

Comparative Chloroplast Genomes Analysis Provided Adaptive Evolution Insights in Medicago ruthenica
 ,
,
Abstract
1. Introduction
2. Results
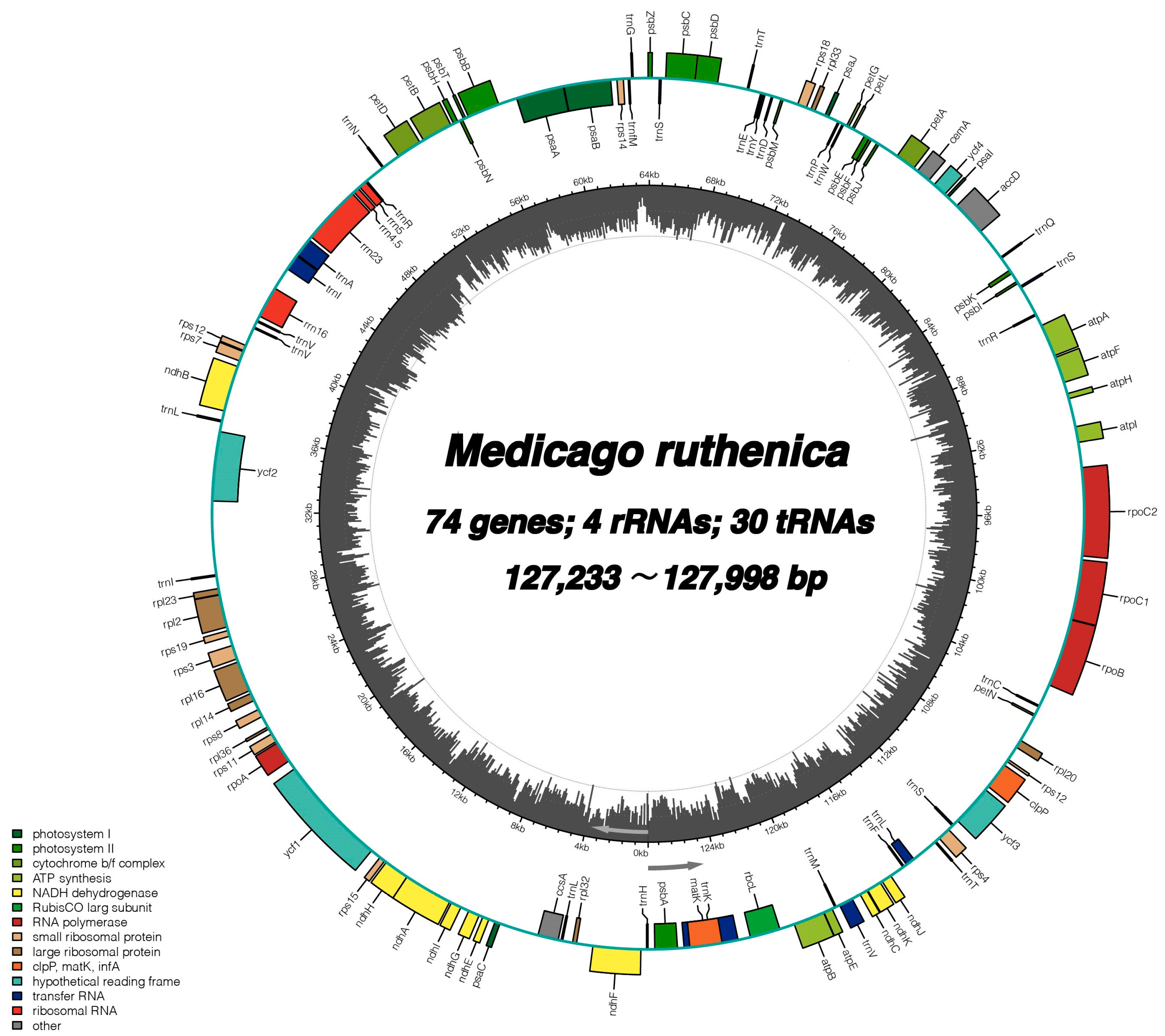
2.1. General Features of Medicago ruthenic Chloroplast Genomes
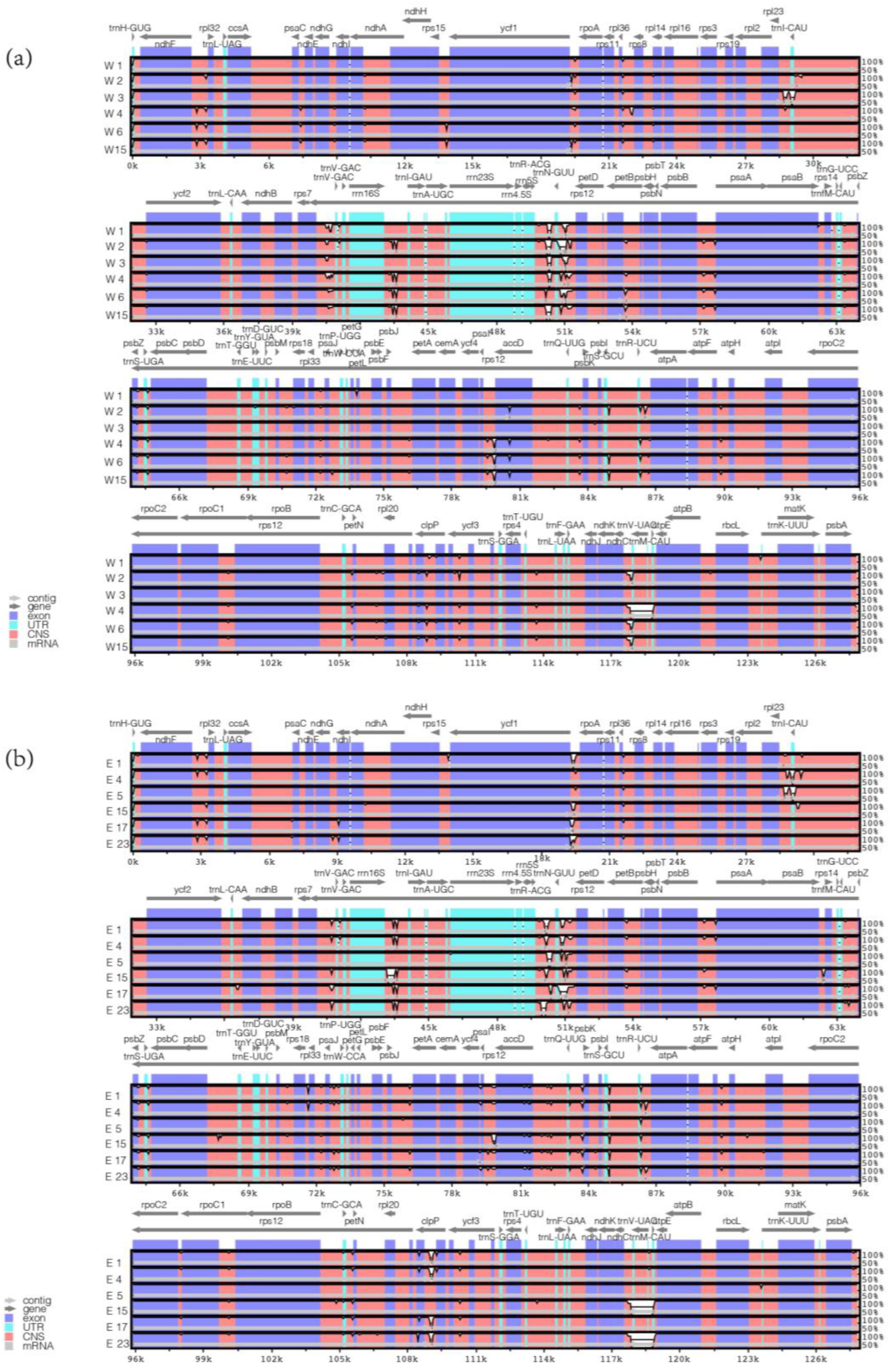
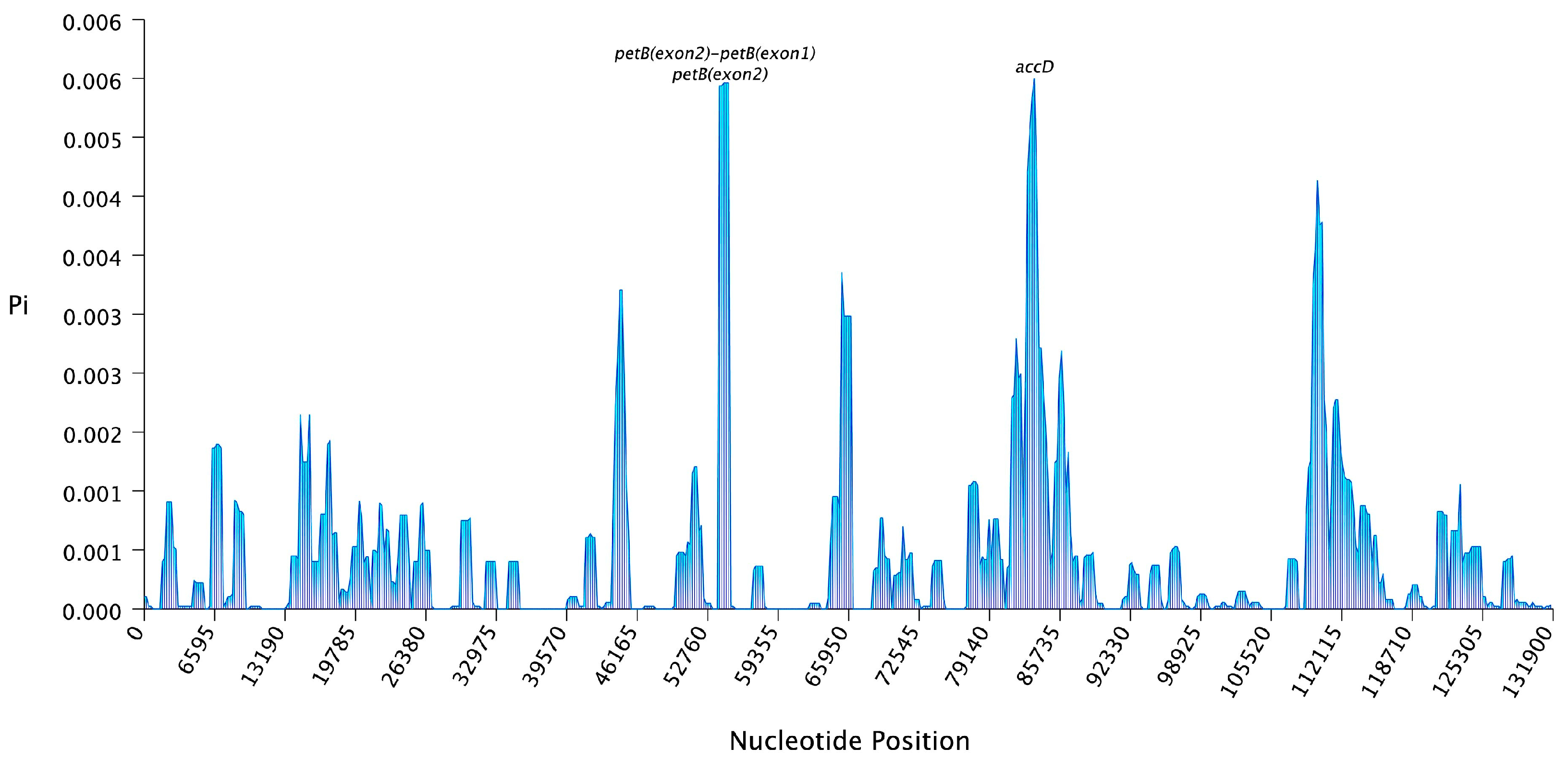
2.2. Comparative Chloroplast Genome Analysis
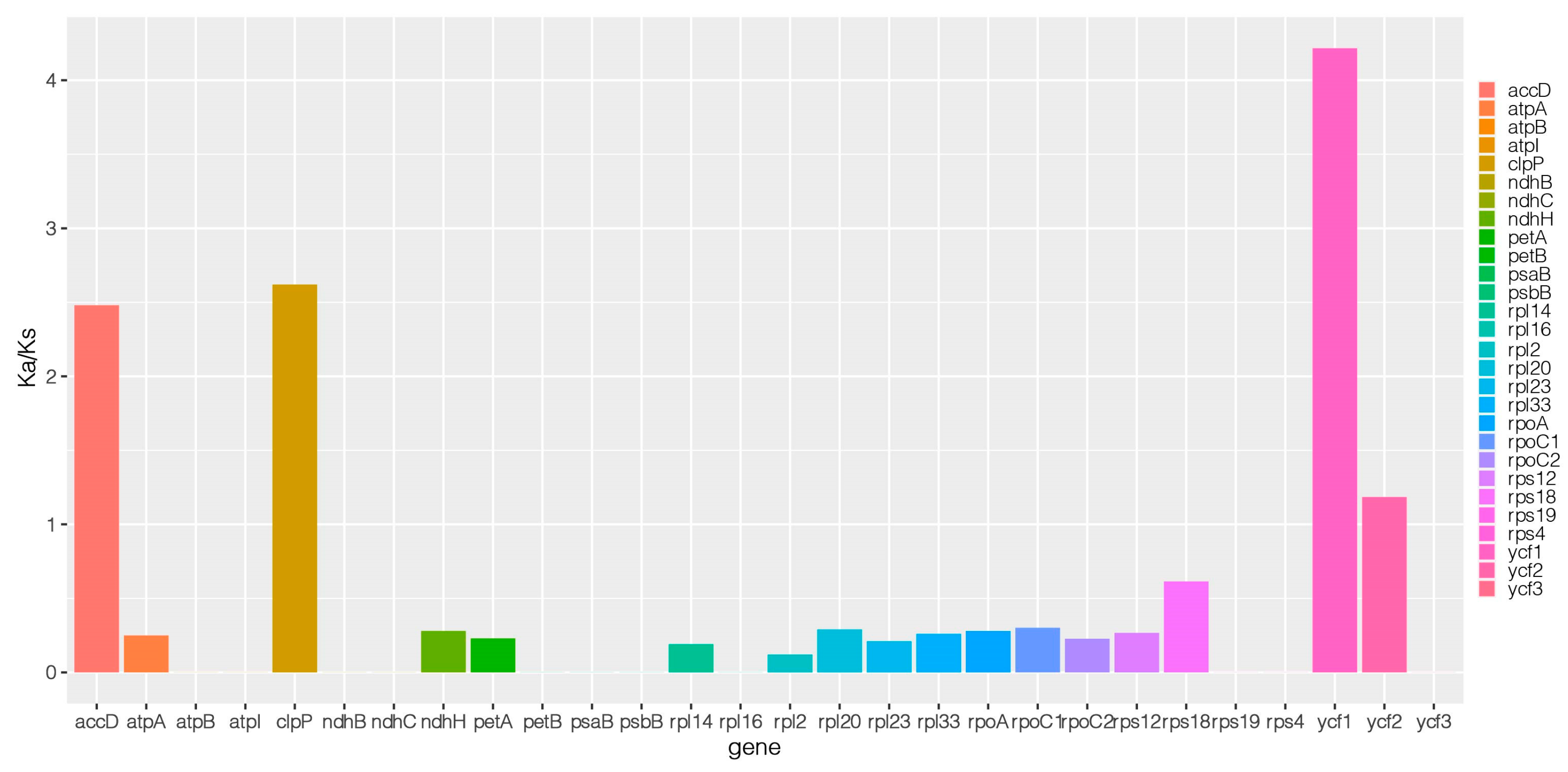
2.3. Adaptive Evolution of the Medicago ruthenica Chloroplast Genomes
2.4. Phylogenetic Analyses and Haplotype of Medicago ruthenica
2.5. Phylogenetic Tree Based on SNPs
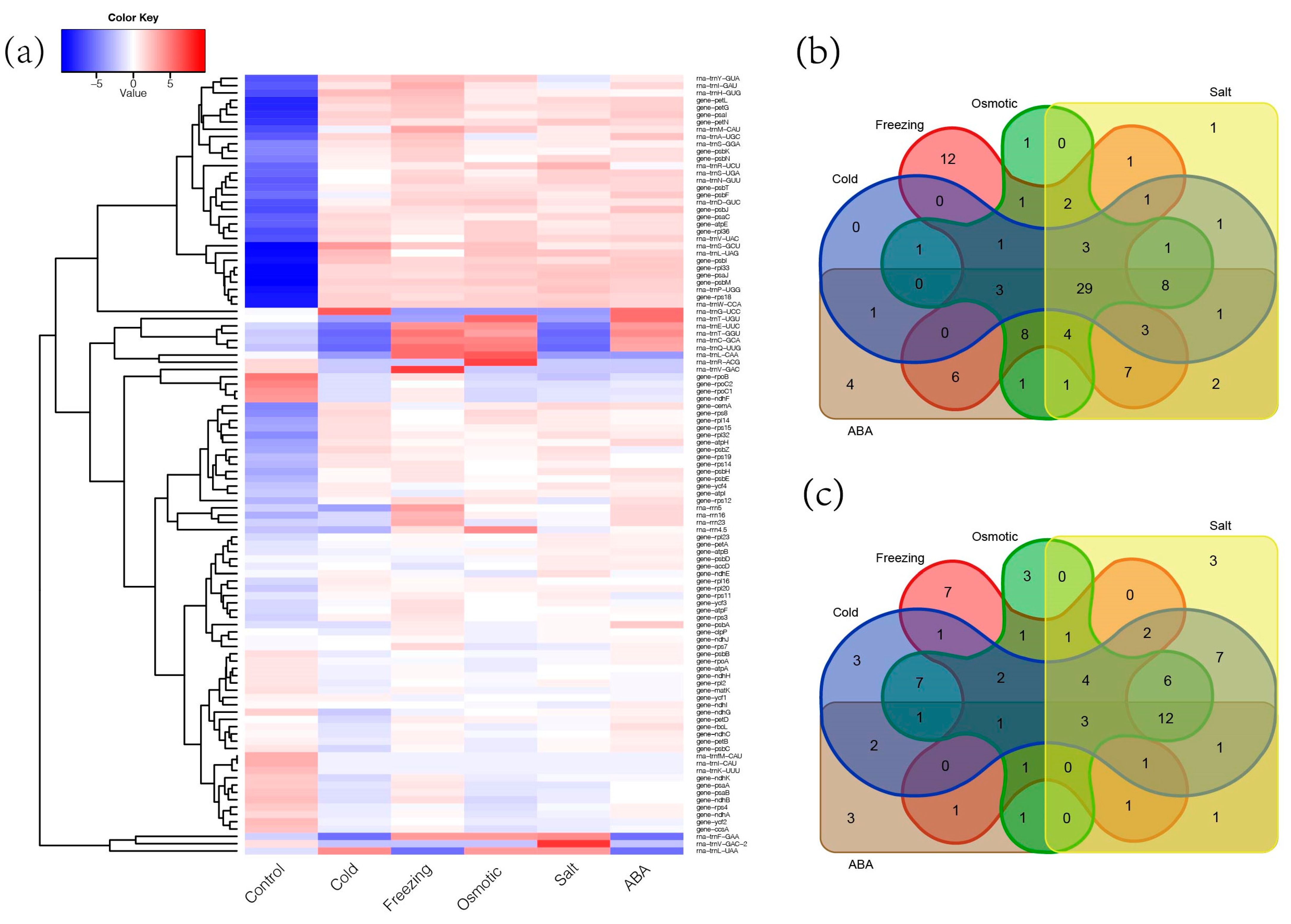
2.6. Response of Chloroplast Genes to Abiotic Stress in Medicago ruthenica
2.7. Analysis of RNA Editing Efficiency
3. Discussion
4. Materials and Methods
4.1. Assembly and Annotation of Chloroplast Genomes
4.2. Divergence Analysis of Medicago ruthenica
4.3. Phylogenetic Tree and Haplotype Analysis
4.4. Variants Calling
4.5. Population Structure and Phylogenetic Tree Based on SNPs
4.6. RNA-Seq Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lesins, K.; Lesins, I. Genus Medicago (Leguminosae): A Taxogenetic Study; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Yang, J.; Zheng, W.; Tian, Y.; Wu, Y.; Zhou, D. Effects of various mixed salt-alkaline stresses on growth, photosynthesis, and photosynthetic pigment concentrations of Medicago ruthenica seedlings. Photosynthetica 2011, 49, 275–284. [Google Scholar] [CrossRef]
- Guan, B.; Zhou, D.; Zhang, H.; Tian, Y.; Japhet, W.; Wang, P. Germination responses of Medicago ruthenica seeds to salinity, alkalinity, and temperature. J. Arid Environ. 2009, 73, 135–138. [Google Scholar] [CrossRef]
- Zhang, Y.; Shi, F.; Qiao, Y. Effects of EMS mutated Medicago ruthenica (L.) Sojak. cv. Zhilixing seeds on seedling emergence and seedling growth during germination stage. Grassl. Prataculture 2019, 31, 24–31. [Google Scholar]
- Wu, R.; Xu, B.; Shi, F. Leaf transcriptome analysis of Medicago ruthenica revealed its response and adaptive strategy to drought and drought recovery. BMC Plant Biol. 2022, 22, 562. [Google Scholar] [CrossRef] [PubMed]
