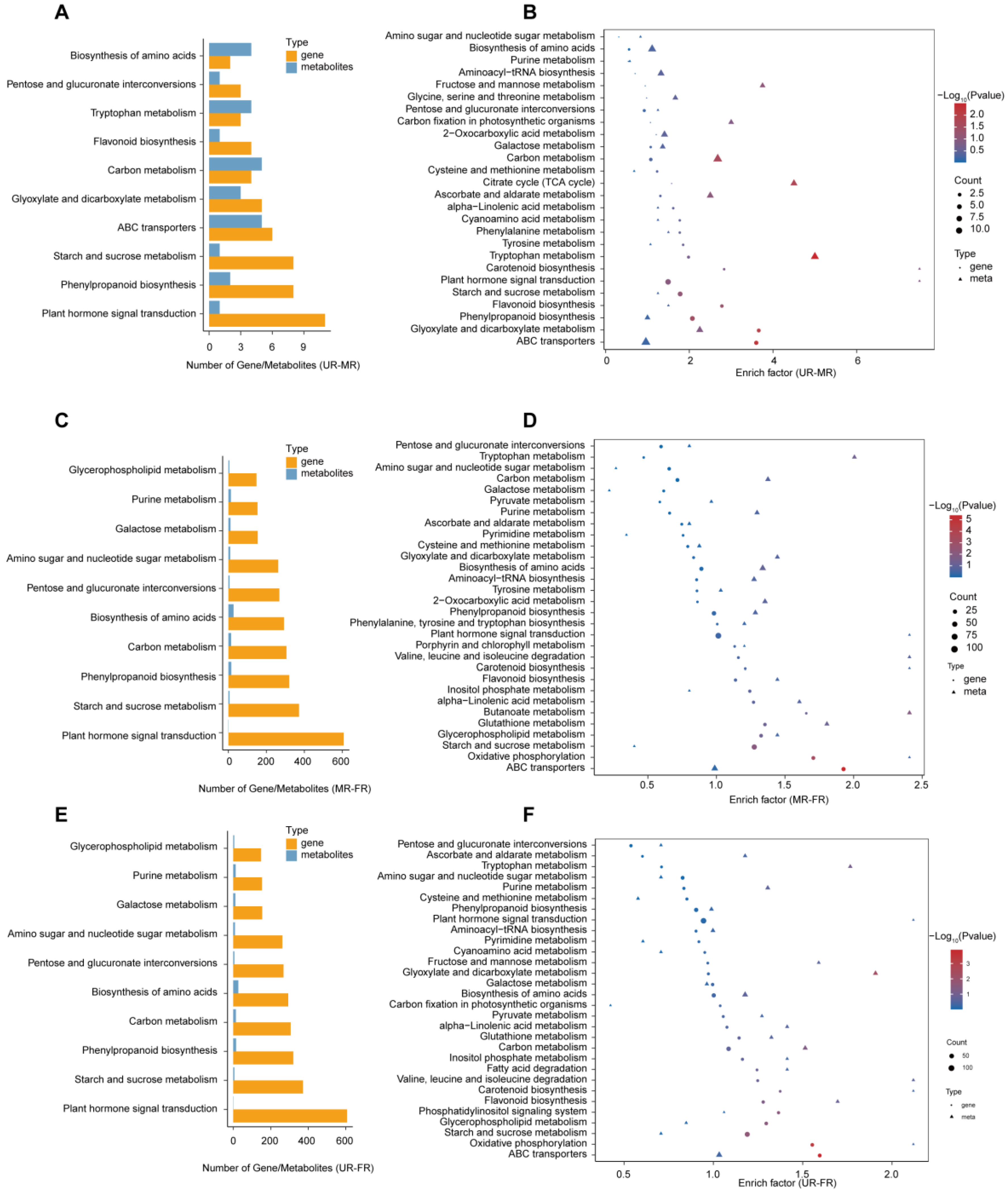
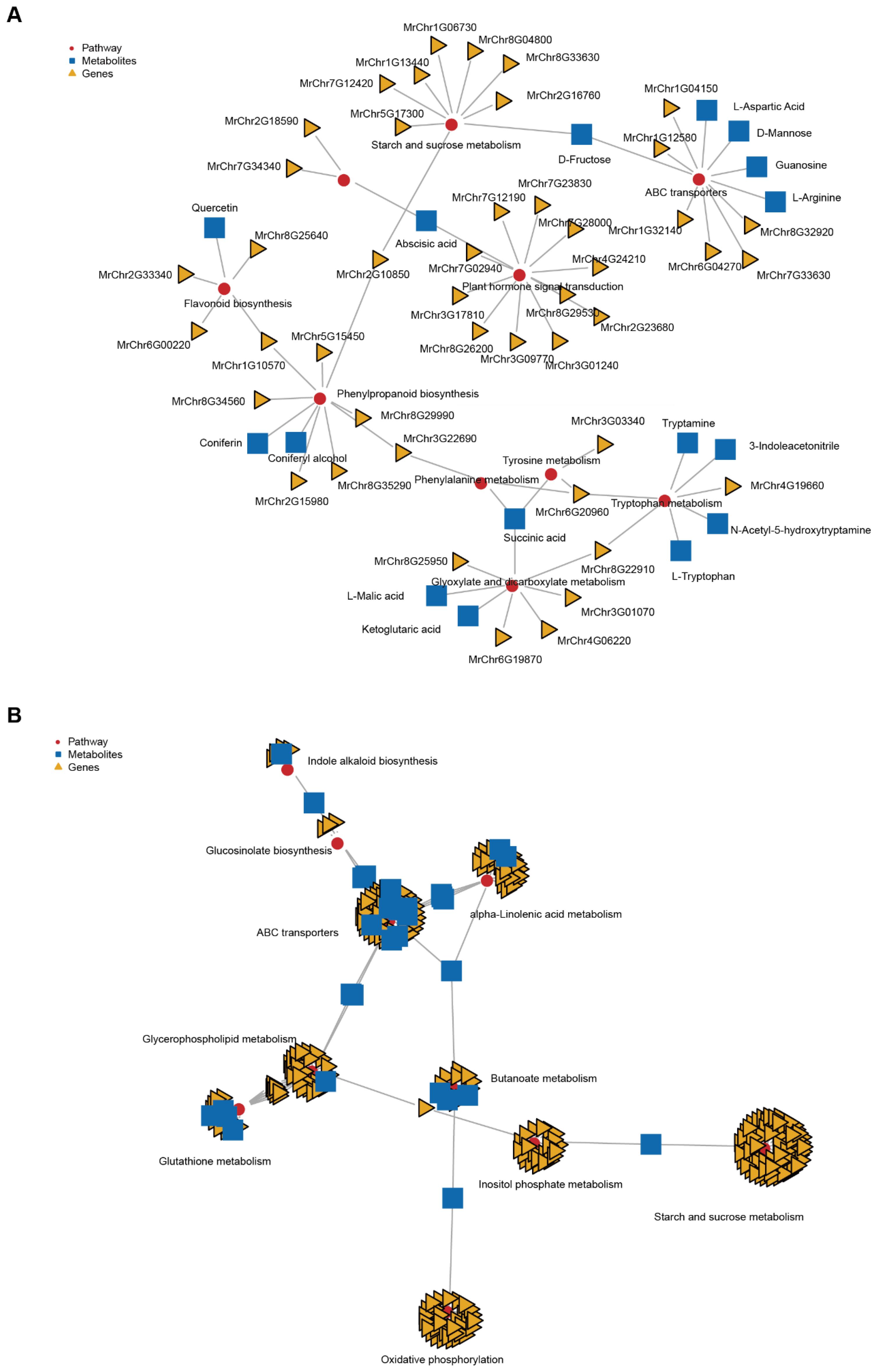
2.4. Conjoint Analysis of Transcriptome and Metabolome Data
2.5. Flavonoid Biosynthesis Changes during Fruit Ripening
2.6. Starch and Sucrose Metabolism Changes during Fruit Ripening
3. Discussion
4. Material and Methods
4.1. Plant Material
4.2. RNA Sequencing
4.3. Analysis of RNA Sequencing Data
4.4. Metabolite Extraction
4.5. LC-MS/MS Analysis
4.6. Data Preprocessing and Annotation
4.7. Metabolome Analysis
4.8. Quantitative Real-Time PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, Z.; Wu, X.; Liu, H.; Zhang, M.; Liao, W. DNA methylation in tomato fruit ripening. Physiol. Plant 2022, 174, e13627. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, L.; Simkin, A.J.; George Priya Doss, C.; Siva, R. Fruit ripening: Dynamics and integrated analysis of carotenoids and anthocyanins. BMC Plant Biol. 2022, 22, 27. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.I.; Hwang, I.; Goswami, G.; Jung, H.J.; Nath, U.K.; Yoo, H.J.; Lee, J.M.; Nou, I.S. Molecular Insights Reveal Psy1, SGR, and SlMYB12 Genes are Associated with Diverse Fruit Color Pigments in Tomato (Solanum lycopersicum L.). Molecules 2017, 22, 2180. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Wang, F.; Lian, Y.; Xiao, H.; Zheng, J. Biosynthesis of citrus flavonoids and their health effects. Crit. Rev. Food Sci. Nutr. 2020, 60, 566–583. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Xu, X.; Hu, X.; Liu, Y.; Cao, H.; Chan, H.; Gong, Z.; Yuan, Y.; Luo, Y.; Feng, B.; et al. SlMYB72 Regulates the Metabolism of Chlorophylls, Carotenoids, and Flavonoids in Tomato Fruit. Plant Physiol. 2020, 183, 854–868. [Google Scholar] [CrossRef] [PubMed]
- Serafini, M.; Peluso, I.; Raguzzini, A. Flavonoids as anti-inflammatory agents. Proc. Nutr. Soc. 2010, 69, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Calis, Z.; Mogulkoc, R.; Baltaci, A.K. The Roles of Flavonols/Flavonoids in Neurodegeneration and Neuroinflammation. Mini Rev. Med. Chem. 2020, 20, 1475–1488. [Google Scholar] [CrossRef]
