

Endoplasmic Reticulum Stress Differently Modulates the Release of IL-6 and IL-8 Cytokines in Human Glial Cells

 , ,
, , 
Abstract

1. Introduction
2. Results
2.1. Effect of Tunicamycin on Gene Expression Related to ER Stress in Glial Cells
2.2. Induction of Different Levels of ER Stress in Astrocytes and Microglia
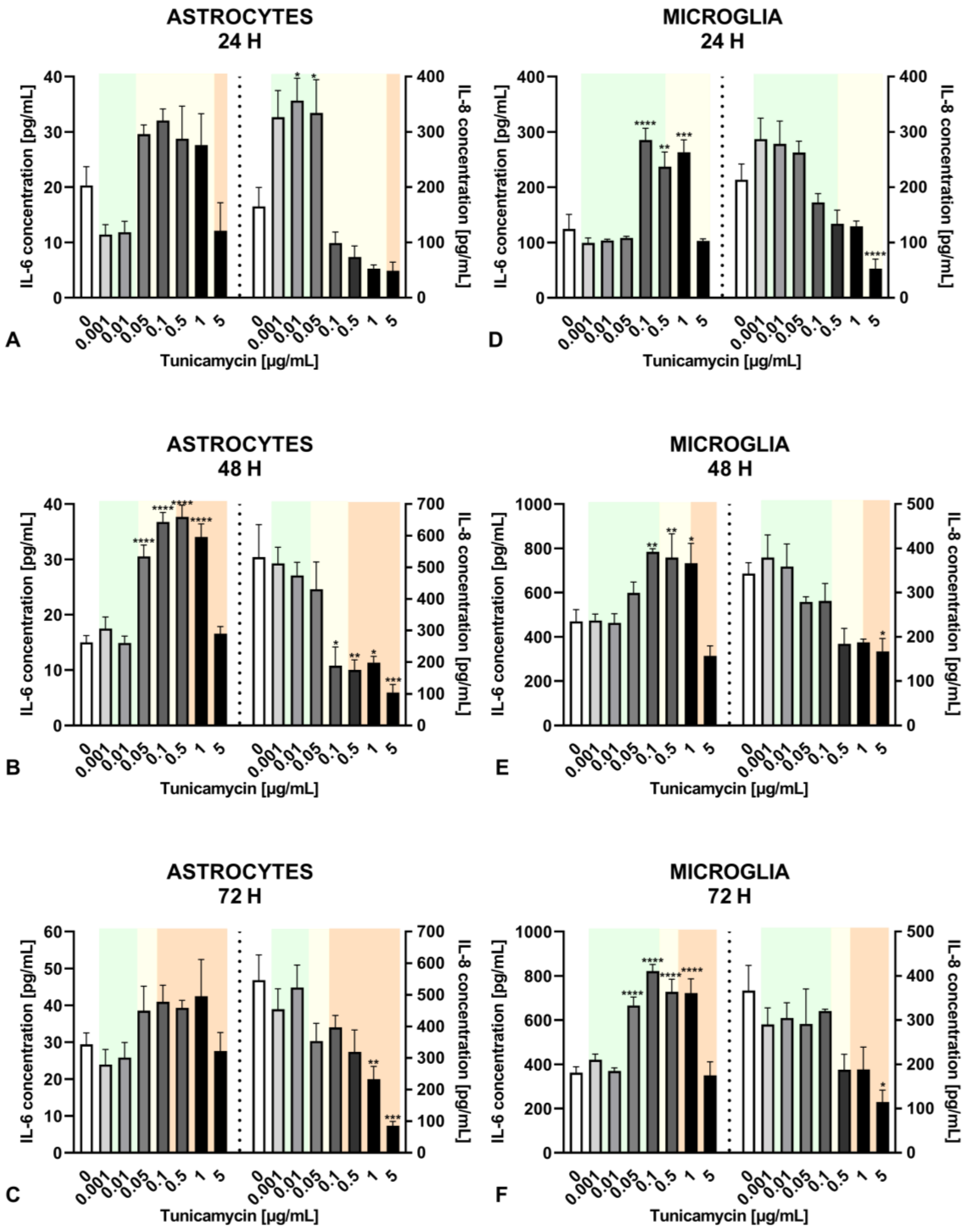
2.3. Impact of Different Levels of ER Stress on Cytokine Release in Astrocytes and Microglia
2.4. Distinct Cytokine Secretion by Astrocytes and Microglia under the Influence of Inflammatory Agents
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Analysis of Gene Expression Related to ER Stress
4.4. Cell Viability
4.5. ELISA Tests
4.6. Data Analysis
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luís, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic Reticulum Stress Signalling—From Basic Mechanisms to Clinical Applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef] [PubMed]
- Merighi, A.; Lossi, L. Endoplasmic Reticulum Stress Signaling and Neuronal Cell Death. Int. J. Mol. Sci. 2022, 23, 15186. [Google Scholar] [CrossRef] [PubMed]
- Read, A.; Schröder, M. The Unfolded Protein Response: An Overview. Biology 2021, 10, 384. [Google Scholar] [CrossRef] [PubMed]
- Oakes, S.A.; Papa, F.R. The Role of Endoplasmic Reticulum Stress in Human Pathology. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 173–194. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhou, Q.; Zhang, X.; Qian, Q.; Xu, J.; Ni, P.; Qian, Y. Mild Endoplasmic Reticulum Stress Ameliorates Lipopolysaccharide-Induced Neuroinflammation and Cognitive Impairment via Regulation of Microglial Polarization. J. Neuroinflamm. 2017, 14, 233. [Google Scholar] [CrossRef]
- Sprenkle, N.T.; Sims, S.G.; Sánchez, C.L.; Meares, G.P. Endoplasmic Reticulum Stress and Inflammation in the Central Nervous System. Mol. Neurodegener. 2017, 12, 42. [Google Scholar] [CrossRef] [PubMed]
- Perner, C.; Krüger, E. Endoplasmic Reticulum Stress and Its Role in Homeostasis and Immunity of Central and Peripheral Neurons. Front. Immunol. 2022, 13, 859703. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Chai, Y.; Zhang, J.; Chen, X. Endoplasmic Reticulum Stress-Associated Neuronal Death and Innate Immune Response in Neurological Diseases. Front. Immunol. 2022, 12, 794580. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-Y.; Wang, Z.; Lu, X.-H.; Kong, X.-X.; Wu, F.-Z.; Lin, L.; Tan, X.; Ye, L.-B.; Xiao, J. Endoplasmic Reticulum Stress: Relevance and Therapeutics in Central Nervous System Diseases. Mol. Neurobiol. 2015, 51, 1343–1352. [Google Scholar] [CrossRef]