- Campbell, T.; Bao, G.; Xia, Z. Completion of the agronomic evaluations of Medicago ruthenica [(L.) Ledebour] germplasm collected in Inner Mongolia. Genet. Resour. Crop Evol. 1999, 46, 477–484. [Google Scholar] [CrossRef]
- Yin, M.; Zhang, S.; Du, X.; Mateo, R.G.; Guo, W.; Li, A.; Wang, Z.; Wu, S.; Chen, J.; Liu, J. Genomic analysis of Medicago ruthenica provides insights into its tolerance to abiotic stress and demographic history. Mol. Ecol. Resour. 2021, 21, 1641–1657. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; LI, H.; LUO, X. Crossbreeding of Melilotoides ruthenicus and Medicago sativa. Acta Agrestia Sin. 2008, 16, 458. [Google Scholar]
- Wang, T.; Ren, L.; Li, C.; Zhang, D.; Zhang, X.; Zhou, G.; Gao, D.; Chen, R.; Chen, Y.; Wang, Z.; et al. The genome of a wild Medicago species provides insights into the tolerant mechanisms of legume forage to environmental stress. BMC Biol. 2021, 19, 96. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Li, Y.; Shang, X.; Zhong, H.; Dong, K.; Xia, F. The complete chloroplast genome of Medicago ruthenica cv. ‘Taihang’ (Fabaceae) from Shanxi, China. Mitochondrial DNA Part B 2021, 6, 2688–2690. [Google Scholar] [CrossRef]
- Xie, J.; Mao, J.; Li, Z.; Liang, C.; Ou, W.; Tang, J.; Liu, H.; Wang, H.; Ji, Z.; Kazhuocairang; et al. Complete chloroplast genome of a high-quality forage in north China, Medicago ruthenica (Fabaceae: Trifolieae). Mitochondrial DNA B Resour. 2021, 6, 29–30. [Google Scholar] [CrossRef]
- Xiao, S.; Xu, P.; Deng, Y.; Dai, X.; Zhao, L.; Heider, B.; Zhang, A.; Zhou, Z.; Cao, Q. Comparative analysis of chloroplast genomes of cultivars and wild species of sweetpotato (Ipomoea batatas [L.] Lam). BMC Genom. 2021, 22, 1–12. [Google Scholar]
- Martin, W.F.; Garg, S.; Zimorski, V. Endosymbiotic theories for eukaryote origin. Philos. Trans. R. Soc. Lond B Biol. Sci. 2015, 370, 20140330. [Google Scholar] [CrossRef] [PubMed]
- Sato, N. Are Cyanobacteria an Ancestor of Chloroplasts or Just One of the Gene Donors for Plants and Algae? Genes 2021, 12, 823. [Google Scholar] [CrossRef]
- Brunkard, J.O.; Runkel, A.M.; Zambryski, P.C. Chloroplasts extend stromules independently and in response to internal redox signals. Proc. Natl. Acad. Sci. USA 2015, 112, 10044–10049. [Google Scholar] [CrossRef]