- Romagnolo, D.F.; Selmin, O.I. Flavonoids and cancer prevention: A review of the evidence. J. Nutr. Gerontol. Geriatr. 2012, 31, 206–238. [Google Scholar] [CrossRef]
- Hertog, M.G.; Feskens, E.J.; Hollman, P.C.; Katan, M.B.; Kromhout, D. Dietary antioxidant flavonoids and risk of coronary heart disease: The Zutphen Elderly Study. Lancet 1993, 342, 1007–1011. [Google Scholar] [CrossRef]
- Hertog, M.G.; Kromhout, D.; Aravanis, C.; Blackburn, H.; Buzina, R.; Fidanza, F.; Giampaoli, S.; Jansen, A.; Menotti, A.; Nedeljkovic, S.; et al. Flavonoid intake and long-term risk of coronary heart disease and cancer in the seven countries study. Arch. Intern. Med. 1995, 155, 381–386. [Google Scholar] [CrossRef]
- Samkumar, A.; Karppinen, K.; McGhie, T.K.; Espley, R.V.; Martinussen, I.; Jaakola, L. Flavonoid biosynthesis is differentially altered in detached and attached ripening bilberries in response to spectral light quality. Front. Plant Sci. 2022, 13, 969934. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; Hou, N.; Xv, X.; Zhang, D.; Fan, T.Q.; Zhang, Q.X.; Huang, Y.J. Flavonoid Synthesis and Metabolism During the Fruit Development in Hickory (Carya cathayensis). Front. Plant Sci. 2022, 13, 896421. [Google Scholar] [CrossRef]
- Morales-Quintana, L.; Ramos, P. A Talk between Flavonoids and Hormones to Reorient the Growth of Gymnosperms. Int. J. Mol. Sci. 2021, 22, 12630. [Google Scholar] [CrossRef]
- Singh, K.; Kumar, A.; Kajal, M.; Singh, B. Characterization and expression analysis of chalcone synthase and chalcone isomerase genes in Phyllanthus emblica (L.). J. Plant Biochem. Biotechnol. 2019, 28, 105–113. [Google Scholar] [CrossRef]
- Muir, S.R.; Collins, G.J.; Robinson, S.; Hughes, S.; Bovy, A.; Ric De Vos, C.H.; van Tunen, A.J.; Verhoeyen, M.E. Overexpression of petunia chalcone isomerase in tomato results in fruit containing increased levels of flavonols. Nat. Biotechnol. 2001, 19, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Moyano, E.; Portero-Robles, I.; Medina-Escobar, N.; Valpuesta, V.; Muñoz-Blanco, J.; Caballero, J.L. A fruit-specific putative dihydroflavonol 4-reductase gene is differentially expressed in strawberry during the ripening process. Plant Physiol. 1998, 117, 711–716. [Google Scholar] [CrossRef]