- Placido, A.I.; Pereira, C.; Duarte, A.; Candeias, E.; Correia, S.; Carvalho, C.; Cardoso, S.; Oliveira, C.; Moreira, P. Modulation of Endoplasmic Reticulum Stress: An Opportunity to Prevent Neurodegeneration? CNS Neurol. Disord. Drug Targets 2015, 14, 518–533. [Google Scholar] [CrossRef]
- Kim, S.; Kim, D.K.; Jeong, S.; Lee, J. The Common Cellular Events in the Neurodegenerative Diseases and the Associated Role of Endoplasmic Reticulum Stress. Int. J. Mol. Sci. 2022, 23, 5894. [Google Scholar] [CrossRef] [PubMed]
- Stefani, I.C.; Wright, D.; Polizzi, K.M.; Kontoravdi, C. The Role of ER Stress-Induced Apoptosis in Neurodegeneration. Curr. Alzheimer Res. 2012, 9, 373–387. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, A.; Kasahara, T.; Kametani, M.; Toyota, T.; Yoshikawa, T.; Kato, T. Aberrant Endoplasmic Reticulum Stress Response in Lymphoblastoid Cells from Patients with Bipolar Disorder. Int. J. Neuropsychopharm. 2009, 12, 33. [Google Scholar] [CrossRef] [PubMed]
- Bown, C. Increased Temporal Cortex ER Stress Proteins in Depressed Subjects Who Died by Suicide. Neuropsychopharmacology 2000, 22, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Sims, S.G.; Cisney, R.N.; Lipscomb, M.M.; Meares, G.P. The Role of Endoplasmic Reticulum Stress in Astrocytes. Glia 2022, 70, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V. Astrocyte Barriers to Neurotoxic Inflammation. Nat. Rev. Neurosci. 2015, 16, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Priller, J. Microglia and Brain Macrophages in the Molecular Age: From Origin to Neuropsychiatric Disease. Nat. Rev. Neurosci. 2014, 15, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Kempuraj, D.; Thangavel, R.; Natteru, P.A.; Selvakumar, G.P.; Saeed, D.; Zahoor, H.; Zaheer, S.; Iyer, S.S.; Zaheer, A. Neuroinflammation Induces Neurodegeneration. J. Neurol. Neurosurg. Spine 2016, 1, 1003. [Google Scholar]
- Darweesh, S.K.L.; Wolters, F.J.; Ikram, M.A.; de Wolf, F.; Bos, D.; Hofman, A. Inflammatory Markers and the Risk of Dementia and Alzheimer’s Disease: A Meta-analysis. Alzheimer’s Dement. 2018, 14, 1450–1459. [Google Scholar] [CrossRef]
- Lai, K.S.P.; Liu, C.S.; Rau, A.; Lanctôt, K.L.; Köhler, C.A.; Pakosh, M.; Carvalho, A.F.; Herrmann, N. Peripheral Inflammatory Markers in Alzheimer’s Disease: A Systematic Review and Meta-Analysis of 175 Studies. J. Neurol. Neurosurg. Psychiatry 2017, 88, 876–882. [Google Scholar] [CrossRef] [PubMed]