- Kaiser, E.; Correa Galvis, V.; Armbruster, U. Efficient photosynthesis in dynamic light environments: A chloroplast’s perspective. Biochem. J. 2019, 476, 2725–2741. [Google Scholar] [CrossRef]
- Medina-Puche, L.; Tan, H.; Dogra, V.; Wu, M.; Rosas-Diaz, T.; Wang, L.; Ding, X.; Zhang, D.; Fu, X.; Kim, C.; et al. A Defense Pathway Linking Plasma Membrane and Chloroplasts and Co-opted by Pathogens. Cell 2020, 182, 1109–1124.e25. [Google Scholar] [CrossRef]
- Llorente, B.; Torres-Montilla, S.; Morelli, L.; Florez-Sarasa, I.; Matus, J.T.; Ezquerro, M.; D’Andrea, L.; Houhou, F.; Majer, E.; Picó, B.; et al. Synthetic conversion of leaf chloroplasts into carotenoid-rich plastids reveals mechanistic basis of natural chromoplast development. Proc. Natl. Acad. Sci. USA 2020, 117, 21796–21803. [Google Scholar] [CrossRef] [PubMed]
- Dobrogojski, J.; Adamiec, M.; Luciński, R. The chloroplast genome: A review. Acta Physiol. Plant. 2020, 42, 98. [Google Scholar] [CrossRef]
- Qu, X.; Zou, D.; Zhang, R.; Stull, G.; Yi, T. Progress, challenge and prospect of plant plastome annotation. Front. Plant Sci. 2023, 14, 1166140. [Google Scholar] [CrossRef]
- Choi, I.; Jansen, R.; Ruhlman, T. Lost and Found: Return of the Inverted Repeat in the Legume Clade Defined by Its Absence. Genome Biol. Evol. 2019, 11, 1321–1333. [Google Scholar] [CrossRef]
- Zhang, T.; Chen, X.; Yan, W.; Li, M.; Huang, W.; Liu, Q.; Li, Y.; Guo, C.; Shu, Y. Comparative Analysis of Chloroplast Pan-Genomes and Transcriptomics Reveals Cold Adaptation in Medicago sativa. Int. J. Mol. Sci. 2024, 25, 1776. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liao, X.; Gu, C.; Xiang, K.; Wang, J.; Li, S.; Tembrock, L.; Wu, Z.; He, W. The Asian lotus (Nelumbo nucifera) pan-plastome: Diversity and divergence in a living fossil grown for seed, rhizome, and aesthetics. Ornam. Plant Res. 2022, 2, 2. [Google Scholar] [CrossRef]
- Chen, Q.; Chen, L.; Teixeira da Silva, J.A.; Yu, X. The plastome reveals new insights into the evolutionary and domestication history of peonies in East Asia. BMC Plant Biol. 2023, 23, 243. [Google Scholar] [CrossRef] [PubMed]