- Singh, P.; Arif, Y.; Bajguz, A.; Hayat, S. The role of quercetin in plants. Plant Physiol. Biochem. 2021, 166, 10–19. [Google Scholar] [CrossRef]
- Wang, R.; Shu, P.; Zhang, C.; Zhang, J.; Chen, Y.; Zhang, Y.; Du, K.; Xie, Y.; Li, M.; Ma, T.; et al. Integrative analyses of metabolome and genome-wide transcriptome reveal the regulatory network governing flavor formation in kiwifruit (Actinidia chinensis). New Phytol. 2022, 233, 373–389. [Google Scholar] [CrossRef]
- Wang, S.; Pei, J.; Li, J.; Tang, G.; Zhao, J.; Peng, X.; Nie, S.; Ding, Y.; Wang, C. Sucrose and starch metabolism during Fargesia yunnanensis shoot growth. Physiol. Plant 2020, 168, 188–204. [Google Scholar] [CrossRef]
- Wang, D.; Yeats, T.H.; Uluisik, S.; Rose, J.K.C.; Seymour, G.B. Fruit Softening: Revisiting the Role of Pectin. Trends Plant Sci. 2018, 23, 302–310. [Google Scholar] [CrossRef]
- Shi, L.; Chen, X.; Wang, K.; Yang, M.; Chen, W.; Yang, Z.; Cao, S. MrMYB6 From Chinese Bayberry (Myrica rubra) Negatively Regulates Anthocyanin and Proanthocyanidin Accumulation. Front. Plant Sci. 2021, 12, 685654. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Huang, H.; Xu, C.; Li, X.; Chen, K. Biological activities of extracts from Chinese bayberry (Myrica rubra Sieb. et Zucc.): A review. Plant Foods Hum. Nutr. 2013, 68, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yu, Z.; Sun, L.; Ren, H.; Zheng, X.; Liang, S.; Qi, X. An overview of the nutritional value, health properties, and future challenges of Chinese bayberry. PeerJ 2022, 10, e13070. [Google Scholar] [CrossRef]
- Yao, W.R.; Wang, H.Y.; Wang, S.T.; Sun, S.L.; Zhou, J.; Luan, Y.Y. Assessment of the antibacterial activity and the antidiarrheal function of flavonoids from bayberry fruit. J. Agric. Food Chem. 2011, 59, 5312–5317. [Google Scholar] [CrossRef]