- Custodero, C.; Ciavarella, A.; Panza, F.; Gnocchi, D.; Lenato, G.M.; Lee, J.; Mazzocca, A.; Sabbà, C.; Solfrizzi, V. Role of Inflammatory Markers in the Diagnosis of Vascular Contributions to Cognitive Impairment and Dementia: A Systematic Review and Meta-Analysis. GeroScience 2022, 44, 1373–1392. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Duan, J.; Luo, F.; Tao, P.; Hu, C. IL-6, IL-8 and IL-10 Polymorphisms May Impact Predisposition of Alzheimer’s Disease: A Meta-Analysis. Acta Neurol. Belg. 2021, 121, 1505–1512. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Chen, S.; Liu, J.; Yang, J.; Ou, R.; Zhang, L.; Chen, X.; Shang, H. Serum Inflammatory Cytokines Levels and the Correlation Analyses in Parkinson’s Disease. Front. Cell Dev. Biol. 2023, 11, 1104393. [Google Scholar] [CrossRef]
- Eide, S.; Misztal, M.; Feng, Z.-P. Interleukin-6 as a Marker of Huntington’s Disease Progression: Systematic Review and Meta-Analysis. Brain Behav. Immun.—Health 2023, 30, 100635. [Google Scholar] [CrossRef]
- Qin, B.; Li, L.; Wang, S.; Wu, J.; Huang, Y.; Zhou, P.; Bai, J.; Zheng, Y. Interleukin-8 Gene Polymorphism –251T>A Contributes to Alzheimer’s Disease Susceptibility. Medicine 2016, 95, e5039. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, Y.; Zhao, A.; Luo, N.; Niu, M.; Kang, W.; Xie, A.; Lu, H.; Chen, L.; Liu, J. Parkinson’s Disease Peripheral Immune Biomarker Profile: A Multicentre, Cross-Sectional and Longitudinal Study. J. Neuroinflamm. 2022, 19, 116. [Google Scholar] [CrossRef]
- Dolcetti, E.; Bruno, A.; Azzolini, F.; Gilio, L.; Pavone, L.; Iezzi, E.; Galifi, G.; Gambardella, S.; Ferese, R.; Buttari, F.; et al. Genetic Regulation of IL-8 Influences Disease Presentation of Multiple Sclerosis. Mult. Scler. 2023, 29, 512–520. [Google Scholar] [CrossRef]
- Lund, B.T.; Ashikian, N.; Ta, H.Q.; Chakryan, Y.; Manoukian, K.; Groshen, S.; Gilmore, W.; Cheema, G.S.; Stohl, W.; Burnett, M.E.; et al. Increased CXCL8 (IL-8) Expression in Multiple Sclerosis. J. Neuroimmunol. 2004, 155, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Ichikawa, S. Tunicamycin: Chemical Synthesis and Biosynthesis. J. Antibiot. 2019, 72, 924–933. [Google Scholar] [CrossRef]
- Jóźwiak-Bębenista, M.; Sokołowska, P.; Siatkowska, M.; Panek, C.A.; Komorowski, P.; Kowalczyk, E.; Wiktorowska-Owczarek, A. The Importance of Endoplasmic Reticulum Stress as a Novel Antidepressant Drug Target and Its Potential Impact on CNS Disorders. Pharmaceutics 2022, 14, 846. [Google Scholar] [CrossRef]
- ISO 10993-5:2009; Biological Evaluation of Medical Devices. Part 5: Tests for In Vitro Cytotoxicity. ISO: Geneva, Switzerland, 2009; 34p.