- Ruf, S.; Forner, J.; Hasse, C.; Kroop, X.; Seeger, S.; Schollbach, L.; Schadach, A.; Bock, R. High-efficiency generation of fertile transplastomic Arabidopsis plants. Nat. Plants 2019, 5, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Burrows, P.; Sazanov, L.; Svab, Z.; Maliga, P.; Nixon, P. Identification of a functional respiratory complex in chloroplasts through analysis of tobacco mutants containing disrupted plastid ndh genes. Embo J. 1998, 17, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Ren, D.; Xie, W.; Xu, Q.; Hu, J.; Zhu, L.; Zhang, G.; Zeng, D.; Qian, Q. LSL1 controls cell death and grain production by stabilizing chloroplast in rice. Sci. China Life Sci. 2022, 65, 2148–2161. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Bai, S.; Zhang, Z.; Zheng, F.; Song, L.; Wen, L.; Guo, M.; Cheng, G.; Yao, W.; Gao, Y.; et al. Comparative analysis of chloroplast genomes of 29 tomato germplasms: Genome structures, phylogenetic relationships, and adaptive evolution. Front. Plant Sci. 2023, 14, 1179009. [Google Scholar] [CrossRef]
- Jin, J.; Yu, W.; Yang, J.; Song, Y.; dePamphilis, C.; Yi, T.; Li, D. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.; Wojciechowski, M.; Sanniyasi, E.; Lee, S.; Daniell, H. Complete plastid genome sequence of the chickpea (Cicer arietinum) and the phylogenetic distribution of rps12 and clpP intron losses among legumes (Leguminosae). Mol. Phylogenet Evol. 2008, 48, 1204–1217. [Google Scholar] [CrossRef]
- Jo, I.; Han, S.; Shim, D.; Ryu, H.; Hyun, T.; Lee, Y.; Kim, D.; So, Y.; Chung, J. Complete Chloroplast Genome of the Inverted Repeat-Lacking Species Vicia bungei and Development of Polymorphic Simple Sequence Repeat Markers. Front. Plant Sci. 2022, 13, 891783. [Google Scholar] [CrossRef]
- Palmer, J. Plastid chromosomes: Structure and evolution. Mol. Biol. Plast. 1991, 7, 5–53. [Google Scholar]
- Huang, X.; Coulibaly, D.; Tan, W.; Ni, Z.; Shi, T.; Li, H.; Hayat, F.; Gao, Z. The analysis of genetic structure and characteristics of the chloroplast genome in different Japanese apricot germplasm populations. BMC Plant Biol. 2022, 22, 354. [Google Scholar] [CrossRef]