- Wu, D.; Cheng, H.; Chen, J.; Ye, X.; Liu, Y. Characteristics changes of Chinese bayberry (Myrica rubra) during different growth stages. J. Food Sci. Technol. 2019, 56, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Yu, Q.; Zhang, S.; Yu, Z.; Liang, S.; Zheng, X.; Ren, H.; Qi, X. Genome-Wide Identification and Expression Analysis of Beta-Galactosidase Family Members in Chinese Bayberry (Myrica rubra). Horticulturae 2024, 10, 225. [Google Scholar] [CrossRef]
- Xia, H.; Zhang, Z.; Luo, C.; Wei, K.; Li, X.; Mu, X.; Duan, M.; Zhu, C.; Jin, L.; He, X.; et al. MultiPrime: A reliable and efficient tool for targeted next-generation sequencing. iMeta 2023, 2, e143. [Google Scholar] [CrossRef]
- Jiang, Y.; Qi, Y.; Chen, X.; Yan, Q.; Chen, J.; Liu, H.; Shi, F.; Wen, Y.; Cai, C.; Ou, L. Combined Metabolome and Transcriptome Analyses Unveil the Molecular Mechanisms of Fruit Acidity Variation in Litchi (Litchi chinensis Sonn.). Int. J. Mol. Sci. 2023, 24, 1871. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wen, H.; Yang, X.; Wu, C.; Ming, J.; Zhang, H.; Chen, J.; Wang, J.; Xu, J. Metabolome and transcriptome profiling revealed the enhanced synthesis of volatile esters in Korla pear. BMC Plant Biol. 2023, 23, 264. [Google Scholar] [CrossRef]
- Li, X.; Qi, L.; Zang, N.; Zhao, L.; Sun, Y.; Huang, X.; Wang, H.; Yin, Z.; Wang, A. Integrated metabolome and transcriptome analysis of the regulatory network of volatile ester formation during fruit ripening in pear. Plant Physiol. Biochem. 2022, 185, 80–90. [Google Scholar] [CrossRef]
- Lin, Q.; Zhong, Q.; Zhang, Z. A comparative metabolomics study of anthocyanins and taste components in Chinese bayberry (Morella rubra) with different flesh colors. PeerJ 2022, 10, e13466. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; Huang, X.; Zhang, Z.; Lin, Q.; Zhong, Q.; Zhao, Y.; Gao, Z.; Xu, C. An Anthocyanin-Related Glutathione S-Transferase, MrGST1, Plays an Essential Role in Fruit Coloration in Chinese Bayberry (Morella rubra). Front. Plant Sci. 2022, 13, 903333. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Cao, S.; Chen, X.; Chen, W.; Zheng, Y.; Yang, Z. Proanthocyanidin Synthesis in Chinese Bayberry (Myrica rubra Sieb. et Zucc.) Fruits. Front. Plant Sci. 2018, 9, 212. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.; Chen, M.; Xu, C.J.; Bai, L.; Yin, X.R.; Li, X.; Allan, A.C.; Ferguson, I.B.; Chen, K.S. Transcriptomic analysis of Chinese bayberry (Myrica rubra) fruit development and ripening using RNA-Seq. BMC Genom. 2012, 13, 19. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Cao, S.; Shao, J.; Chen, W.; Zheng, Y.; Jiang, Y.; Yang, Z. Relationship between sucrose metabolism and anthocyanin biosynthesis during ripening in Chinese bayberry fruit. J. Agric. Food Chem. 2014, 62, 10522–10528. [Google Scholar] [CrossRef] [PubMed]
- Niu, S.S.; Xu, C.J.; Zhang, W.S.; Zhang, B.; Li, X.; Lin-Wang, K.; Ferguson, I.B.; Allan, A.C.; Chen, K.S. Coordinated regulation of anthocyanin biosynthesis in Chinese bayberry (Myrica rubra) fruit by a R2R3 MYB transcription factor. Planta 2010, 231, 887–899. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Yang, J.; Lu, Q.; Tang, Q.; Chen, S.; Jia, G. The RNA N(6)-methyladenosine demethylase ALKBH9B modulates ABA responses in Arabidopsis. J. Integr. Plant Biol. 2022, 64, 2361–2373. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, L.; Zhang, S.; Yu, Z.; Zheng, X.; Liang, S.; Ren, H.; Qi, X. Transcription-Associated Metabolomic Analysis Reveals the Mechanism of Fruit Ripening during the Development of Chinese Bayberry. Int. J. Mol. Sci. 2024, 25, 8654. https://doi.org/10.3390/ijms25168654
Sun L, Zhang S, Yu Z, Zheng X, Liang S, Ren H, Qi X. Transcription-Associated Metabolomic Analysis Reveals the Mechanism of Fruit Ripening during the Development of Chinese Bayberry. International Journal of Molecular Sciences. 2024; 25(16):8654. https://doi.org/10.3390/ijms25168654
Chicago/Turabian StyleSun, Li, Shuwen Zhang, Zheping Yu, Xiliang Zheng, Senmiao Liang, Haiying Ren, and Xingjiang Qi. 2024. "Transcription-Associated Metabolomic Analysis Reveals the Mechanism of Fruit Ripening during the Development of Chinese Bayberry" International Journal of Molecular Sciences 25, no. 16: 8654. https://doi.org/10.3390/ijms25168654