- Erguler, K.; Pieri, M.; Deltas, C. A Mathematical Model of the Unfolded Protein Stress Response Reveals the Decision Mechanism for Recovery, Adaptation and Apoptosis. BMC Syst. Biol. 2013, 7, 16. [Google Scholar] [CrossRef] [PubMed]
- Inagi, R.; Kumagai, T.; Nishi, H.; Kawakami, T.; Miyata, T.; Fujita, T.; Nangaku, M. Preconditioning with Endoplasmic Reticulum Stress Ameliorates Mesangioproliferative Glomerulonephritis. J. Am. Soc. Nephrol. 2008, 19, 915–922. [Google Scholar] [CrossRef] [PubMed]
- Fouillet, A.; Levet, C.; Virgone, A.; Robin, M.; Dourlen, P.; Rieusset, J.; Belaidi, E.; Ovize, M.; Touret, M.; Nataf, S.; et al. ER Stress Inhibits Neuronal Death by Promoting Autophagy. Autophagy 2012, 8, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, Y.; Zhou, Q.; Xu, J.; Qian, Q.; Ni, P.; Qian, Y. Mild Endoplasmic Reticulum Stress Protects Against Lipopolysaccharide-Induced Astrocytic Activation and Blood-Brain Barrier Hyperpermeability. Front. Cell. Neurosci. 2018, 12, 222. [Google Scholar] [CrossRef] [PubMed]
- Uciechowski, P.; Dempke, W.C.M. Interleukin-6: A Masterplayer in the Cytokine Network. Oncology 2020, 98, 131–137. [Google Scholar] [CrossRef]
- Erta, M.; Quintana, A.; Hidalgo, J. Interleukin-6, a Major Cytokine in the Central Nervous System. Int. J. Biol. Sci. 2012, 8, 1254–1266. [Google Scholar] [CrossRef] [PubMed]
- Kummer, K.K.; Zeidler, M.; Kalpachidou, T.; Kress, M. Role of IL-6 in the Regulation of Neuronal Development, Survival and Function. Cytokine 2021, 144, 155582. [Google Scholar] [CrossRef]
- Meares, G.P.; Liu, Y.; Rajbhandari, R.; Qin, H.; Nozell, S.E.; Mobley, J.A.; Corbett, J.A.; Benveniste, E.N. PERK-Dependent Activation of JAK1 and STAT3 Contributes to Endoplasmic Reticulum Stress-Induced Inflammation. Mol. Cell. Biol. 2014, 34, 3911–3925. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Plenge, R. JAK and STAT Signaling Molecules in Immunoregulation and Immune-Mediated Disease. Immunity 2012, 36, 542–550. [Google Scholar] [CrossRef]
- Studencka-Turski, M.; Çetin, G.; Junker, H.; Ebstein, F.; Krüger, E. Molecular Insight Into the IRE1α-Mediated Type I Interferon Response Induced by Proteasome Impairment in Myeloid Cells of the Brain. Front. Immunol. 2019, 10, 2900. [Google Scholar] [CrossRef] [PubMed]
- Bickel, M. The Role of Interleukin-8 in Inflammation and Mechanisms of Regulation. J. Periodontol. 1993, 64, 456–460. [Google Scholar] [PubMed]
- Mosiołek, A.; Pięta, A.; Jakima, S.; Zborowska, N.; Mosiołek, J.; Szulc, A. Effects of Antidepressant Treatment on Peripheral Biomarkers in Patients with Major Depressive Disorder (MDD). J. Clin. Med. 2021, 10, 1706. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Cui, G.; Zhu, M.; Kang, X.; Guo, H. Neuroinflammation in Alzheimer’s Disease: Chemokines Produced by Astrocytes and Chemokine Receptors. Int. J. Clin. Exp. Pathol. 2014, 7, 8342–8355. [Google Scholar] [PubMed]
- Joo, J.; Jeong, J.; Park, H.J. Blood Biomarkers in Patients with Parkinson’s Disease: A Review in Context of Anesthetic Care. Diagnostics 2023, 13, 693. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, L.C.; Hu, S.; Sheng, W.S.; Sutton, R.L.; Rockswold, G.L.; Peterson, P.K.; Chao, C.C. Cytokine Regulation of Human Microglial Cell IL-8 Production. J. Immunol. 1998, 160, 1944–1948. [Google Scholar] [CrossRef] [PubMed]