- Cai, X.; Landis, J.; Wang, H.; Wang, J.; Zhu, Z.; Wang, H. Plastome structure and phylogenetic relationships of Styracaceae (Ericales). BMC Ecol. Evol. 2021, 21, 103. [Google Scholar] [CrossRef]
- Kode, V.; Mudd, E.; Iamtham, S.; Day, A. The tobacco plastid accD gene is essential and is required for leaf development. Plant J. 2005, 44, 237–244. [Google Scholar] [CrossRef]
- Katayama, H.; Tachibana, M.; Iketani, H.; Zhang, S.; Uematsu, C. Phylogenetic utility of structural alterations found in the chloroplast genome of pear: Hypervariable regions in a highly conserved genome. Tree Genet. Genomes 2012, 8, 313–326. [Google Scholar] [CrossRef]
- Erixon, P.; Oxelman, B. Whole-gene positive selection, elevated synonymous substitution rates, duplication, and indel evolution of the chloroplast clpP1 gene. PLoS ONE 2008, 3, e1386. [Google Scholar] [CrossRef]
- Zhou, T.; Ning, K.; Mo, Z.; Zhang, F.; Zhou, Y.; Chong, X.; Zhang, D.; El-Kassaby, Y.; Bian, J.; Chen, H. Complete chloroplast genome of Ilex dabieshanensis: Genome structure, comparative analyses with three traditional Ilex tea species, and its phylogenetic relationships within the family Aquifoliaceae. PLoS ONE 2022, 17, e0268679. [Google Scholar] [CrossRef]
- Jin, Y.; Wang, Y.; Gao, Y.; Zhou, L.; Wang, Y.; Yuan, Q.; Dong, W. Complete chloroplast genome of Ligustrum lucidum and highly variable marker identification for Ligustrum. Zhongguo Zhong Yao Za Zhi 2022, 47, 1847–1856. [Google Scholar]
- Raman, G.; Nam, G.; Park, S. Extensive reorganization of the chloroplast genome of Corydalis platycarpa: A comparative analysis of their organization and evolution with other Corydalis plastomes. Front. Plant Sci. 2022, 13, 1043740. [Google Scholar] [CrossRef]
- Jiang, H.; Tian, J.; Yang, J.; Dong, X.; Zhong, Z.; Mwachala, G.; Zhang, C.; Hu, G.; Wang, Q. Comparative and phylogenetic analyses of six Kenya Polystachya (Orchidaceae) species based on the complete chloroplast genome sequences. BMC Plant Biol. 2022, 22, 177. [Google Scholar] [CrossRef]
- Lawrie, D.; Messer, P.; Hershberg, R.; Petrov, D. Strong purifying selection at synonymous sites in D. melanogaster. PLoS Genet. 2013, 9, e1003527. [Google Scholar] [CrossRef]
- Li, H.; Xiao, W.; Tong, T.; Li, Y.; Zhang, M.; Lin, X.; Zou, X.; Wu, Q.; Guo, X. The specific DNA barcodes based on chloroplast genes for species identification of Orchidaceae plants. Sci. Rep. 2021, 11, 1424. [Google Scholar] [CrossRef]
- Xia, L.; Wang, H.; Zhao, X.; Obel, H.O.; Yu, X.; Lou, Q.; Chen, J.; Cheng, C. Chloroplast Pan-Genomes and Comparative Transcriptomics Reveal Genetic Variation and Temperature Adaptation in the Cucumber. Int. J. Mol. Sci. 2023, 24, 8943. [Google Scholar] [CrossRef]
- Abdel-Ghany, S.; LaManna, L.; Harroun, H.; Maliga, P.; Sloan, D. Rapid sequence evolution is associated with genetic incompatibilities in the plastid Clp complex. Plant Mol. Biol. 2022, 108, 277–287. [Google Scholar] [CrossRef]
- Hu, Y.; Woeste, K.; Zhao, P. Completion of the Chloroplast Genomes of Five Chinese Juglans and Their Contribution to Chloroplast Phylogeny. Front. Plant Sci. 2016, 7, 1955. [Google Scholar] [CrossRef]
- Li, C.; Liu, Y.; Lin, F.; Zheng, Y.; Huang, P. Characterization of the complete chloroplast genome sequences of six Dalbergia species and its comparative analysis in the subfamily of Papilionoideae (Fabaceae). PeerJ 2022, 10, e13570. [Google Scholar] [CrossRef]
- Li, H.; Li, Z.; Cai, L.; Shi, W.; Mi, F.; Shi, F. Analysis of genetic diversity of Ruthenia Medic (Medicago ruthenica (L.) Trautv.) in Inner Mongolia using ISSR and SSR markers. Genet. Resour. Crop Evol. 2013, 60, 1687–1694. [Google Scholar] [CrossRef]
- Zhang, A.; Jiang, X.; Zhang, F.; Wang, T.; Zhang, X. Dynamic response of RNA editing to temperature in grape by RNA deep sequencing. Funct. Integr. Genom. 2020, 20, 421–432. [Google Scholar] [CrossRef]
- Nishimura, K.; Wijk, K. Organization, function and substrates of the essential Clp protease system in plastids. Biochim. Biophys. Acta 2015, 1847, 915–930. [Google Scholar] [CrossRef]
- Lu, C.; Li, L.; Liu, X.; Chen, M.; Wan, S.; Li, G. Salt Stress Inhibits Photosynthesis and Destroys Chloroplast Structure by Downregulating Chloroplast Development-Related Genes in Robinia pseudoacacia Seedlings. Plants 2023, 12, 1283. [Google Scholar] [CrossRef]