- Kronfol, Z. Cytokines and the Brain: Implications for Clinical Psychiatry. Am. J. Psychiatry 2000, 157, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.-J. Role of Interleukin 8 in Depression and Other Psychiatric Disorders. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2021, 106, 110173. [Google Scholar] [CrossRef]
- Krupkova, O.; Sadowska, A.; Kameda, T.; Hitzl, W.; Hausmann, O.N.; Klasen, J.; Wuertz-Kozak, K. P38 MAPK Facilitates Crosstalk Between Endoplasmic Reticulum Stress and IL-6 Release in the Intervertebral Disc. Front. Immunol. 2018, 9, 1706. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Studied Gene | Alternative Name | Encoded Protein | MICROGLIA | ASTROCYTES [31] * |
|---|---|---|---|---|
| Fold Change | Fold Change | |||
| Tunicamycin [0.5 µg/mL] | Tunicamycin [0.5 µg/mL] | |||
| ATF4 | CREB-2 | Activating transcription factor 4 | 2.33 ± 0.14 | 2.87 ± 0.28 |
| ATF6 | - | Activating transcription factor 6 | 2.00 ± 0.52 | 2.50 ± 0.31 |
| CREB3L1 | Oasis | CAMP responsive element binding protein 3 like 1 | 1.17 ± 0.37 | 1.49 ± 0.13 |
| DDIT3 | CHOP | DNA damage-inducible transcript 3/C/EBP-homologous protein | 12.22 ± 2.29 | 13.75 ± 1.08 |
| EDEM1 | EDEM | ER degradation enhancing alpha-mannosidase-like protein 1 | 1.83 ± 0.34 | 3.85 ± 0.35 |
| ERN1 | IRE1 | Endoplasmic reticulum to nucleus signaling 1/Inositol-requiring enzyme 1 | 1.41 ± 0.45 | 3.61 ± 0.37 |
| HSPA5 | GRP78 | Heat shock protein family A (Hsp70) member 5 | 10.14 ± 2.04 | 19.36 ± 1.26 |
 —increase;
—increase;  —decrease;
—decrease;  —no effect vs. untreated control.
—increase; —decrease; —no effect vs. untreated control.
—no effect vs. untreated control.
—increase; —decrease; —no effect vs. untreated control.| ER Stress | MILD | MODERATE | SEVERE | TNF-α/LPS | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ASTROCYTES | ||||||||||||
| Hours | 24 | 48 | 72 | 24 | 48 | 72 | 24 | 48 | 72 | 24 | 48 | 72 |
| IL-6 |  |  | |  | | |  |   | |  | | |
| IL-8 |  | | |  | | | | | | | | |
| MICROGLIA | ||||||||||||
| Hours | 24 | 48 | 72 | 24 | 48 | 72 | 24 | 48 | 72 | 24 | 48 | 72 |
| IL-6 | | | |  | | | - | | | | | |
| IL-8 | | | | | | | - | | | | | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sokołowska, P.; Wiktorowska-Owczarek, A.; Tambor, J.; Gawlak-Socka, S.; Kowalczyk, E.; Jóźwiak-Bębenista, M. Endoplasmic Reticulum Stress Differently Modulates the Release of IL-6 and IL-8 Cytokines in Human Glial Cells. Int. J. Mol. Sci. 2024, 25, 8687. https://doi.org/10.3390/ijms25168687
Sokołowska P, Wiktorowska-Owczarek A, Tambor J, Gawlak-Socka S, Kowalczyk E, Jóźwiak-Bębenista M. Endoplasmic Reticulum Stress Differently Modulates the Release of IL-6 and IL-8 Cytokines in Human Glial Cells. International Journal of Molecular Sciences. 2024; 25(16):8687. https://doi.org/10.3390/ijms25168687
Chicago/Turabian StyleSokołowska, Paulina, Anna Wiktorowska-Owczarek, Jakub Tambor, Sebastian Gawlak-Socka, Edward Kowalczyk, and Marta Jóźwiak-Bębenista. 2024. "Endoplasmic Reticulum Stress Differently Modulates the Release of IL-6 and IL-8 Cytokines in Human Glial Cells" International Journal of Molecular Sciences 25, no. 16: 8687. https://doi.org/10.3390/ijms25168687
APA StyleSokołowska, P., Wiktorowska-Owczarek, A., Tambor, J., Gawlak-Socka, S., Kowalczyk, E., & Jóźwiak-Bębenista, M. (2024). Endoplasmic Reticulum Stress Differently Modulates the Release of IL-6 and IL-8 Cytokines in Human Glial Cells. International Journal of Molecular Sciences, 25(16), 8687. https://doi.org/10.3390/ijms25168687