- Czarnocka, W.; Karpiński, S. Friend or foe? Reactive oxygen species production, scavenging and signaling in plant response to environmental stresses. Free Radic. Biol. Med. 2018, 122, 4–20. [Google Scholar] [CrossRef]
- Gao, Y.; Thiele, W.; Saleh, O.; Scossa, F.; Arabi, F.; Zhang, H.; Sampathkumar, A.; Kühn, K.; Fernie, A.; Bock, R.; et al. Chloroplast translational regulation uncovers nonessential photosynthesis genes as key players in plant cold acclimation. Plant Cell 2022, 34, 2056–2079. [Google Scholar] [CrossRef]
- Wang, S.; Blumwald, E. Stress-induced chloroplast degradation in Arabidopsis is regulated via a process independent of autophagy and senescence-associated vacuoles. Plant Cell 2014, 26, 4875–4888. [Google Scholar] [CrossRef]
- Liu, H.; Ren, D.; Jiang, L.; Li, X.; Yao, Y.; Mi, L.; Chen, W.; Mo, A.; Jiang, N.; Yang, J.; et al. A Natural Variation in PLEIOTROPIC DEVELOPMENTAL DEFECTS Uncovers a Crucial Role for Chloroplast tRNA Modification in Translation and Plant Development. Plant Cell 2020, 32, 2345–2366. [Google Scholar] [CrossRef]
- Bolger, A.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Marçais, G.; Delcher, A.; Phillippy, A.; Coston, R.; Salzberg, S.; Zimin, A. MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 2018, 14, e1005944. [Google Scholar] [CrossRef]
- Shi, L.; Chen, H.; Jiang, M.; Wang, L.; Wu, X.; Huang, L.; Liu, C. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 2019, 47, W65–W73. [Google Scholar] [CrossRef]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef]
- Zheng, S.; Poczai, P.; Hyvnen, J.; Tang, J.; Amiryousefi, A. Chloroplot: An Online Program for the Versatile Plotting of Organelle Genomes. Front. Genet. 2020, 11, 576124. [Google Scholar] [CrossRef]
- Frazer, K.; Pachter, L.; Poliakov, A.; Rubin, E.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.; Schmidt, H.; Chernomor, O.; Schrempf, D.; Woodhams, M.; Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A. FigTree, a Graphical Viewer of Phylogenetic Trees. 2009. Available online: http://tree.bio.ed.ac.uk/software/figtree (accessed on 5 August 2024).
- Leigh, J.; Bryant, D.; Nakagawa, S. POPART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Zhang, Z. KaKs_Calculator 3.0: Calculating Selective Pressure on Coding and Non-coding Sequences. Genom. Proteom. Bioinform. 2022, 20, 536–540. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Subgroup, G.P.D.P. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.; Irizarry, R.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Liu, W.; Aljohi, H.; Alromaih, S.; Alanazi, I.; Lin, Q.; Yu, J.; Hu, S. REDO: RNA Editing Detection in Plant Organelles Based on Variant Calling Results. J. Comput. Biol. 2018, 25, 509–516. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Category | Functional Group | Gene Name |
|---|---|---|
| Photosynthesis | Subunits of photosystem I | psaA, psaB, psaC, psaI, psaJ |
| Subunits of photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbM, psbN, psbT, psbZ | |
| Subunits of NADH dehydrogenase | ndhA *, ndhB *, ndhC, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK | |
| Subunits of cytochrome b/f complex | petA, petB *, petD *, petG, petL, petN | |
| Subunits of ATP synthase | atpA, atpB, atpE, atpF *, atpH, atpI | |
| Large subunit of rubisco | rbcL | |
| Subunits photochlorophyllide reductase | - | |
| Self-replication | Proteins of large ribosomal subunit | rpl14, rpl16 *, rpl2 *, rpl20, rpl23, rpl32, rpl33, rpl36 |
| Proteins of small ribosomal subunit | rps11, rps12 *, rps14, rps15, rps18, rps19, rps3, rps4, rps7, rps8 | |
| Subunits of RNA polymerase | rpoA, rpoB, rpoC1 *, rpoC2 | |
| Ribosomal RNAs | rrn16S, rrn23S, rrn4.5S, rrn5S | |
| Transfer RNAs | trnA-UGC *, trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnG-UCC, trnH-GUG, trnI-CAU, trnI-GAU *, trnK-UUU *, trnL-CAA, trnL-UAA *, trnL-UAG, trnM-CAU, trnN-GUU, trnP-UGG, trnQ-UUG, trnR-ACG, trnR-UCU, trnS-GCU, trnS-GGA, trnS-UGA, trnT-GGU, trnT-UGU, trnV-GAC(2), trnV-UAC *, trnW-CCA, trnY-GUA, trnfM-CAU | |
| Other genes | Maturase | matK |
| Protease | clpP * | |
| Envelope membrane protein | cemA | |
| Acetyl-CoA carboxylase | accD | |
| c-type cytochrome synthesis gene | ccsA | |
| Translation initiation factor | - | |
| other | - | |
| Genes of unknown function | Conserved hypothetical chloroplast ORF | ycf1, ycf2, ycf3 **, ycf4 |
| Category | Number | Varieties |
|---|---|---|
| Hap_33 | 2 | W5, W6 |
| Hap_37 | 2 | W14, W29 |
| Hap_45 | 4 | W10, W12, W19, W20 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, T.; Li, M.; Zhu, X.; Li, S.; Guo, M.; Guo, C.; Shu, Y. Comparative Chloroplast Genomes Analysis Provided Adaptive Evolution Insights in Medicago ruthenica. Int. J. Mol. Sci. 2024, 25, 8689. https://doi.org/10.3390/ijms25168689
Zhang T, Li M, Zhu X, Li S, Guo M, Guo C, Shu Y. Comparative Chloroplast Genomes Analysis Provided Adaptive Evolution Insights in Medicago ruthenica. International Journal of Molecular Sciences. 2024; 25(16):8689. https://doi.org/10.3390/ijms25168689
Chicago/Turabian StyleZhang, Tianxiang, Manman Li, Xiaoyue Zhu, Shuaixian Li, Meiyan Guo, Changhong Guo, and Yongjun Shu. 2024. "Comparative Chloroplast Genomes Analysis Provided Adaptive Evolution Insights in Medicago ruthenica" International Journal of Molecular Sciences 25, no. 16: 8689. https://doi.org/10.3390/ijms25168689
APA StyleZhang, T., Li, M., Zhu, X., Li, S., Guo, M., Guo, C., & Shu, Y. (2024). Comparative Chloroplast Genomes Analysis Provided Adaptive Evolution Insights in Medicago ruthenica. International Journal of Molecular Sciences, 25(16), 8689. https://doi.org/10.3390/ijms25168689